
Carboplatin-Induced Thrombocytopenia through JAK2 Downregulation, S-Phase Cell Cycle Arrest and Apoptosis in Megakaryocytes

 , and
, and
Abstract
:
1. Introduction
2. Results
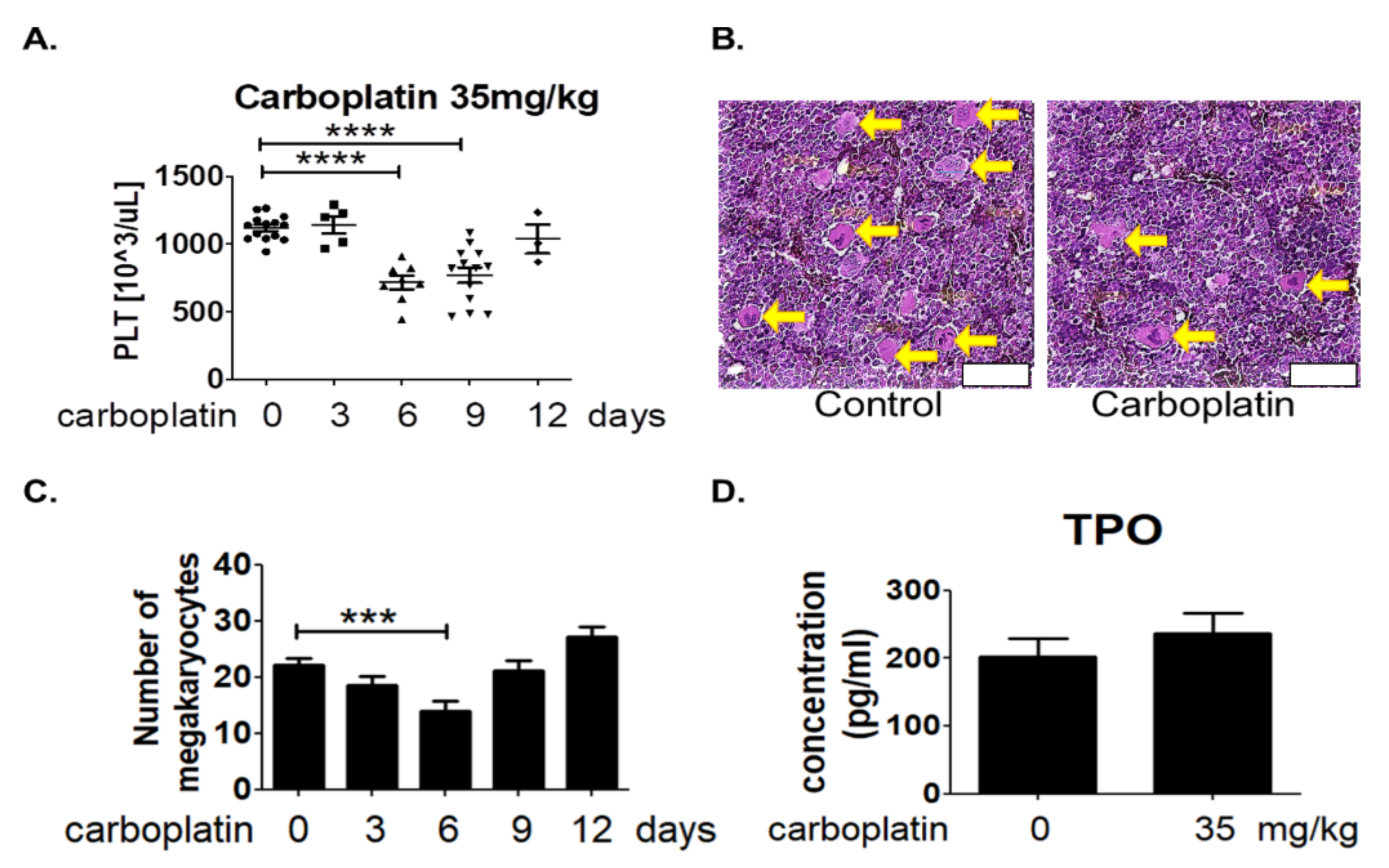
2.1. Carboplatin Induced Thrombocytopenia in SD Rats
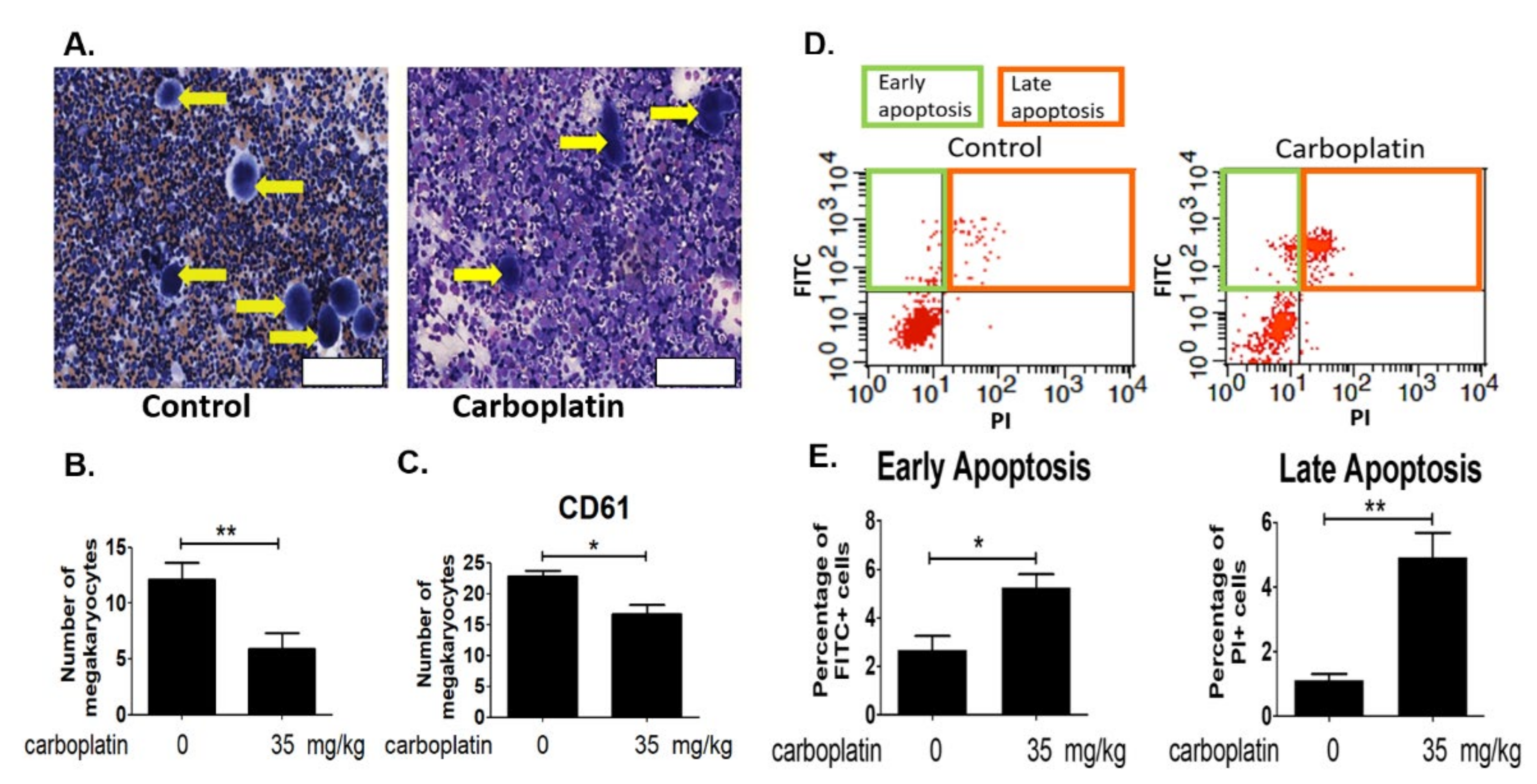
2.2. Carboplatin Significantly Reduced the Number of Megakaryocytes in Rat Bone Marrow
2.3. Carboplatin Induced Apoptosis of Megakaryocytes in Rat Bone Marrow
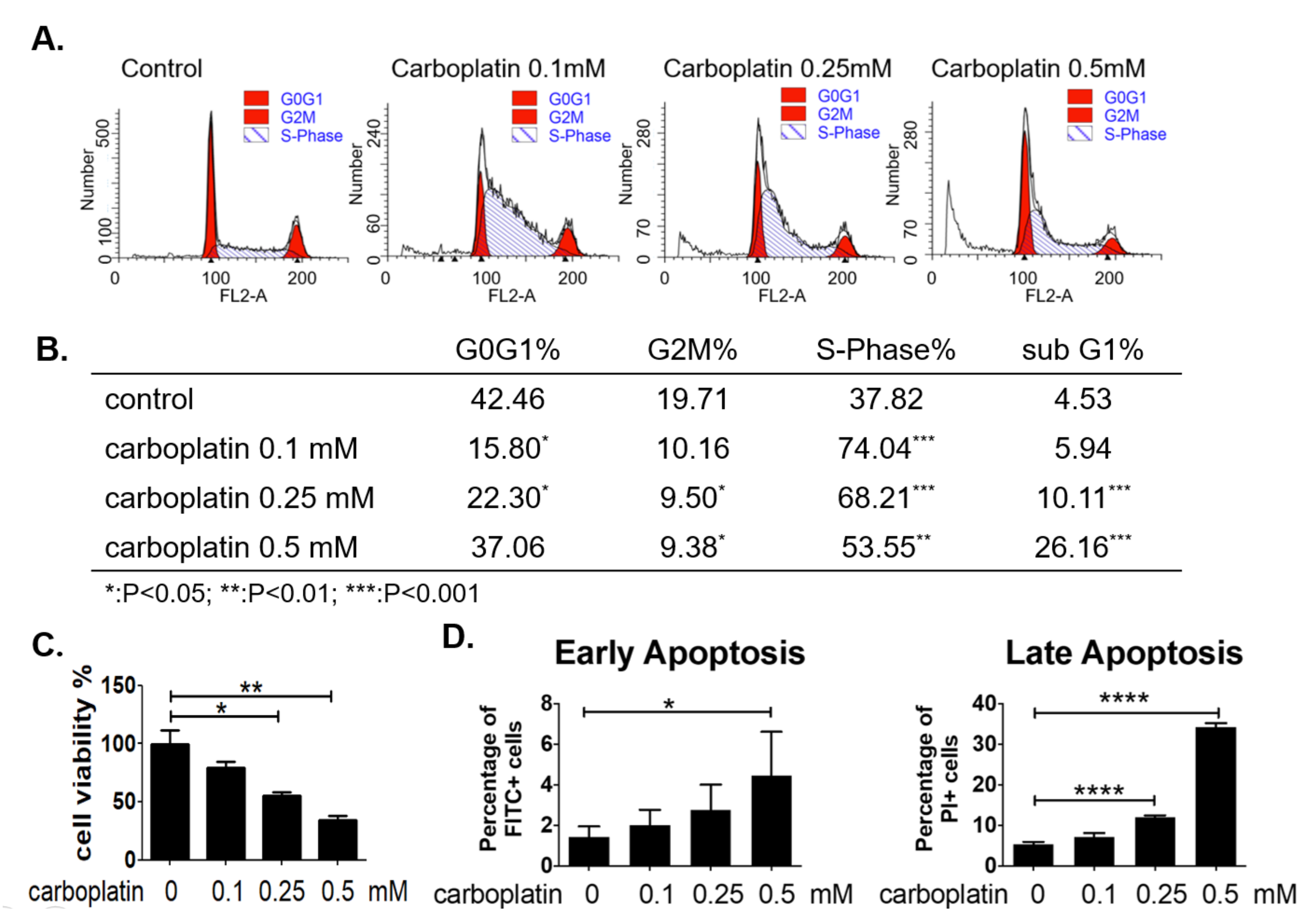
2.4. Carboplatin Caused S-Phase Arrest and Apoptosis of Human Megakaryocytic Cells In Vitro
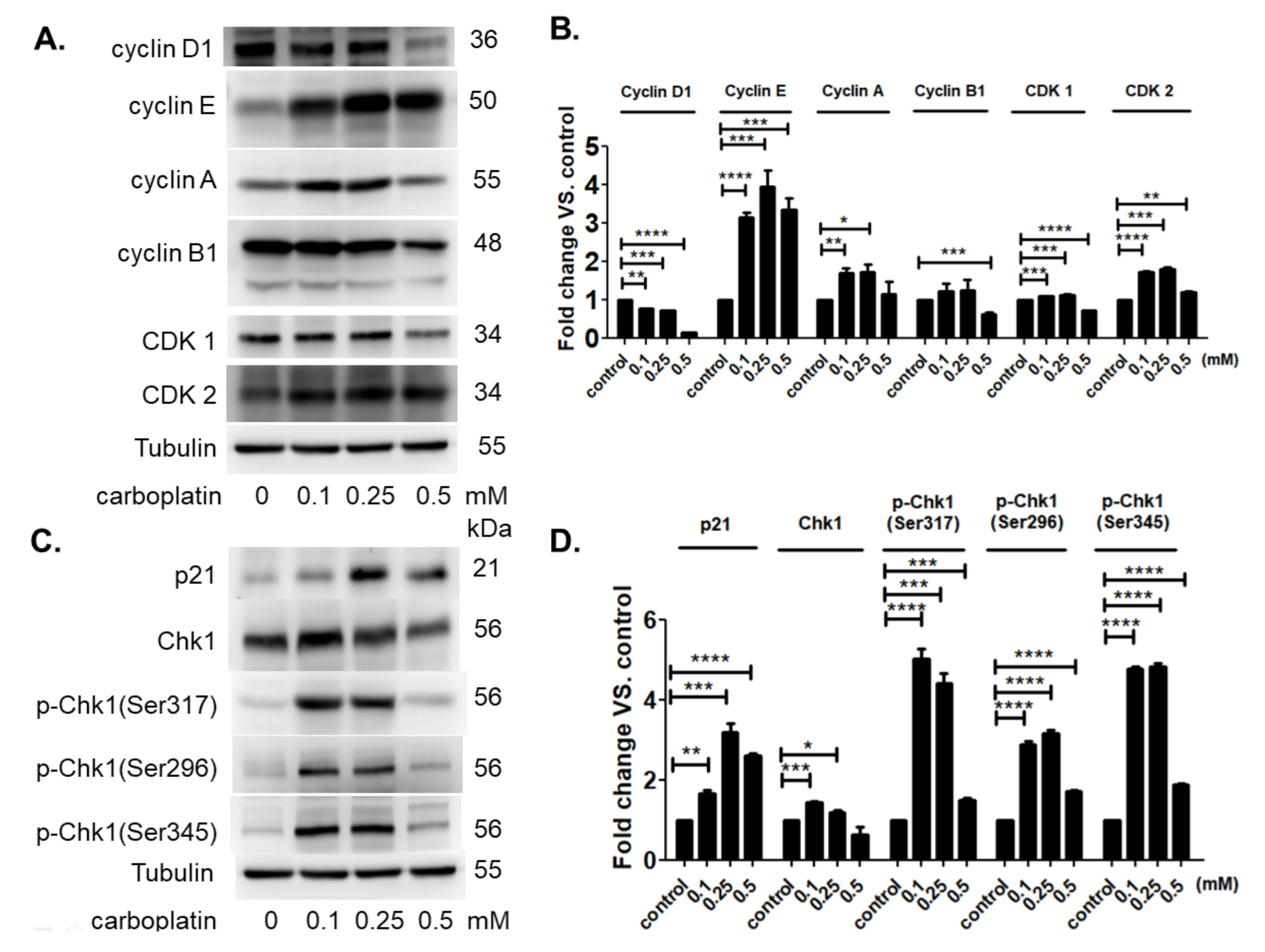
2.5. Carboplatin Modulated the Protein Expression of Cell Cycle-Dependent Genes and Cell Cycle Inhibitors
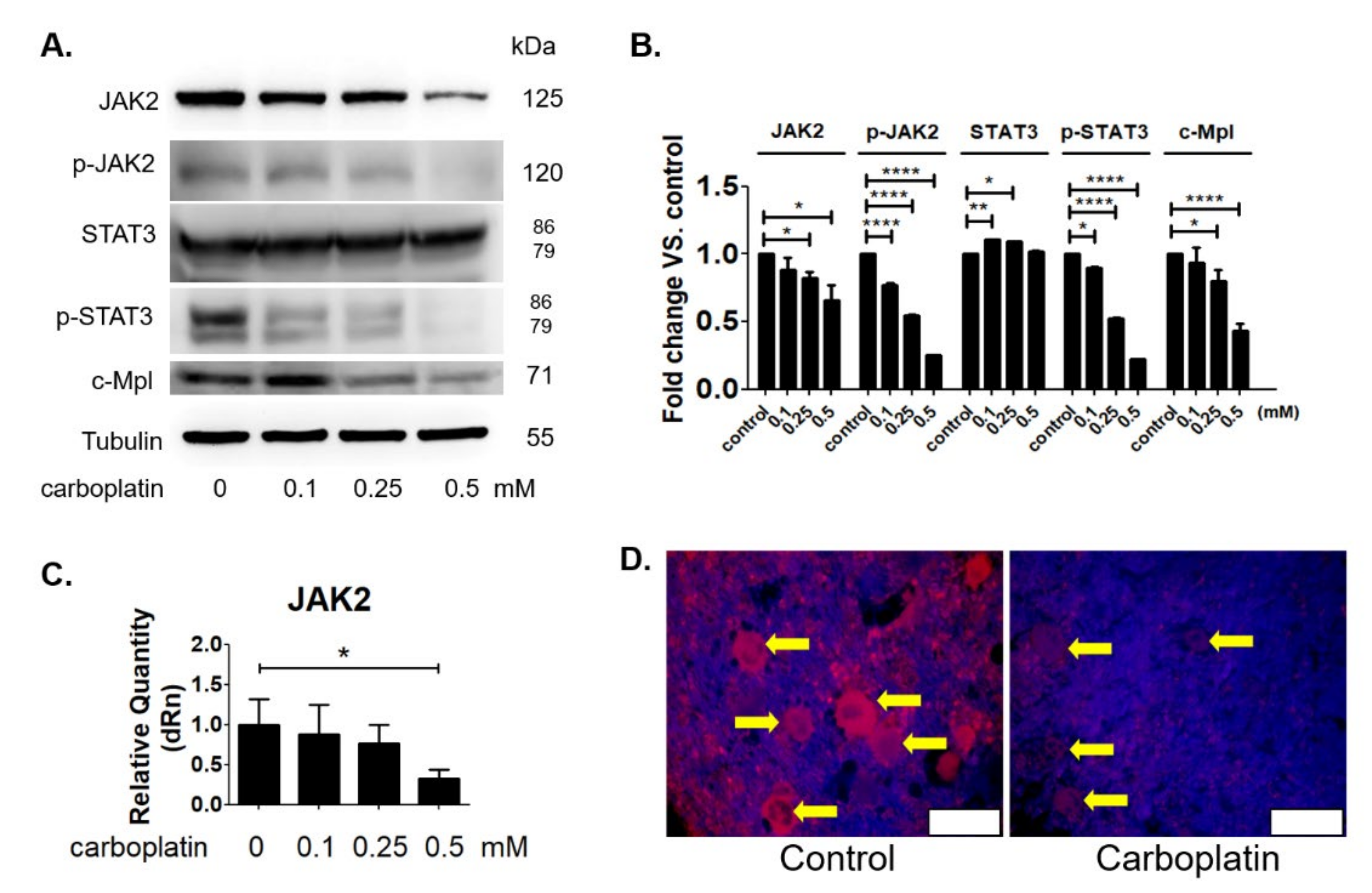
2.6. Carboplatin Suppressed the JAK2 Protein Expression Both In Vitro and In Vivo
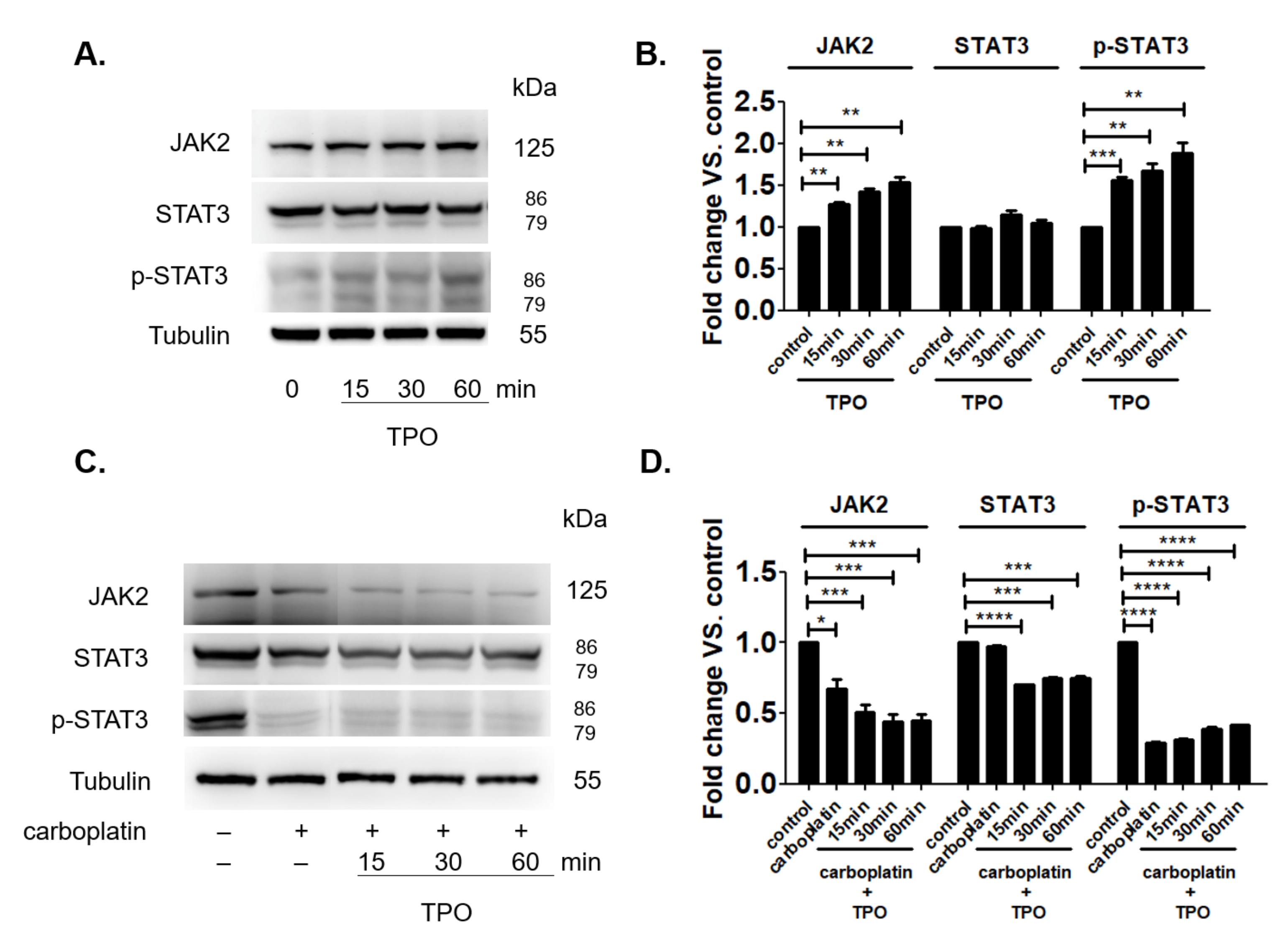
2.7. TPO Could Not Activate the JAK2 Signal Pathway in the Presence of Carboplatin
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Animal Experiments
4.3. ELISA
4.4. Cell Culture
4.5. Flow Cytometry
4.6. Western Blot Analysis
4.7. RNA Isolation and RT/Real-Time PCR
4.8. Immunohistochemistry
4.9. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kuter, D.J. Managing thrombocytopenia associated with cancer chemotherapy. Oncology 2015, 29, 282–294. [Google Scholar] [PubMed]
- Elting, L.S.; Rubenstein, E.B.; Martin, C.G.; Kurtin, D.; Rodriguez, S.; Laiho, E.; Kanesan, K.; Cantor, S.B.; Benjamin, R.S. Incidence, cost, and outcomes of bleeding and chemotherapy dose modification among solid tumor patients with chemotherapy-induced thrombocytopenia. J. Clin. Oncol. 2001, 19, 1137–1146. [Google Scholar] [CrossRef] [PubMed]
- Joseph, N.; Clark, R.M.; Dizon, D.S.; Lee, M.S.; Goodman, A.; Boruta, D., Jr.; Schorge, J.O.; Del Carmen, M.G.; Growdon, W.B. Delay in chemotherapy administration impacts survival in elderly patients with epithelial ovarian cancer. Gynecol. Oncol. 2015, 137, 401–405. [Google Scholar] [CrossRef] [PubMed]
- Liutkauskiene, S.; Janciauskiene, R.; Jureniene, K.; Grizas, S.; Malonyte, R.; Juozaityte, E. Retrospective analysis of the impact of platinum dose reduction and chemotherapy delays on the outcomes of stage III ovarian cancer patients. BMC Cancer 2015, 15, 105. [Google Scholar] [CrossRef] [PubMed]
- Tamamyan, G.; Danielyan, S.; Lambert, M.P. Chemotherapy induced thrombocytopenia in pediatric oncology. Crit. Rev. Oncol. Hematol. 2016, 99, 299–307. [Google Scholar] [CrossRef]
- Ten Berg, M.J.; van den Bemt, P.M.; Shantakumar, S.; Bennett, D.; Voest, E.E.; Huisman, A.; van Solinge, W.W.; Egberts, T.C. Thrombocytopenia in adult cancer patients receiving cytotoxic chemotherapy: Results from a retrospective hospital-based cohort study. Drug Saf. 2011, 34, 1151–1160. [Google Scholar] [CrossRef]
- Rabik, C.A.; Dolan, M.E. Molecular mechanisms of resistance and toxicity associated with platinating agents. Cancer Treat. Rev. 2007, 33, 9–23. [Google Scholar] [CrossRef]
- McManus, P.M.; Weiss, L. Busulfan-induced chronic bone marrow failure: Changes in cortical bone, marrow stromal cells, and adherent cell colonies. Blood 1984, 64, 1036–1041. [Google Scholar] [CrossRef]
- DeZern, A.E.; Petri, M.; Drachman, D.B.; Kerr, D.; Hammond, E.R.; Kowalski, J.; Tsai, H.L.; Loeb, D.M.; Anhalt, G.; Wigley, F.; et al. High-dose cyclophosphamide without stem cell rescue in 207 patients with aplastic anemia and other autoimmune diseases. Medicine 2011, 90, 89–98. [Google Scholar] [CrossRef]
- Lonial, S.; Waller, E.K.; Richardson, P.G.; Jagannath, S.; Orlowski, R.Z.; Giver, C.R.; Jaye, D.L.; Francis, D.; Giusti, S.; Torre, C.; et al. Risk factors and kinetics of thrombocytopenia associated with bortezomib for relapsed, refractory multiple myeloma. Blood 2005, 106, 3777–3784. [Google Scholar] [CrossRef]
- White, M.J.; Schoenwaelder, S.M.; Josefsson, E.C.; Jarman, K.E.; Henley, K.J.; James, C.; Debrincat, M.A.; Jackson, S.P.; Huang, D.C.; Kile, B.T. Caspase-9 mediates the apoptotic death of megakaryocytes and platelets, but is dispensable for their generation and function. Blood 2012, 119, 4283–4290. [Google Scholar] [CrossRef] [PubMed]
- Josefsson, E.C.; White, M.J.; Dowling, M.R.; Kile, B.T. Platelet life span and apoptosis. Methods Mol. Biol. 2012, 788, 59–71. [Google Scholar] [PubMed]
- Zhang, H.; Nimmer, P.M.; Tahir, S.K.; Chen, J.; Fryer, R.M.; Hahn, K.R.; Iciek, L.A.; Morgan, S.J.; Nasarre, M.C.; Nelson, R.; et al. Bcl-2 family proteins are essential for platelet survival. Cell Death Differ. 2007, 14, 943–951. [Google Scholar] [CrossRef] [PubMed]
- Nimbalkar, D.; Henry, M.K.; Quelle, F.W. Cytokine activation of phosphoinositide 3-kinase sensitizes hematopoietic cells to cisplatin-induced death. Cancer Res. 2003, 63, 1034–1039. [Google Scholar] [PubMed]
- Mitchell, W.B.; Pinheiro, M.P.; Boulad, N.; Kaplan, D.; Edison, M.N.; Psaila, B.; Karpoff, M.; White, M.J.; Josefsson, E.C.; Kile, B.T.; et al. Effect of thrombopoietin receptor agonists on the apoptotic profile of platelets in patients with chronic immune thrombocytopenia. Am. J. Hematol. 2014, 89, E228–E234. [Google Scholar] [CrossRef] [PubMed]
- Kirito, K.; Watanabe, T.; Sawada, K.; Endo, H.; Ozawa, K.; Komatsu, N. Thrombopoietin regulates Bcl-xL gene expression through Stat5 and phosphatidylinositol 3-kinase activation pathways. J. Biol. Chem. 2002, 277, 8329–8337. [Google Scholar] [CrossRef]
- Sim, X.; Poncz, M.; Gadue, P.; French, D.L. Understanding platelet generation from megakaryocytes: Implications for in vitro-derived platelets. Blood 2016, 127, 1227–1233. [Google Scholar] [CrossRef]
- Sanz, C.; Benet, I.; Richard, C.; Badia, B.; Andreu, E.J.; Prosper, F.; Fernández-Luna, J.L. Antiapoptotic protein Bcl-xL is up-regulated during megakaryocytic differentiation of CD34+ progenitors but is absent from senescent megakaryocytes. Exp. Hematol. 2001, 29, 728–735. [Google Scholar] [CrossRef]
- Zhang, X.; Chuai, Y.; Nie, W.; Wang, A.; Dai, G. Thrombopoietin receptor agonists for prevention and treatment of chemotherapy-induced thrombocytopenia in patients with solid tumours. Cochrane Database Syst. Rev. 2017, 11, CD012035. [Google Scholar] [CrossRef]
- Eto, K.; Kunishima, S. Linkage between the mechanisms of thrombocytopenia and thrombopoiesis. Blood 2016, 127, 1234–1241. [Google Scholar] [CrossRef]
- Grisouard, J.; Hao-Shen, H.; Dirnhofer, S.; Wagner, K.U.; Skoda, R.C. Selective deletion of Jak2 in adult mouse hematopoietic cells leads to lethal anemia and thrombocytopenia. Haematologica 2014, 99, e52–e54. [Google Scholar] [CrossRef] [PubMed]
- Kuter, D.J. The physiology of platelet production. Stem Cells 1996, 14 (Suppl. S1), 88–101. [Google Scholar] [CrossRef] [PubMed]
- Akkerman, J.W. Thrombopoietin and platelet function. Semin. Thromb. Hemost. 2006, 32, 295–304. [Google Scholar] [CrossRef] [PubMed]
- Kuter, D.J. New thrombopoietic growth factors. Blood 2007, 109, 4607–4616. [Google Scholar] [CrossRef]
- Besancenot, R.; Roos-Weil, D.; Tonetti, C.; Abdelouahab, H.; Lacout, C.; Pasquier, F.; Willekens, C.; Rameau, P.; Lecluse, Y.; Micol, J.B.; et al. JAK2 and MPL protein levels determine TPO-induced megakaryocyte proliferation vs differentiation. Blood 2014, 124, 2104–2115. [Google Scholar] [CrossRef]
- Meyer, S.C.; Keller, M.D.; Woods, B.A.; LaFave, L.M.; Bastian, L.; Kleppe, M.; Bhagwat, N.; Marubayashi, S.; Levine, R.L. Genetic studies reveal an unexpected negative regulatory role for Jak2 in thrombopoiesis. Blood 2014, 124, 2280–2284. [Google Scholar] [CrossRef]
- Downward, J. PI 3-kinase, Akt and cell survival. Semin. Cell Dev. Biol. 2004, 15, 177–182. [Google Scholar] [CrossRef]
- Grundschober, E.; Hoelbl-Kovacic, A.; Bhagwat, N.; Kovacic, B.; Scheicher, R.; Eckelhart, E.; Kollmann, K.; Keller, M.; Grebien, F.; Wagner, K.U.; et al. Acceleration of Bcr-Abl+ leukemia induced by deletion of JAK2. Leukemia 2014, 28, 1918–1922. [Google Scholar] [CrossRef]
- Kagoya, Y.; Tanaka, S.; Guo, T.; Anczurowski, M.; Wang, C.H.; Saso, K.; Butler, M.O.; Minden, M.D.; Hirano, N. A novel chimeric antigen receptor containing a JAK-STAT signaling domain mediates superior antitumor effects. Nat. Med. 2018, 24, 352–359. [Google Scholar] [CrossRef]
- Piatek, C.; Akhtari, M. Thrombocytopenia: Optimizing approaches in cancer patients. Oncology 2015, 29, 297–298. [Google Scholar]
- Hassan, M.N.; Waller, E.K. Treating chemotherapy-induced thrombocytopenia: Is it time for oncologists to use thrombopoietin agonists? Oncology 2015, 29, 295–296. [Google Scholar]
- Tepler, I.; Elias, L.; Smith, J.W., 2nd; Hussein, M.; Rosen, G.; Chang, A.Y.; Moore, J.O.; Gordon, M.S.; Kuca, B.; Beach, K.J.; et al. A randomized placebo-controlled trial of recombinant human interleukin-11 in cancer patients with severe thrombocytopenia due to chemotherapy. Blood 1996, 87, 3607–3614. [Google Scholar] [PubMed]
- Basser, R.L.; O’Flaherty, E.; Green, M.; Edmonds, M.; Nichol, J.; Menchaca, D.M.; Cohen, B.; Begley, C.G. Development of pancytopenia with neutralizing antibodies to thrombopoietin after multicycle chemotherapy supported by megakaryocyte growth and development factor. Blood 2002, 99, 2599–2602. [Google Scholar] [CrossRef] [PubMed]
- Bussel, J.B.; Provan, D.; Shamsi, T.; Cheng, G.; Psaila, B.; Kovaleva, L.; Salama, A.; Jenkins, J.M.; Roychowdhury, D.; Mayer, B.; et al. Effect of eltrombopag on platelet counts and bleeding during treatment of chronic idiopathic thrombocytopenic purpura: A randomised, double-blind, placebo-controlled trial. Lancet 2009, 373, 641–648. [Google Scholar] [CrossRef]
- Townsley, D.M.; Scheinberg, P.; Winkler, T.; Desmond, R.; Dumitriu, B.; Rios, O.; Weinstein, B.; Valdez, J.; Lotter, J.; Feng, X.; et al. Eltrombopag Added to Standard Immunosuppression for Aplastic Anemia. N. Engl. J. Med. 2017, 376, 1540–1550. [Google Scholar] [CrossRef]
- Shinjo, K.; Takeshita, A.; Nakamura, S.; Naitoh, K.; Yanagi, M.; Tobita, T.; Ohnishi, K.; Ohno, R. Serum thrombopoietin levels in patients correlate inversely with platelet counts during chemotherapy-induced thrombocytopenia. Leukemia 1998, 12, 295–300. [Google Scholar] [CrossRef]
- Engel, C.; Loeffler, M.; Franke, H.; Schmitz, S. Endogenous thrombopoietin serum levels during multicycle chemotherapy. Br. J. Haematol. 1999, 105, 832–838. [Google Scholar] [CrossRef]
- McElroy, P.L.; Wei, P.; Buck, K.; Sinclair, A.M.; Eschenberg, M.; Sasu, B.; Molineux, G. Romiplostim promotes platelet recovery in a mouse model of multicycle chemotherapy-induced thrombocytopenia. Exp. Hematol. 2015, 43, 479–487. [Google Scholar] [CrossRef]
- Ng, A.P.; Kauppi, M.; Metcalf, D.; Hyland, C.D.; Josefsson, E.C.; Lebois, M.; Zhang, J.G.; Baldwin, T.M.; Di Rago, L.; Hilton, D.J.; et al. Mpl expression on megakaryocytes and platelets is dispensable for thrombopoiesis but essential to prevent myeloproliferation. Proc. Natl. Acad. Sci. USA 2014, 111, 5884–5889. [Google Scholar] [CrossRef]
- Cruet-Hennequart, S.; Villalan, S.; Kaczmarczyk, A.; O’Meara, E.; Sokol, A.M.; Carty, M.P. Characterization of the effects of cisplatin and carboplatin on cell cycle progression and DNA damage response activation in DNA polymerase eta-deficient human cells. Cell Cycle 2009, 8, 3039–3050. [Google Scholar] [CrossRef]
- Vitrat, N.; Cohen-Solal, K.; Pique, C.; Le Couedic, J.P.; Norol, F.; Larsen, A.K.; Katz, A.; Vainchenker, W.; Debili, N. Endomitosis of human megakaryocytes are due to abortive mitosis. Blood 1998, 91, 3711–3723. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, Z.; Ravid, K. The cell cycle in polyploid megakaryocytes is associated with reduced activity of cyclin B1-dependent cdc2 kinase. J. Biol. Chem. 1996, 271, 4266–4272. [Google Scholar] [CrossRef] [PubMed]
- Williams, N.; Jackson, H. Regulation of proliferation of murine megakaryocyte progenitor cells by cell cycle. Blood 1978, 52, 163–170. [Google Scholar] [CrossRef] [PubMed]
- Cen, D.; Levin, J. Cell cycle status of murine megakaryocyte and granulocyte-macrophage colony-forming cells in bone marrow and spleen. Exp. Hematol. 1992, 20, 1094–1100. [Google Scholar]
- Mao, Z.; Bozzella, M.; Seluanov, A.; Gorbunova, V. DNA repair by nonhomologous end joining and homologous recombination during cell cycle in human cells. Cell Cycle 2008, 7, 2902–2906. [Google Scholar] [CrossRef]
- Xiong, H.; Zhang, Z.G.; Tian, X.Q.; Sun, D.F.; Liang, Q.C.; Zhang, Y.J.; Lu, R.; Chen, Y.X.; Fang, J.Y. Inhibition of JAK1, 2/STAT3 signaling induces apoptosis, cell cycle arrest, and reduces tumor cell invasion in colorectal cancer cells. Neoplasia 2008, 10, 287–297. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Control | Carboplatin Day 3 | Carboplatin Day 6 | Carboplatin Day 9 | Carboplatin Day 12 | p-Value | |
|---|---|---|---|---|---|---|
| Weight (g) | 214.0 ± 21.3 | 244.2 ± 54.1 | 240.3 ± 42.0 | 223.7 ± 38.2 | 249.0 ± 57.4 | 0.28 |
| WBC | 5.8 ± 1.6 | 5.9 ± 1.6 | 4.8 ± 1.6 | 5.6 ± 1.9 | 5.0 ± 1.3 | 0.56 |
| RBC | 6.7 ± 0.5 | 6.5 ± 0.8 | 6.4 ± 0.5 | 6.5 ± 0.6 | 6.0 ± 1.0 | 0.25 |
| HGB | 12.6 ± 0.5 | 12.4 ± 0.8 | 12.2 ± 0.7 | 12.1 ± 0.9 | 12.2 ± 1.8 | 0.53 |
| HCT | 41.7 ± 1.1 | 39.7 ± 2.3 | 39.9 ± 1.9 | 39.8 ± 2.8 | 39.2 ± 5.4 | 0.16 |
| MCV | 62.4 ± 1.9 | 61.4 ± 5.2 | 62.2 ± 3.3 | 61.8 ± 3.1 | 59.6 ± 3.3 | 0.51 |
| MCH | 18.9 ± 0.6 | 19.1 ± 1.2 | 18.9 ± 0.9 | 18.8 ± 0.8 | 18.6 ± 1.2 | 0.87 |
| MCHC | 30.3 ± 0.5 | 31.3 ± 0.9 | 30.4 ± 0.4 | 30.5 ± 0.6 | 31.1 ± 1.0 | <0.05 |
| PLT | 1117.5 ± 179.7 | 1142.2 ± 141.4 | 731.2 ± 141.7 | 756.7 ± 287.9 | 982.2 ± 366.2 | <0.05 |
| RDW-SD | 29.8 ± 1.0 | 28.7 ± 2.2 | 29.7 ± 2.2 | 30.5 ± 2.4 | 29.9 ± 2.0 | 0.51 |
| RDW-CV | 13.4 ± 0.5 | 13.4 ± 0.4 | 13.9 ± 0.8 | 14.3 ± 0.8 | 15.1 ± 1.0 | <0.05 |
| PDW | 7.0 ± 0.5 | 6.9 ± 0.2 | 7.0 ± 0.4 | 7.0 ± 0.8 | 7.4 ± 1.1 | 0.66 |
| MPV | 6.9 ± 0.4 | 6.8 ± 0.2 | 7.0 ± 0.2 | 7.0 ± 0.5 | 7.3 ± 0.7 | 0.37 |
| P-LCR | 5.8 ± 1.8 | 5.3 ± 0.9 | 6.4 ± 1.5 | 7.0 ± 2.9 | 8.4 ± 4.8 | 0.22 |
| PCT | 0.8 ± 0.1 | 0.8 ± 0.1 | 0.6 ± 0.2 | 0.5 ± 0.2 | 0.7 ± 0.2 | <0.05 |
| NEUT | 0.7 ± 0.3 | 0.7 ± 0.2 | 0.9 ± 0.4 | 0.9 ± 0.5 | 0.6 ± 0.3 | 0.35 |
| LYMPH | 4.4 ± 1.4 | 5.1 ± 1.7 | 3.6 ± 1.1 | 4.4 ± 1.7 | 4.3 ± 1.2 | 0.61 |
| MONO | 0.1 ± 0.1 | 0.1 ± 0.1 | 0.0 ± 0.0 | 0.1 ± 0.1 | 0.1 ± 0.0 | 0.18 |
| EO | 0.04 ± 0.01 | 0.03 ± 0.02 | 0.02 ± 0.01 | 0.03 ± 0.02 | 0.04 ± 0.03 | 0.14 |
| BASO | 0 ± 0 | 0 ± 0 | 0 ± 0 | 0.001 ± 0.003 | 0.002 ± 0.004 | 0.32 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, Y.-H.; Chen, H.-Y.; Hong, W.-C.; Wei, C.-Y.; Pang, J.-H.S. Carboplatin-Induced Thrombocytopenia through JAK2 Downregulation, S-Phase Cell Cycle Arrest and Apoptosis in Megakaryocytes. Int. J. Mol. Sci. 2022, 23, 6290. https://doi.org/10.3390/ijms23116290
Wu Y-H, Chen H-Y, Hong W-C, Wei C-Y, Pang J-HS. Carboplatin-Induced Thrombocytopenia through JAK2 Downregulation, S-Phase Cell Cycle Arrest and Apoptosis in Megakaryocytes. International Journal of Molecular Sciences. 2022; 23(11):6290. https://doi.org/10.3390/ijms23116290
Chicago/Turabian StyleWu, Yi-Hong, Hsing-Yu Chen, Wei-Chin Hong, Chen-Ying Wei, and Jong-Hwei Su Pang. 2022. "Carboplatin-Induced Thrombocytopenia through JAK2 Downregulation, S-Phase Cell Cycle Arrest and Apoptosis in Megakaryocytes" International Journal of Molecular Sciences 23, no. 11: 6290. https://doi.org/10.3390/ijms23116290