
Dealing with Macrophage Plasticity to Address Therapeutic Challenges in Head and Neck Cancers

 ,
, 
Abstract
:1. Introduction
2. Results
2.1. Macrophage Characterization in the HNSCC TME
2.1.1. Clinical Cohort Composition
2.1.2. Macrophages Infiltration in a Clinical Series of 60 HNSCCs
2.2. THP1 Monocyte Differentiation in M1 and M2 Macrophage Phenotypes
2.2.1. Analysis of Morphological Changes during Macrophage Polarization
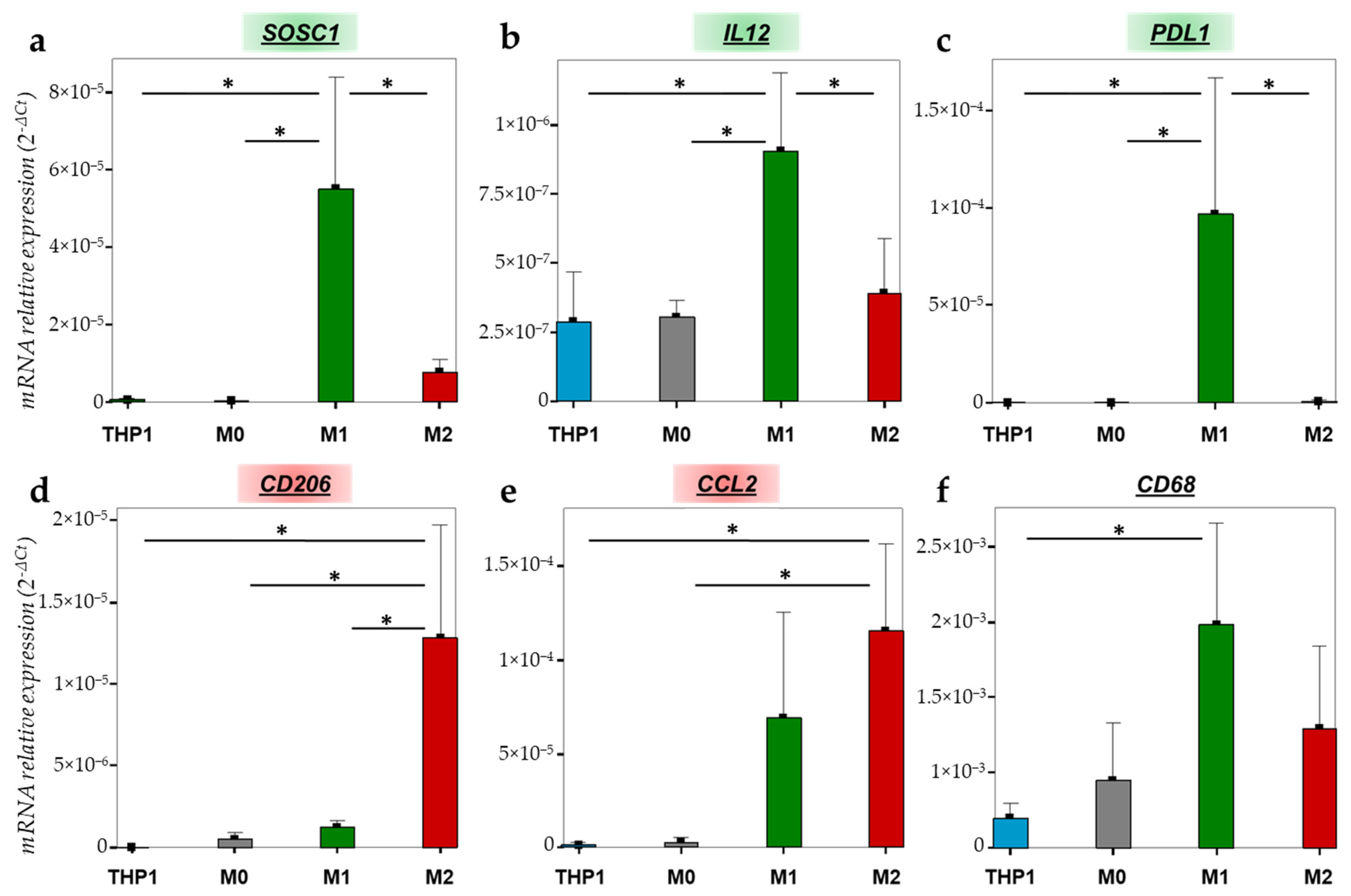
2.2.2. Gene Expression Variations during the Macrophage Polarization
2.2.3. Differential Expression of M1 and M2 Specific Proteins
2.3. Validation of Macrophage Polarization Process on PBMC Differentiation
2.4. Characterization of M1 versus M2 Macrophage Phenotype
2.4.1. Metabolism Variations
2.4.2. Oxidative Stress Comparison
2.5. Macrophage Editing as a Target for Cancer Therapy
3. Discussion
4. Materials and Methods
4.1. Patients and Clinical Data
4.2. Immunohistochemistry
4.3. Cell Culture
4.4. PBMC Purification and Isolation
4.5. Macrophages Polarization
4.6. RNA Extraction, cDNA Synthesis and qPCR
4.7. Immunofluorescence
4.8. Flow Cytometry
4.9. ROS Evaluation
4.10. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Mathers, C.; Parkin, D.M.; Piñeros, M.; Znaor, A.; Bray, F. Estimating the Global Cancer Incidence and Mortality in 2018: GLOBOCAN Sources and Methods. Int. J. Cancer 2019, 144, 1941–1953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, E.; Mazul, A.L.; Farquhar, D.; Brennan, P.; Anantharaman, D.; Abedi-Ardekani, B.; Weissler, M.C.; Hayes, D.N.; Olshan, A.F.; Zevallos, J.P. Long-Term Survival in Head and Neck Cancer: Impact of Site, Stage, Smoking, and Human Papillomavirus Status. Laryngoscope 2019, 129, 2506–2513. [Google Scholar] [CrossRef] [PubMed]
- Koo, H.Y.; Han, K.; Shin, D.W.; Yoo, J.E.; Cho, M.H.; Jeon, K.H.; Kim, D.; Hong, S.; Jun, J.K. Alcohol Drinking Pattern and Risk of Head and Neck Cancer: A Nationwide Cohort Study. Int. J. Environ. Res. Public Health 2021, 18, 11204. [Google Scholar] [CrossRef] [PubMed]
- Miranda-Galvis, M.; Loveless, R.; Kowalski, L.P.; Teng, Y. Impacts of Environmental Factors on Head and Neck Cancer Pathogenesis and Progression. Cells 2021, 10, 389. [Google Scholar] [CrossRef] [PubMed]
- Brewczyński, A.; Jabłońska, B.; Kentnowski, M.; Mrowiec, S.; Składowski, K.; Rutkowski, T. The Association between Carotenoids and Head and Neck Cancer Risk. Nutrients 2021, 14, 88. [Google Scholar] [CrossRef]
- Mazilu, L.; Suceveanu, A.-I.; Stanculeanu, D.-L.; Gheorghe, A.-D.; Fricatel, G.; Negru, S.-M. Tumor Microenvironment Is Not an ‘Innocent Bystander’ in the Resistance to Treatment of Head and Neck Cancers (Review). Exp. Ther. Med. 2021, 22, 1128. [Google Scholar] [CrossRef]
- Peltanova, B.; Raudenska, M.; Masarik, M. Effect of Tumor Microenvironment on Pathogenesis of the Head and Neck Squamous Cell Carcinoma: A Systematic Review. Mol. Cancer 2019, 18, 63. [Google Scholar] [CrossRef]
- Seminerio, I.; Kindt, N.; Descamps, G.; Bellier, J.; Lechien, J.R.; Mat, Q.; Pottier, C.; Journé, F.; Saussez, S. High Infiltration of CD68+ Macrophages Is Associated with Poor Prognoses of Head and Neck Squamous Cell Carcinoma Patients and Is Influenced by Human Papillomavirus. Oncotarget 2018, 9, 11046–11059. [Google Scholar] [CrossRef] [Green Version]
- Seminerio, I.; Descamps, G.; Dupont, S.; de Marrez, L.; Laigle, J.-A.; Lechien, J.R.; Kindt, N.; Journe, F.; Saussez, S. Infiltration of FoxP3+ Regulatory T Cells Is a Strong and Independent Prognostic Factor in Head and Neck Squamous Cell Carcinoma. Cancers 2019, 11, 227. [Google Scholar] [CrossRef] [Green Version]
- Kindt, N.; Descamps, G.; Seminerio, I.; Bellier, J.; Lechien, J.R.; Mat, Q.; Pottier, C.; Delvenne, P.; Journé, F.; Saussez, S. High Stromal Foxp3-Positive T Cell Number Combined to Tumor Stage Improved Prognosis in Head and Neck Squamous Cell Carcinoma. Oral Oncol. 2017, 67, 183–191. [Google Scholar] [CrossRef]
- Nordfors, C.; Grün, N.; Tertipis, N.; Ährlund-Richter, A.; Haeggblom, L.; Sivars, L.; Du, J.; Nyberg, T.; Marklund, L.; Munck-Wikland, E.; et al. CD8+ and CD4+ Tumour Infiltrating Lymphocytes in Relation to Human Papillomavirus Status and Clinical Outcome in Tonsillar and Base of Tongue Squamous Cell Carcinoma. Eur. J. Cancer 2013, 49, 2522–2530. [Google Scholar] [CrossRef] [PubMed]
- Ohashi, T.; Aoki, M.; Tomita, H.; Akazawa, T.; Sato, K.; Kuze, B.; Mizuta, K.; Hara, A.; Nagaoka, H.; Inoue, N.; et al. M2-like Macrophage Polarization in High Lactic Acid-Producing Head and Neck Cancer. Cancer Sci. 2017, 108, 1128–1134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, B.; Ren, M.; Zhou, X.; Han, Q.; Cheng, L. Targeting Tumor-Associated Macrophages in Head and Neck Squamous Cell Carcinoma. Oral Oncol. 2020, 106, 104723. [Google Scholar] [CrossRef] [PubMed]
- Evrard, D.; Szturz, P.; Tijeras-Raballand, A.; Astorgues-Xerri, L.; Abitbol, C.; Paradis, V.; Raymond, E.; Albert, S.; Barry, B.; Faivre, S. Macrophages in the Microenvironment of Head and Neck Cancer: Potential Targets for Cancer Therapy. Oral Oncol. 2019, 88, 29–38. [Google Scholar] [CrossRef]
- Troiano, G.; Caponio, V.C.A.; Adipietro, I.; Tepedino, M.; Santoro, R.; Laino, L.; Lo Russo, L.; Cirillo, N.; Lo Muzio, L. Prognostic Significance of CD68+ and CD163+ Tumor Associated Macrophages in Head and Neck Squamous Cell Carcinoma: A Systematic Review and Meta-Analysis. Oral Oncol. 2019, 93, 66–75. [Google Scholar] [CrossRef]
- Lechien, J.R.; Descamps, G.; Seminerio, I.; Furgiuele, S.; Dequanter, D.; Mouawad, F.; Badoual, C.; Journe, F.; Saussez, S. HPV Involvement in the Tumor Microenvironment and Immune Treatment in Head and Neck Squamous Cell Carcinomas. Cancers 2020, 12, 1060. [Google Scholar] [CrossRef]
- Fu, E.; Liu, T.; Yu, S.; Chen, X.; Song, L.; Lou, H.; Ma, F.; Zhang, S.; Hussain, S.; Guo, J.; et al. M2 Macrophages Reduce the Radiosensitivity of Head and Neck Cancer by Releasing HB-EGF. Oncol. Rep. 2020, 44, 698–710. [Google Scholar] [CrossRef]
- Palma, A.; Jarrah, A.S.; Tieri, P.; Cesareni, G.; Castiglione, F. Gene Regulatory Network Modeling of Macrophage Differentiation Corroborates the Continuum Hypothesis of Polarization States. Front. Physiol. 2018, 9, 1659. [Google Scholar] [CrossRef] [Green Version]
- Aras, S.; Zaidi, M.R. TAMeless Traitors: Macrophages in Cancer Progression and Metastasis. Br. J. Cancer 2017, 117, 1583–1591. [Google Scholar] [CrossRef] [Green Version]
- Chávez-Galán, L.; Olleros, M.L.; Vesin, D.; Garcia, I. Much More than M1 and M2 Macrophages, There Are Also CD169(+) and TCR(+) Macrophages. Front. Immunol. 2015, 6, 263. [Google Scholar] [CrossRef]
- Mantovani, A.; Marchesi, F.; Malesci, A.; Laghi, L.; Allavena, P. Tumor-Associated Macrophages as Treatment Targets in Oncology. Nat. Rev. Clin. Oncol. 2017, 14, 399–416. [Google Scholar] [CrossRef] [PubMed]
- Koo, S.; Garg, N.J. Metabolic Programming of Macrophage Functions and Pathogens Control. Redox Biol. 2019, 24, 101198. [Google Scholar] [CrossRef] [PubMed]
- Locati, M.; Curtale, G.; Mantovani, A. Diversity, Mechanisms and Significance of Macrophage Plasticity. Annu. Rev. Pathol. 2020, 15, 123–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Yan, B.; Lou, H.; Shen, Z.; Tong, F.; Zhai, A.; Wei, L.; Zhang, F. Immunological Network Analysis in HPV Associated Head and Neck Squamous Cancer and Implications for Disease Prognosis. Mol. Immunol. 2018, 96, 28–36. [Google Scholar] [CrossRef] [PubMed]
- Fujii, N.; Shomori, K.; Shiomi, T.; Nakabayashi, M.; Takeda, C.; Ryoke, K.; Ito, H. Cancer-Associated Fibroblasts and CD163-Positive Macrophages in Oral Squamous Cell Carcinoma: Their Clinicopathological and Prognostic Significance. J. Oral Pathol. Med. 2012, 41, 444–451. [Google Scholar] [CrossRef] [PubMed]
- Shiratori, H.; Feinweber, C.; Luckhardt, S.; Linke, B.; Resch, E.; Geisslinger, G.; Weigert, A.; Parnham, M.J. THP-1 and Human Peripheral Blood Mononuclear Cell-Derived Macrophages Differ in Their Capacity to Polarize in Vitro. Mol. Immunol. 2017, 88, 58–68. [Google Scholar] [CrossRef]
- Lund, M.E.; To, J.; O’Brien, B.A.; Donnelly, S. The Choice of Phorbol 12-Myristate 13-Acetate Differentiation Protocol Influences the Response of THP-1 Macrophages to a pro-Inflammatory Stimulus. J. Immunol. Methods 2016, 430, 64–70. [Google Scholar] [CrossRef]
- Baxter, E.W.; Graham, A.E.; Re, N.A.; Carr, I.M.; Robinson, J.I.; Mackie, S.L.; Morgan, A.W. Standardized Protocols for Differentiation of THP-1 Cells to Macrophages with Distinct M(IFNγ+LPS), M(IL-4) and M(IL-10) Phenotypes. J. Immunol. Methods 2020, 478, 112721. [Google Scholar] [CrossRef]
- Kashfi, K.; Kannikal, J.; Nath, N. Macrophage Reprogramming and Cancer Therapeutics: Role of INOS-Derived NO. Cells 2021, 10, 3194. [Google Scholar] [CrossRef]
- Costa, N.L.; Valadares, M.C.; Souza, P.P.C.; Mendonça, E.F.; Oliveira, J.C.; Silva, T.A.; Batista, A.C. Tumor-Associated Macrophages and the Profile of Inflammatory Cytokines in Oral Squamous Cell Carcinoma. Oral Oncol. 2013, 49, 216–223. [Google Scholar] [CrossRef]
- Pettersen, J.S.; Fuentes-Duculan, J.; Suárez-Fariñas, M.; Pierson, K.C.; Pitts-Kiefer, A.; Fan, L.; Belkin, D.A.; Wang, C.Q.F.; Bhuvanendran, S.; Johnson-Huang, L.M.; et al. Tumor-Associated Macrophages in the Cutaneous SCC Microenvironment Are Heterogeneously Activated. J. Investig. Dermatol. 2011, 131, 1322–1330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mori, K.; Haraguchi, S.; Hiori, M.; Shimada, J.; Ohmori, Y. Tumor-Associated Macrophages in Oral Premalignant Lesions Coexpress CD163 and STAT1 in a Th1-Dominated Microenvironment. BMC Cancer 2015, 15, 573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, T.D.; Tse, M.J.; Read, E.L.; Liu, W.F. Regulation of Macrophage Polarization and Plasticity by Complex Activation Signals. Integr. Biol. Quant. Biosci. Nano Macro 2016, 8, 946–955. [Google Scholar] [CrossRef] [PubMed]
- Martinez, F.O.; Gordon, S. The M1 and M2 Paradigm of Macrophage Activation: Time for Reassessment. F1000Prime Rep. 2014, 6, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ballini, A.; Dipalma, G.; Isacco, C.G.; Boccellino, M.; Di Domenico, M.; Santacroce, L.; Nguyễn, K.C.D.; Scacco, S.; Calvani, M.; Boddi, A.; et al. Oral Microbiota and Immune System Crosstalk: A Translational Research. Biology 2020, 9, 131. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Zhou, R.; Wang, H.; Li, W.; Pan, M.; Yao, X.; Zhan, W.; Yang, S.; Xu, L.; Ding, Y.; et al. Gut Microbiota-Stimulated Cathepsin K Secretion Mediates TLR4-Dependent M2 Macrophage Polarization and Promotes Tumor Metastasis in Colorectal Cancer. Cell Death Differ. 2019, 26, 2447–2463. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Fan, L.; Lin, Y.; Shen, W.; Qi, Y.; Zhang, Y.; Chen, Z.; Wang, L.; Long, Y.; Hou, T.; et al. Fusobacterium Nucleatum Promotes Colorectal Cancer Metastasis through MiR-1322/CCL20 Axis and M2 Polarization. Gut Microbes 2021, 13, 1980347. [Google Scholar] [CrossRef]
- Yang, C.-Y.; Yeh, Y.-M.; Yu, H.-Y.; Chin, C.-Y.; Hsu, C.-W.; Liu, H.; Huang, P.-J.; Hu, S.-N.; Liao, C.-T.; Chang, K.-P.; et al. Oral Microbiota Community Dynamics Associated With Oral Squamous Cell Carcinoma Staging. Front. Microbiol. 2018, 9, 862. [Google Scholar] [CrossRef] [Green Version]
- Fan, X.; Peters, B.A.; Jacobs, E.J.; Gapstur, S.M.; Purdue, M.P.; Freedman, N.D.; Alekseyenko, A.V.; Wu, J.; Yang, L.; Pei, Z.; et al. Drinking Alcohol Is Associated with Variation in the Human Oral Microbiome in a Large Study of American Adults. Microbiome 2018, 6, 59. [Google Scholar] [CrossRef] [Green Version]
- Heideveld, E.; Horcas-Lopez, M.; Lopez-Yrigoyen, M.; Forrester, L.M.; Cassetta, L.; Pollard, J.W. Chapter Eight-Methods for Macrophage Differentiation and in Vitro Generation of Human Tumor Associated-like Macrophages. In Methods in Enzymology; Galluzzi, L., Rudqvist, N.-P., Eds.; Tumor Immunology and Immunotherapy–Cellular Methods Part B; Academic Press: Cambridge, MA, USA, 2020; Volume 632, pp. 113–131. [Google Scholar]
- Genard, G.; Lucas, S.; Michiels, C. Reprogramming of Tumor-Associated Macrophages with Anticancer Therapies: Radiotherapy versus Chemo- and Immunotherapies. Front. Immunol. 2017, 8, 828. [Google Scholar] [CrossRef] [Green Version]
- Ivashkiv, L.B. IFNγ: Signalling, Epigenetics and Roles in Immunity, Metabolism, Disease and Cancer Immunotherapy. Nat. Rev. Immunol. 2018, 18, 545–558. [Google Scholar] [CrossRef] [PubMed]
- Kang, K.; Bachu, M.; Park, S.H.; Kang, K.; Bae, S.; Park-Min, K.-H.; Ivashkiv, L.B. IFN-γ Selectively Suppresses a Subset of TLR4-Activated Genes and Enhancers to Potentiate Macrophage Activation. Nat. Commun. 2019, 10, 3320. [Google Scholar] [CrossRef] [PubMed]
- Genin, M.; Clement, F.; Fattaccioli, A.; Raes, M.; Michiels, C. M1 and M2 Macrophages Derived from THP-1 Cells Differentially Modulate the Response of Cancer Cells to Etoposide. BMC Cancer 2015, 15, 577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujihara, M.; Muroi, M.; Tanamoto, K.; Suzuki, T.; Azuma, H.; Ikeda, H. Molecular Mechanisms of Macrophage Activation and Deactivation by Lipopolysaccharide: Roles of the Receptor Complex. Pharmacol. Ther. 2003, 100, 171–194. [Google Scholar] [CrossRef] [PubMed]
- Liau, N.P.D.; Laktyushin, A.; Lucet, I.S.; Murphy, J.M.; Yao, S.; Whitlock, E.; Callaghan, K.; Nicola, N.A.; Kershaw, N.J.; Babon, J.J. The Molecular Basis of JAK/STAT Inhibition by SOCS1. Nat. Commun. 2018, 9, 1558. [Google Scholar] [CrossRef]
- Qiu, N.; Wang, G.; Wang, J.; Zhou, Q.; Guo, M.; Wang, Y.; Hu, X.; Zhou, H.; Bai, R.; You, M.; et al. Tumor-Associated Macrophage and Tumor-Cell Dually Transfecting Polyplexes for Efficient Interleukin-12 Cancer Gene Therapy. Adv. Mater. (Deerfield Beach Fla.) 2021, 33, e2006189. [Google Scholar] [CrossRef]
- Wang, Q.; Cheng, F.; Ma, T.; Xiong, H.-Y.; Li, Z.-W.; Xie, C.-L.; Liu, C.-Y.; Tu, Z.-G. Interleukin-12 Inhibits the Hepatocellular Carcinoma Growth by Inducing Macrophage Polarization to the M1-like Phenotype through Downregulation of Stat-3. Mol. Cell. Biochem. 2016, 415, 157–168. [Google Scholar] [CrossRef]
- Oguejiofor, K.; Galletta-Williams, H.; Dovedi, S.J.; Roberts, D.L.; Stern, P.L.; West, C.M.L. Distinct Patterns of Infiltrating CD8+ T Cells in HPV+ and CD68 Macrophages in HPV- Oropharyngeal Squamous Cell Carcinomas Are Associated with Better Clinical Outcome but PD-L1 Expression Is Not Prognostic. Oncotarget 2017, 8, 14416–14427. [Google Scholar] [CrossRef] [Green Version]
- Raggi, F.; Pelassa, S.; Pierobon, D.; Penco, F.; Gattorno, M.; Novelli, F.; Eva, A.; Varesio, L.; Giovarelli, M.; Bosco, M.C. Regulation of Human Macrophage M1–M2 Polarization Balance by Hypoxia and the Triggering Receptor Expressed on Myeloid Cells-1. Front. Immunol. 2017, 8, 1097. [Google Scholar] [CrossRef]
- Nielsen, M.C.; Andersen, M.N.; Møller, H.J. Monocyte Isolation Techniques Significantly Impact the Phenotype of Both Isolated Monocytes and Derived Macrophages in Vitro. Immunology 2020, 159, 63–74. [Google Scholar] [CrossRef]
- Bhattacharjee, A.; Shukla, M.; Yakubenko, V.P.; Mulya, A.; Kundu, S.; Cathcart, M.K. IL-4 and IL-13 Employ Discrete Signaling Pathways for Target Gene Expression in Alternatively Activated Monocytes/Macrophages. Free Radic. Biol. Med. 2013, 54, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Genard, G.; Wera, A.-C.; Huart, C.; Le Calve, B.; Penninckx, S.; Fattaccioli, A.; Tabarrant, T.; Demazy, C.; Ninane, N.; Heuskin, A.-C.; et al. Proton Irradiation Orchestrates Macrophage Reprogramming through NFκB Signaling. Cell Death Dis. 2018, 9, 728. [Google Scholar] [CrossRef] [PubMed]
- Shiraishi, D.; Fujiwara, Y.; Horlad, H.; Saito, Y.; Iriki, T.; Tsuboki, J.; Cheng, P.; Nakagata, N.; Mizuta, H.; Bekki, H.; et al. CD163 Is Required for Protumoral Activation of Macrophages in Human and Murine Sarcoma. Cancer Res. 2018, 78, 3255–3266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jimenez-Duran, G.; Luque-Martin, R.; Patel, M.; Koppe, E.; Bernard, S.; Sharp, C.; Buchan, N.; Rea, C.; de Winther, M.P.J.; Turan, N.; et al. Pharmacological Validation of Targets Regulating CD14 during Macrophage Differentiation. EBioMedicine 2020, 61, 103039. [Google Scholar] [CrossRef] [PubMed]
- Spano, A.; Barni, S.; Sciola, L. PMA Withdrawal in PMA-treated Monocytic THP-1 Cells and Subsequent Retinoic Acid Stimulation, Modulate Induction of Apoptosis and Appearance of Dendritic Cells. Cell Prolif. 2013, 46, 328–347. [Google Scholar] [CrossRef] [PubMed]
- Oyarce, C.; Vizcaino-Castro, A.; Chen, S.; Boerma, A.; Daemen, T. Re-Polarization of Immunosuppressive Macrophages to Tumor-Cytotoxic Macrophages by Repurposed Metabolic Drugs. Oncoimmunology 2021, 10, 1898753. [Google Scholar] [CrossRef]
- Chen, Y.; Song, Y.; Du, W.; Gong, L.; Chang, H.; Zou, Z. Tumor-Associated Macrophages: An Accomplice in Solid Tumor Progression. J. Biomed. Sci. 2019, 26, 78. [Google Scholar] [CrossRef]
- Tavakoli, S.; Downs, K.; Short, J.D.; Nguyen, H.N.; Lai, Y.; Jerabek, P.A.; Goins, B.; Toczek, J.; Sadeghi, M.M.; Asmis, R. Characterization of Macrophage Polarization States Using Combined Measurement of 2-Deoxyglucose and Glutamine Accumulation. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 1840–1848. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, T.; Nguyen-Tien, D.; Sorimachi, Y.; Sugiura, Y.; Suzuki, T.; Karyu, H.; Shimabukuro-Demoto, S.; Uemura, T.; Okamura, T.; Taguchi, T.; et al. SLC15A4 Mediates M1-Prone Metabolic Shifts in Macrophages and Guards Immune Cells from Metabolic Stress. Proc. Natl. Acad. Sci. USA 2021, 118, e2100295118. [Google Scholar] [CrossRef]
- Freemerman, A.J.; Johnson, A.R.; Sacks, G.N.; Milner, J.J.; Kirk, E.L.; Troester, M.A.; Macintyre, A.N.; Goraksha-Hicks, P.; Rathmell, J.C.; Makowski, L. Metabolic Reprogramming of Macrophages. J. Biol. Chem. 2014, 289, 7884–7896. [Google Scholar] [CrossRef] [Green Version]
- Tannir, N.M.; Fan, A.C.; Lee, R.J.; Carthon, B.C.; Iliopoulos, O.; Mier, J.W.; Patel, M.R.; Meric-Bernstam, F.; DeMichele, A.; Voss, M.H.; et al. Phase 1 Study of Glutaminase (GLS) Inhibitor CB-839 Combined with Either Everolimus (E) or Cabozantinib (Cabo) in Patients (Pts) with Clear Cell (Cc) and Papillary (Pap) Metastatic Renal Cell Cancer (MRCC). J. Clin. Oncol. 2018, 36, 603. [Google Scholar] [CrossRef]
- Vidal, G.; Kalinsky, K.; Stringer-Reasor, E.; Lynce, F.; Cole, J.; Valdes-Albini, F.; Soliman, H.; Nikolinakos, P.; Silber, A.; DeMichele, A.; et al. Abstract P6-20-07: Efficacy and Safety of CB-839, a Small Molecule Inhibitor of Glutaminase, in Combination with Paclitaxel in Patients with Advanced Triple Negative Breast Cancer (TNBC): Initial Findings from a Multicenter, Open-Label Phase 2 Study. Cancer Res. 2019, 79, P6-20–07. [Google Scholar] [CrossRef]
- Mills, E.L.; Kelly, B.; Logan, A.; Costa, A.S.H.; Varma, M.; Bryant, C.E.; Tourlomousis, P.; Däbritz, J.H.M.; Gottlieb, E.; Latorre, I.; et al. Repurposing Mitochondria from ATP Production to ROS Generation Drives a Pro-Inflammatory Phenotype in Macrophages That Depends on Succinate Oxidation by Complex II. Cell 2016, 167, 457–470.e13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, H.-Y.; Wang, N.; Li, S.; Hong, M.; Wang, X.; Feng, Y. The Reactive Oxygen Species in Macrophage Polarization: Reflecting Its Dual Role in Progression and Treatment of Human Diseases. Oxidative Med. Cell. Longev. 2016, 2016, e2795090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rojas, J.M.; Sanz-Ortega, L.; Mulens-Arias, V.; Gutiérrez, L.; Pérez-Yagüe, S.; Barber, D.F. Superparamagnetic Iron Oxide Nanoparticle Uptake Alters M2 Macrophage Phenotype, Iron Metabolism, Migration and Invasion. Nanomed. Nanotechnol. Biol. Med. 2016, 12, 1127–1138. [Google Scholar] [CrossRef]
- Mulens-Arias, V.; Rojas, J.M.; Pérez-Yagüe, S.; Morales, M.P.; Barber, D.F. Polyethylenimine-Coated SPIONs Trigger Macrophage Activation through TLR-4 Signaling and ROS Production and Modulate Podosome Dynamics. Biomaterials 2015, 52, 494–506. [Google Scholar] [CrossRef]
- Lu, M.; Cohen, M.H.; Rieves, D.; Pazdur, R. FDA Report: Ferumoxytol for Intravenous Iron Therapy in Adult Patients with Chronic Kidney Disease. Am. J. Hematol. 2010, 85, 315–319. [Google Scholar] [CrossRef]
- Zanganeh, S.; Hutter, G.; Spitler, R.; Lenkov, O.; Mahmoudi, M.; Shaw, A.; Pajarinen, J.S.; Nejadnik, H.; Goodman, S.; Moseley, M.; et al. Iron Oxide Nanoparticles Inhibit Tumour Growth by Inducing Pro-Inflammatory Macrophage Polarization in Tumour Tissues. Nat. Nanotechnol. 2016, 11, 986–994. [Google Scholar] [CrossRef]
- Mulens-Arias, V.; Rojas, J.M.; Barber, D.F. The Use of Iron Oxide Nanoparticles to Reprogram Macrophage Responses and the Immunological Tumor Microenvironment. Front. Immunol. 2021, 12, 693709. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variables | Number of Cases |
|---|---|
| n = 60 | |
| Age (years) | |
| Median (range) | 62 (42–89) |
| Gender | |
| Male | 43 |
| Female | 17 |
| Anatomic site | |
| Oral cavity | 22 |
| Oropharynx | 19 |
| Larynx | 16 |
| Hypopharynx | 2 |
| Nasopharynx | 1 |
| Tumor stage | |
| I-II | 35 |
| III-IV | 18 |
| Unknown | 7 |
| Histological grade | |
| Poorly differentiated | 24 |
| Well differentiated | 30 |
| Unknown | 6 |
| Lymph nodes invasion | |
| Yes | 47 |
| No | 10 |
| Unknown | 3 |
| Risk factors | |
| Tobacco | |
| Smoker | 51 |
| Non-smoker | 9 |
| Alcohol | |
| Drinker | 37 |
| Non-drinker | 23 |
| p16 status | |
| Positive | 29 |
| Negative | 31 |
| Recurrence (RFS) (months) | |
| Median (range) | 14 (1–106) |
| Yes | 26 |
| No | 32 |
| Unknown | 2 |
| Overall survival (OS) (months) | |
| Median (range) | 24 (1–294) |
| Alive | 31 |
| Dead | 28 |
| Unknown | 1 |
| Genes | Forward Sequences | Reverse Sequences |
|---|---|---|
| SOCS1 | TTTTCGCCCTTAGCGTGAA | CATCCAGGTGAAAGCGGC |
| IL-12 | AAAATAGATGCGTGCAAGAGAGG | GGGGAAGACCTGTGACTTGAG |
| PD-L1 | AAATGGAACCTGGCGAAAGC | GATGAGCCCCTCAGGCATTT |
| CD206 | CTACAAGGGATCGGGTTTATGGA | TTGGCATTGCCTAGTAGCGTA |
| CCL2 | CTCTCGCCTCCAGCATGAAA | TTTGCTTGTCCAGGTGGTCC |
| CD68 | CTTCTCTCATTCCCCTATGGACA | GAAGGACACATTGTACTCCACC |
| KGA | GGTCTCCTCCTCTGGATAAGATGG | CCCGTTGTCAGAATCTCCTTGAGG |
| SLC1A5 | TCATGTGGTACGCCCCTGT | GCGGGCAAAGAGTAAACCCA |
| NOX2 | CCTAAGATAGCGGTTGATGG | GACTTGAGAATGGATGCGAA |
| SOD2 | CACTGCAAGGAACAACAGGC | ACCAGGCTTGATGCACATCTT |
| 18S | CATTTAGGTGACACTATAGAAGACGATCAGATACCGTCGTAGTTCC | GGATCCTAATACGACTCACTATAGGCCTTTAAGTTTCAGCTTTGCAACC |
| Targets | Antibodies | Blocking Solutions | Primary Antibody Dilution and Incubation | Secondary Antibodies |
|---|---|---|---|---|
| CD68 | Anti-human, rabbit monoclonal, anti-CD68, Cell Signaling | PBS/NGS 5%/Triton 0.3% (1 h) | 1/800, overnight 4 °C | Goat anti-Rabbit IgG (H + L) Highly Cross-Absorbed Secondary Antibody, Alexa Fluor Plus 488 |
| CD14 | Anti-human, mouse monoclonal, anti-CD14, Miltenyi Biotec | PBS/BSA 2% (20 min) | 1/100, overnight 4 °C | Goat anti-Mouse IgG (H + L) Highly Cross-Absorbed Secondary Antibody, Alexa Fluor Plus 555 |
| CD36 | Anti-human, rabbit monoclonal, anti-CD36, ThermoFisher Scientific | PBS/BSA 2% (20 min) | 1/100, overnight 4 °C | Goat anti-Rabbit IgG (H + L) Highly Cross-Absorbed Secondary Antibody, Alexa Fluor Plus 488 |
| CD80 | Anti-human, mouse monoclonal, anti-CD80, R&D Systems | PBS/casein 0.5% (1 h) | 1/50, overnight 4 °C | Goat anti-Mouse IgG (H + L) Highly Cross-Absorbed Secondary Antibody, Alexa Fluor Plus 555 |
| CD86 | Anti-human, rabbit monoclonal, anti-CD86, Cell Signaling | PBS/NGS 5%/Triton 0.3% (1 h) | 1/100, overnight 4 °C | Goat anti-Rabbit IgG (H + L) Highly Cross-Absorbed Secondary Antibody, Alexa Fluor Plus 488 |
| CD163 | Anti-human, mouse monoclonal, anti-CD163, Sanbio | PBS/BSA 2% (20 min) | 1/50, overnight 4 °C | Goat anti-Mouse IgG (H + L) Highly Cross-Absorbed Secondary Antibody, Alexa Fluor Plus 555 |
| CD206 | Anti-human, mouse monoclonal, anti-CD206, Miltenyi Biotec | PBS/BSA 2% (20 min) | 1/100, overnight 4 °C | Goat anti-Mouse IgG (H + L) Highly Cross-Absorbed Secondary Antibody, Alexa Fluor Plus 555 |
| Nrf2 | Anti-human, rabbit monoclonal, anti-Nrf2, Cell Signaling | PBS/NGS 5%/Triton 0.3% (1 h) | 1/200, overnight 4 °C | Goat anti-Rabbit IgG (H + L) Highly Cross-Absorbed Secondary Antibody, Alexa Fluor Plus 488 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Furgiuele, S.; Descamps, G.; Cascarano, L.; Boucq, A.; Dubois, C.; Journe, F.; Saussez, S. Dealing with Macrophage Plasticity to Address Therapeutic Challenges in Head and Neck Cancers. Int. J. Mol. Sci. 2022, 23, 6385. https://doi.org/10.3390/ijms23126385
Furgiuele S, Descamps G, Cascarano L, Boucq A, Dubois C, Journe F, Saussez S. Dealing with Macrophage Plasticity to Address Therapeutic Challenges in Head and Neck Cancers. International Journal of Molecular Sciences. 2022; 23(12):6385. https://doi.org/10.3390/ijms23126385
Chicago/Turabian StyleFurgiuele, Sonia, Géraldine Descamps, Lorena Cascarano, Ambre Boucq, Christine Dubois, Fabrice Journe, and Sven Saussez. 2022. "Dealing with Macrophage Plasticity to Address Therapeutic Challenges in Head and Neck Cancers" International Journal of Molecular Sciences 23, no. 12: 6385. https://doi.org/10.3390/ijms23126385
APA StyleFurgiuele, S., Descamps, G., Cascarano, L., Boucq, A., Dubois, C., Journe, F., & Saussez, S. (2022). Dealing with Macrophage Plasticity to Address Therapeutic Challenges in Head and Neck Cancers. International Journal of Molecular Sciences, 23(12), 6385. https://doi.org/10.3390/ijms23126385