
Human Brain Models of Intellectual Disability: Experimental Advances and Novelties



{kind=link}
{kind=link}
Abstract
:1. Intellectual Disability
- 1
- Deficits in intellectual functions, such as reasoning, problem solving, planning, abstract thinking, judgment, academic learning, and learning from experience, confirmed by both clinical assessment and individualized, standardized intelligence testing.
- 2
- Deficits in adaptive functioning that result in failure to meet developmental and sociocultural standards for personal independence and social responsibility. Without ongoing support, the adaptive deficits limit functioning in one or more activities of daily life, such as communication, social participation and independent living, across multiple environments, such as home, school, work and community.
- 3
- The onset of intellectual and adaptive deficits during the developmental period. ID applies a heavy burden on the affected individuals, families, the society and the health care system. The extremely heterogeneous nature of ID is a consequence of the wide-ranging underlying etiology. Severe ID can be identified by delayed motor, language and social milestones during the first 2 years of life, while mild ID may not be identifiable until school age when academic learning starts [1].
2. Novel Models of ID
2.1. Pluripotent Stem Cells
2.2. Human Brain Models In Vitro
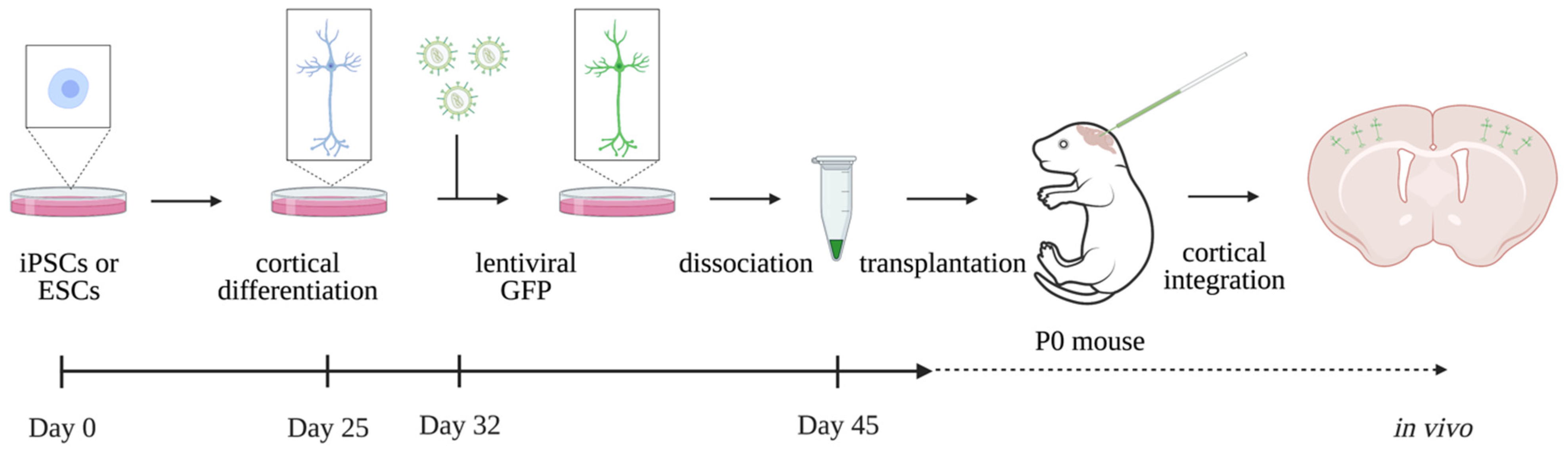
2.3. Human Brain Models In Vivo—Xenotransplantation
2.4. Gene Editing Tools
3. Modelling ID in Practice
3.1. Fragile X Syndrome
3.2. Rett Syndrome
3.3. MECP2 Duplication Syndrome
3.4. Williams–Beuren Syndrome
3.5. Prader–Willi Syndrome
3.6. Angelman Syndrome
4. Challenges and Future Perspectives
- Individuals suffering from ID often carry unique or rare mutations. Reprogramming and differentiating patient derived cell lines involve high costs, time and expertise [20].
- Given that ID comprises cognitive testing and other psychiatric comorbidities, behavioral assays still rely on animal models. However, animal models lack specific cellular features, such as the complexity and slow maturation properties, that are unique to the human brain [72].
- Late timepoints of ID etiology are not obtainable due to the incomplete maturation of hiPSC-derived neurons. Especially for diseases with late onset of symptoms, such as RTT, this poses a problem, as the underlying mechanisms preceding late onset cannot be studied up until now.
- Although iPSCs can be used to model imprinted diseases, one question is whether the genome-wide imprinting status is conserved during the epigenetic rewiring that takes place during somatic reprogramming. iPSCs are known to have defective imprinting, even using different reprogramming procedures. This should be taken into account when using iPSCs to model diseases caused by imprinting defects [69,73].
- In vitro models experience metabolic and endoplasmic reticulum (ER) stress on the cultured cells, which might hamper correct analyses and interpretation of findings. Moreover, in vitro models do not reproduce the complexity of the in vivo brain. Xenotransplantation of in vitro cultured hiPSC-derived neurons into the mouse brain, where cells can fully integrate into the neuronal circuits and initiate action potentials, is known to rescue the cellular stress. Grafting human neurons in the mouse brain additionally enables the study of human neurons up to later stages and in a more physiological setting. Modelling ID using the existing in vivo xenotransplantation model might decode disease etiology to a further extent than in vitro models can [32,33].
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, 5th ed.; American Psychiatric Association: Arlington, VA, USA, 2013; pp. 33–39. ISBN 978-0-89042-554-1. [Google Scholar]
- Maia, N.; Nabais Sá, M.J.; Melo-Pires, M.; de Brouwer, A.P.M.; Jorge, P. Intellectual disability genomics: Current state, pitfalls and future challenges. BMC Genom. 2021, 22, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, S.; Love-Nichols, J.A.; Dies, K.A.; Ledbetter, D.H.; Martin, C.L.; Chung, W.K.; Firth, H.V.; Frazier, T.; Hansen, R.L.; Prock, L.; et al. Meta-analysis and multidisciplinary consensus statement: Exome sequencing is a first-tier clinical diagnostic test for individuals with neurodevelopmental disorders. Genet. Med. 2019, 21, 2413–2421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Luca, C.; Race, V.; Keldermans, L.; Bauters, M.; Van Esch, H. Challenges in molecular diagnosis of X-linked Intellectual disability. Br. Med. Bull. 2020, 133, 36–48. [Google Scholar] [CrossRef] [PubMed]
- Kim, C. iPSC technology-Powerful hand for disease modeling and therapeutic screen. BMB Rep. 2015, 48, 256–265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nikolakopoulou, P.; Rauti, R.; Voulgaris, D.; Shlomy, I.; Maoz, B.M.; Herland, A. Recent progress in translational engineered in vitro models of the central nervous system. Brain 2020, 143, 3181–3213. [Google Scholar] [CrossRef]
- Evans, M.J.; Kaufman, M.H. Establishment in culture of pluripotential cells from mouse embryos. Nature 1981, 292, 154–156. [Google Scholar] [CrossRef] [PubMed]
- Martin, G.R. Isolation of a pluripotent cell line from early mouse embryos cultured in medium conditioned by teratocarcinoma stem cells. Proc. Natl. Acad. Sci. USA 1981, 78, 7634–7638. [Google Scholar] [CrossRef] [Green Version]
- Lian, X.; Hsiao, C.; Wilson, G.; Zhu, K.; Hazeltine, L.B.; Azarin, S.M.; Raval, K.K.; Zhang, J.; Kamp, T.J.; Palecek, S.P. Robust cardiomyocyte differentiation from human pluripotent stem cells via temporal modulation of canonical Wnt signaling. Proc. Natl. Acad. Sci. USA 2012, 109, 1848–1857. [Google Scholar] [CrossRef] [Green Version]
- Hannan, N.R.F.; Segeritz, C.P.; Touboul, T.; Vallier, L. Production of hepatocyte-like cells from human pluripotent stem cells. Nat. Protoc. 2013, 8, 430–437. [Google Scholar] [CrossRef] [Green Version]
- Karumbayaram, S.; Novitch, B.G.; Patterson, M.; Umbach, J.A.; Richter, L.; Lindgren, A.; Conway, A.E.; Clark, A.T.; Goldman, S.A.; Plath, K.; et al. Directed differentiation of human-induced pluripotent stem cells generates active motor neurons. Stem Cells 2009, 27, 806–811. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y.; Kirwan, P.; Livesey, F.J. Directed differentiation of human pluripotent stem cells to cerebral cortex neurons and neural networks. Nat. Protoc. 2012, 7, 1836–1846. [Google Scholar] [CrossRef] [PubMed]
- Qi, Y.; Zhang, X.J.; Renier, N.; Wu, Z.; Atkin, T.; Sun, Z.; Ozair, M.Z.; Tchieu, J.; Zimmer, B.; Fattahi, F.; et al. Combined small-molecule inhibition accelerates the derivation of functional, early-born, cortical neurons from human pluripotent stem cells. Nat. Biotechnol. 2017, 35, 154–163. [Google Scholar] [CrossRef] [PubMed]
- Gaspard, N.; Bouschet, T.; Hourez, R.; Dimidschstein, J.; Naeije, G.; Van Den Ameele, J.; Espuny-Camacho, I.; Herpoel, A.; Passante, L.; Schiffmann, S.N.; et al. An intrinsic mechanism of corticogenesis from embryonic stem cells. Nature 2008, 455, 351–357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Espuny-Camacho, I.; Michelsen, K.A.; Gall, D.; Linaro, D.; Hasche, A.; Bonnefont, J.; Bali, C.; Orduz, D.; Bilheu, A.; Herpoel, A.; et al. Pyramidal Neurons Derived from Human Pluripotent Stem Cells Integrate Efficiently into Mouse Brain Circuits In Vivo. Neuron 2013, 77, 440–456. [Google Scholar] [CrossRef] [Green Version]
- Doss, M.X.; Sachinidis, A. Current Challenges of iPSC-Based Disease Modeling and Therapeutic Implications. Cells 2019, 8, 403. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Adult Human Fibroblasts by Defined Factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Mouse Embryonic and Adult Fibroblast Cultures by Defined Factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [Green Version]
- Chandrasekaran, A.; Avci, H.X.; Ochalek, A.; Rösingh, L.N.; Molnár, K.; László, L.; Bellák, T.; Téglási, A.; Pesti, K.; Mike, A.; et al. Comparison of 2D and 3D neural induction methods for the generation of neural progenitor cells from human induced pluripotent stem cells. Stem Cell Res. 2017, 25, 139–151. [Google Scholar] [CrossRef]
- Sabitha, K.R.; Shetty, A.K.; Upadhya, D. Patient-derived iPSC modeling of rare neurodevelopmental disorders: Molecular pathophysiology and prospective therapies. Neurosci. Biobehav. Rev. 2021, 121, 201–219. [Google Scholar] [CrossRef]
- Sloan, S.A.; Andersen, J.; Pașca, A.M.; Birey, F.; Pașca, S.P. Generation and Assembly of Human Brain Region-Specific Three-Dimensional Cultures. Nat. Protoc. 2018, 13, 2062–2085. [Google Scholar] [CrossRef]
- Lancaster, M.A.; Knoblich, J.A. Generation of cerebral organoids from human pluripotent stem cells. Nat. Protoc. 2014, 9, 2329–2340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhattacharya, A.; Choi, W.W.Y.; Muffat, J.; Li, Y. Modeling Developmental Brain Diseases Using Human Pluripotent Stem Cells-Derived Brain Organoids—Progress and Perspective. J. Mol. Biol. 2022, 434, 167386. [Google Scholar] [CrossRef] [PubMed]
- Marton, R.M.; Pașca, S.P. Organoid and Assembloid Technologies for Investigating Cellular Crosstalk in Human Brain Development and Disease. Trends Cell Biol. 2020, 30, 133–143. [Google Scholar] [CrossRef] [PubMed]
- Lancaster, M.A.; Renner, M.; Martin, C.A.; Wenzel, D.; Bicknell, L.S.; Hurles, M.E.; Homfray, T.; Penninger, J.M.; Jackson, A.P.; Knoblich, J.A. Cerebral organoids model human brain development and microcephaly. Nature 2013, 501, 373–379. [Google Scholar] [CrossRef] [PubMed]
- Gordon, A.; Yoon, S.J.; Tran, S.S.; Makinson, C.D.; Park, J.Y.; Andersen, J.; Valencia, A.M.; Horvath, S.; Xiao, X.; Huguenard, J.R.; et al. Long-term maturation of human cortical organoids matches key early postnatal transitions. Nat. Neurosci. 2021, 24, 331–342. [Google Scholar] [CrossRef] [PubMed]
- Passaro, A.P.; Stice, S.L. Electrophysiological Analysis of Brain Organoids: Current Approaches and Advancements. Front. Neurosci. 2021, 14, 622137. [Google Scholar] [CrossRef]
- Grienberger, C.; Konnerth, A. Imaging Calcium in Neurons. Neuron 2012, 73, 862–885. [Google Scholar] [CrossRef] [Green Version]
- Trujillo, C.A.; Muotri, A.R. Brain Organoids and the Study of Neurodevelopment. Trends Mol. Med. 2018, 24, 982–990. [Google Scholar] [CrossRef]
- Nakai, J.; Ohkura, M.; Imoto, K. A high signal-to-noise Ca(2+) probe composed of a single green fluorescent protein. Nat. Biotechnol. 2001, 19, 137–141. [Google Scholar] [CrossRef]
- Kelava, I.; Lancaster, M.A. Dishing out mini-brains: Current progress and future prospects in brain organoid research. Dev. Biol. 2016, 420, 199–209. [Google Scholar] [CrossRef] [Green Version]
- Linaro, D.; Vermaercke, B.; Iwata, R.; Ramaswamy, A.; Libé-Philippot, B.; Boubakar, L.; Davis, B.A.; Wierda, K.; Davie, K.; Poovathingal, S.; et al. Xenotransplanted Human Cortical Neurons Reveal Species-Specific Development and Functional Integration into Mouse Visual Circuits. Neuron 2019, 104, 972–986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhaduri, A.; Andrews, M.G.; Mancia Leon, W.; Jung, D.; Shin, D.; Allen, D.; Jung, D.; Schmunk, G.; Haeussler, M.; Salma, J.; et al. Cell stress in cortical organoids impairs molecular subtype specification. Nature 2020, 578, 142–148. [Google Scholar] [CrossRef] [PubMed]
- Espuny-Camacho, I.; Michelsen, K.A.; Linaro, D.; Bilheu, A.; Acosta-Verdugo, S.; Herpoel, A.; Giugliano, M.; Gaillard, A.; Vanderhaeghen, P. Human Pluripotent Stem-Cell-Derived Cortical Neurons Integrate Functionally into the Lesioned Adult Murine Visual Cortex in an Area-Specific Way. Cell Rep. 2018, 23, 2732–2743. [Google Scholar] [CrossRef] [Green Version]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef]
- Huong Le, T.T.; Tran, N.T.; Lan Dao, T.M.; Nguyen, D.D.; Do, H.D.; Ha, T.L.; Kühn, R.; Nguyen, T.L.; Rajewsky, K.; Chu, V.T. Efficient and precise CRISPR/Cas9-mediated MECP2 modifications in human-induced pluripotent stem cells. Front. Genet. 2019, 10, 625. [Google Scholar] [CrossRef]
- Xie, N.; Gong, H.; Suhl, J.A.; Chopra, P.; Wang, T.; Warren, S.T. Reactivation of FMR1 by CRISPR/Cas9-Mediated Deletion of the Expanded CGG-Repeat of the Fragile X Chromosome. PLoS ONE 2016, 11, e0165499. [Google Scholar] [CrossRef] [Green Version]
- Gardiner, K.L.; Cideciyan, A.V.; Swider, M.; Dufour, V.L.; Sumaroka, A.; Komáromy, A.M.; Hauswirth, W.W.; Iwabe, S.; Jacobson, S.G.; Beltran, W.A.; et al. Long-Term Structural Outcomes of Late-Stage RPE65 Gene Therapy. Mol. Ther. 2020, 28, 266–278. [Google Scholar] [CrossRef]
- Ozlu, C.; Bailey, R.M.; Sinnett, S.; Goodspeed, K.D. Gene Transfer Therapy for Neurodevelopmental Disorders. Dev. Neurosci. 2021, 43, 230–240. [Google Scholar] [CrossRef]
- Ilyas, M.; Mir, A.; Efthymiou, S.; Houlden, H. The genetics of intellectual disability: Advancing technology and gene editing. F1000Research 2020, 9, 22. [Google Scholar] [CrossRef] [Green Version]
- Hunter, J.E.; Berry-Kravis, E.; Hipp, H.; Todd, P.K. FMR1 Disorders, GeneReviews®; University of Washington: Seattle, WA, USA, 2019. [Google Scholar]
- Telias, M. Molecular Mechanisms of Synaptic Dysregulation in Fragile X Syndrome and Autism Spectrum Disorders. Front. Mol. Neurosci. 2019, 12, 51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.Y. Using an Isogenic Human Pluripotent Stem Cell Model for Better Understanding Neurodevelopmental Defects in Fragile X Syndrome. Biol. Psychiatry 2020, 88, e25–e27. [Google Scholar] [CrossRef] [PubMed]
- NIH National Library of Medicine (USA). Pharmaceuticals N. Clinical Study to Assess the Pharmacokinetics, Safety and Tolerability of Single and Multiple Oral Doses of AFQ056 in Children with Fragile X Syndrome (FXS). Identifier NCT01482143. Available online: https://clinicaltrials.gov/ct2/show/NCT01482143 (accessed on 20 April 2022).
- Achuta, V.S.; Möykkynen, T.; Peteri, U.K.; Turconi, G.; Rivera, C.; Keinänen, K.; Castrén, M.L. Functional changes of AMPA responses in human induced pluripotent stem cell-derived neural progenitors in fragile X syndrome. Sci. Signal. 2018, 11, eaan8784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brighi, C.; Salaris, F.; Soloperto, A.; Cordella, F.; Ghirga, S.; de Turris, V.; Rosito, M.; Porceddu, P.F.; D’Antoni, C.; Reggiani, A.; et al. Novel fragile X syndrome 2D and 3D brain models based on human isogenic FMRP-KO iPSCs. Cell Death Dis. 2021, 12, 498. [Google Scholar] [CrossRef]
- Amir, R.E.; Van Den Veyver, I.B.; Wan, M.; Tran, C.Q.; Francke, U.; Zoghbi, H.Y. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl- CpG-binding protein 2. Nat. Genet. 1999, 23, 185–188. [Google Scholar] [CrossRef]
- Marchetto, M.C.N.; Carromeu, C.; Acab, A.; Yu, D.; Yeo, G.W.; Mu, Y.; Chen, G.; Gage, F.H.; Muotri, A.R. A model for neural development and treatment of rett syndrome using human induced pluripotent stem cells. Cell 2010, 143, 527–539. [Google Scholar] [CrossRef] [Green Version]
- Farra, N.; Zhang, W.B.; Pasceri, P.; Eubanks, J.H.; Salter, M.W.; Ellis, J. Rett syndrome induced pluripotent stem cell-derived neurons reveal novel neurophysiological alterations. Mol. Psychiatry 2012, 17, 1261–1271. [Google Scholar] [CrossRef] [Green Version]
- Ohashi, M.; Korsakova, E.; Allen, D.; Lee, P.; Fu, K.; Vargas, B.S.; Cinkornpumin, J.; Salas, C.; Park, J.C.; Germanguz, I.; et al. Loss of MECP2 Leads to Activation of P53 and Neuronal Senescence. Stem Cell Rep. 2018, 10, 1453–1463. [Google Scholar] [CrossRef]
- Trujillo, C.A.; Adams, J.W.; Negraes, P.D.; Carromeu, C.; Tejwani, L.; Acab, A.; Tsuda, B.; Thomas, C.A.; Sodhi, N.; Fichter, K.M.; et al. Pharmacological reversal of synaptic and network pathology in human MECP2 -KO neurons and cortical organoids. EMBO Mol. Med. 2021, 13, e12523. [Google Scholar] [CrossRef]
- Haase, F.D.; Coorey, B.; Riley, L.; Cantrill, L.C.; Tam, P.P.L.; Gold, W.A. Pre-clinical Investigation of Rett Syndrome Using Human Stem Cell-Based Disease Models. Front. Neurosci. 2021, 15, 994. [Google Scholar] [CrossRef]
- Gomes, A.R.; Fernandes, T.G.; Vaz, S.H.; Silva, T.P.; Bekman, E.P.; Xapelli, S.; Duarte, S.; Ghazvini, M.; Gribnau, J.; Muotri, A.R.; et al. Modeling Rett Syndrome With Human Patient-Specific Forebrain Organoids. Front. Cell Dev. Biol. 2020, 8, 610427. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, H.; Muffat, J.; Cheng, A.W.; Orlando, D.A.; Lovén, J.; Kwok, S.M.; Feldman, D.A.; Bateup, H.S.; Gao, Q.; et al. Global transcriptional and translational repression in human embryonic stem cells-derived Rett Syndrome neurons. Cell Stem Cell 2013, 13, 446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guy, J.; Gan, J.; Selfridge, J.; Cobb, S.; Bird, A. Reversal of neurological defects in a mouse model of Rett syndrome. Science 2007, 315, 1143–1147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Esch, H. MECP2 Duplication Syndrome. Mol. Syndromol. 2020, 2, 128–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Esch, H.; Bauters, M.; Ignatius, J.; Jansen, M.; Raynaud, M.; Hollanders, K.; Lugtenberg, D.; Bienvenu, T.; Jensen, L.R.; Gécz, J.; et al. Duplication of the MECP2 region is a frequent cause of severe mental retardation and progressive neurological symptoms in males. Am. J. Hum. Genet. 2005, 77, 442–453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nageshappa, S.; Carromeu, C.; Trujillo, C.A.; Mesci, P.; Espuny-Camacho, I.; Pasciuto, E.; Vanderhaeghen, P.; Verfaillie, C.M.; Raitano, S.; Kumar, A.; et al. Altered neuronal network and rescue in a human MECP2 duplication model. Mol. Psychiatry 2016, 21, 178–188. [Google Scholar] [CrossRef] [Green Version]
- Morris, C.A. Williams Syndrome, GeneReviews®; University of Washington Seattle: Seattle, WA, USA, 2017. [Google Scholar]
- Osborne, L.R. Animal models of Williams syndrome. Am. J. Med. Genet. C. Semin. Med. Genet. 2010, 154C, 209–219. [Google Scholar] [CrossRef] [Green Version]
- Henrichsen, C.N.; Csárdi, G.; Zabot, M.T.; Fusco, C.; Bergmann, S.; Merla, G.; Reymond, A. Using Transcription Modules to Identify Expression Clusters Perturbed in Williams-Beuren Syndrome. PLoS Comput. Biol. 2011, 7, 1001054. [Google Scholar] [CrossRef]
- Antonell, A.; Vilardell, M.; Jurado, L.A.P. Transcriptome profile in Williams-Beuren syndrome lymphoblast cells reveals gene pathways implicated in glucose intolerance and visuospatial construction deficits. Hum. Genet. 2010, 128, 27–37. [Google Scholar] [CrossRef]
- Khattak, S.; Brimble, E.; Zhang, W.; Zaslavsky, K.; Strong, E.; Ross, P.J.; Hendry, J.; Mital, S.; Salter, M.W.; Osborne, L.R.; et al. Human induced pluripotent stem cell derived neurons as a model for Williams-Beuren syndrome. Mol. Brain 2015, 8, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Chailangkarn, T.; Trujillo, C.A.; Freitas, B.C.; Hrvoj-Mihic, B.; Herai, R.H.; Yu, D.X.; Brown, T.T.; Marchetto, M.C.; Bardy, C.; McHenry, L.; et al. A human neurodevelopmental model for Williams syndrome. Nature 2016, 536, 338–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kinnear, C.; Chang, W.Y.; Khattak, S.; Hinek, A.; Thompson, T.; de Carvalho Rodrigues, D.; Kennedy, K.; Mahmut, N.; Pasceri, P.; Stanford, W.L.; et al. Modeling and rescue of the vascular phenotype of Williams-Beuren syndrome in patient induced pluripotent stem cells. Stem Cells Transl. Med. 2013, 2, 2–15. [Google Scholar] [CrossRef] [PubMed]
- Driscoll, D.J.; Miller, J.L.; Schwartz, S.; Cassidy, S.B. Prader-Willi Syndrome, GeneReviews®; University of Washington Seattle: Seattle, WA, USA, 2017. [Google Scholar]
- Chen, H.; Kaitlyn Victor, A.; Klein, J.; Tacer, K.F.; Tai, D.J.C.; de Esch, C.; Nuttle, A.; Temirov, J.; Burnett, L.C.; Rosenbaum, M.; et al. Loss of MAGEL2 in Prader-Willi syndrome leads to decreased secretory granule and neuropeptide production. JCI Insight 2020, 5, e138576. [Google Scholar] [CrossRef] [PubMed]
- Pólvora-Brandão, D.; Joaquim, M.; Godinho, I.; Aprile, D.; Álvaro, A.R.; Onofre, I.; Raposo, A.C.; Pereira de Almeida, L.; Duarte, S.T.; da Rocha, S.T. Loss of hierarchical imprinting regulation at the Prader–Willi/Angelman syndrome locus in human iPSCs. Hum. Mol. Genet. 2018, 27, 3999–4011. [Google Scholar] [CrossRef] [PubMed]
- Fink, J.J.; Robinson, T.M.; Germain, N.D.; Sirois, C.L.; Bolduc, K.A.; Ward, A.J.; Rigo, F.; Chamberlain, S.J.; Levine, E.S. Disrupted neuronal maturation in Angelman syndrome-derived induced pluripotent stem cells. Nat. Commun. 2017, 8, 1–14. [Google Scholar] [CrossRef]
- Dagli, A.I.; Mathews, J.; Williams, C.A. Angelman Syndrome, GeneReviews®; University of Washington Seattle: Seattle, WA, USA, 2021. [Google Scholar]
- Libé-Philippot, B.; Vanderhaeghen, P. Cellular and Molecular Mechanisms Linking Human Cortical Development and Evolution. Annu. Rev. Genet. 2021, 55, 555–581. [Google Scholar] [CrossRef]
- Yang, J.; Cai, J.; Zhang, Y.; Wang, X.; Li, W.; Xu, J.; Li, F.; Guo, X.; Deng, K.; Zhong, M.; et al. Induced Pluripotent Stem Cells Can Be Used to Model the Genomic Imprinting Disorder Prader-Willi Syndrome. J. Biol. Chem. 2010, 285, 40303. [Google Scholar] [CrossRef] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Merckx, N.L.L.; Van Esch, H. Human Brain Models of Intellectual Disability: Experimental Advances and Novelties. Int. J. Mol. Sci. 2022, 23, 6476. https://doi.org/10.3390/ijms23126476
Merckx NLL, Van Esch H. Human Brain Models of Intellectual Disability: Experimental Advances and Novelties. International Journal of Molecular Sciences. 2022; 23(12):6476. https://doi.org/10.3390/ijms23126476
Chicago/Turabian StyleMerckx, Nona Laura Lisa, and Hilde Van Esch. 2022. "Human Brain Models of Intellectual Disability: Experimental Advances and Novelties" International Journal of Molecular Sciences 23, no. 12: 6476. https://doi.org/10.3390/ijms23126476
APA StyleMerckx, N. L. L., & Van Esch, H. (2022). Human Brain Models of Intellectual Disability: Experimental Advances and Novelties. International Journal of Molecular Sciences, 23(12), 6476. https://doi.org/10.3390/ijms23126476