
Particulate Matter Exacerbates the Death of Dopaminergic Neurons in Parkinson’s Disease through an Inflammatory Response


Abstract
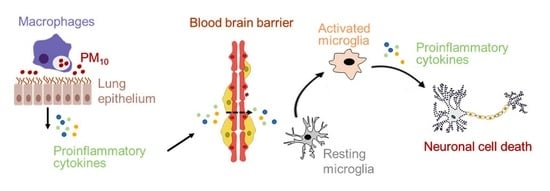
:
1. Introduction
2. Results
2.1. Chronic Intratracheal Instillation of PM10 Aggravated Motor Deficits in Mice with MPTP-induced PD
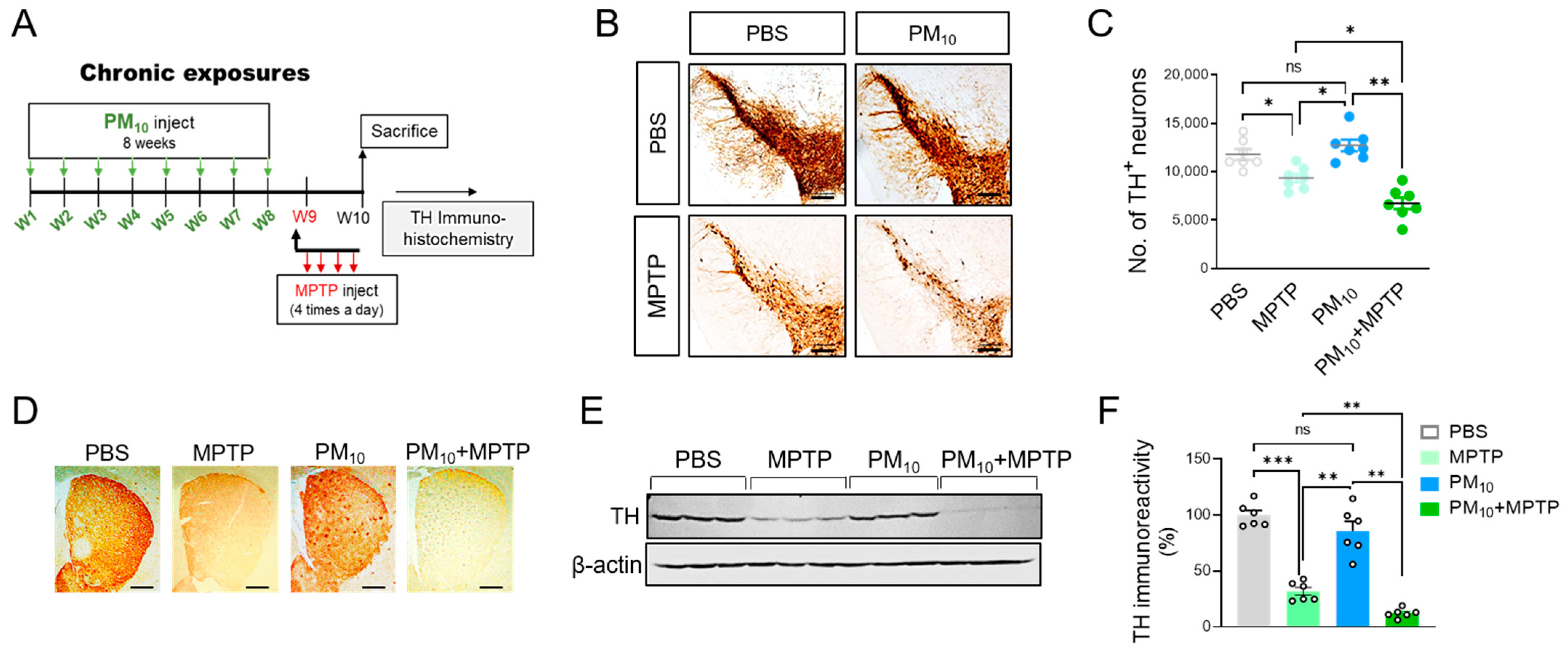
2.2. Chronic PM10 Exposure Enhanced the Degeneration of DA Neurons in PD Mice
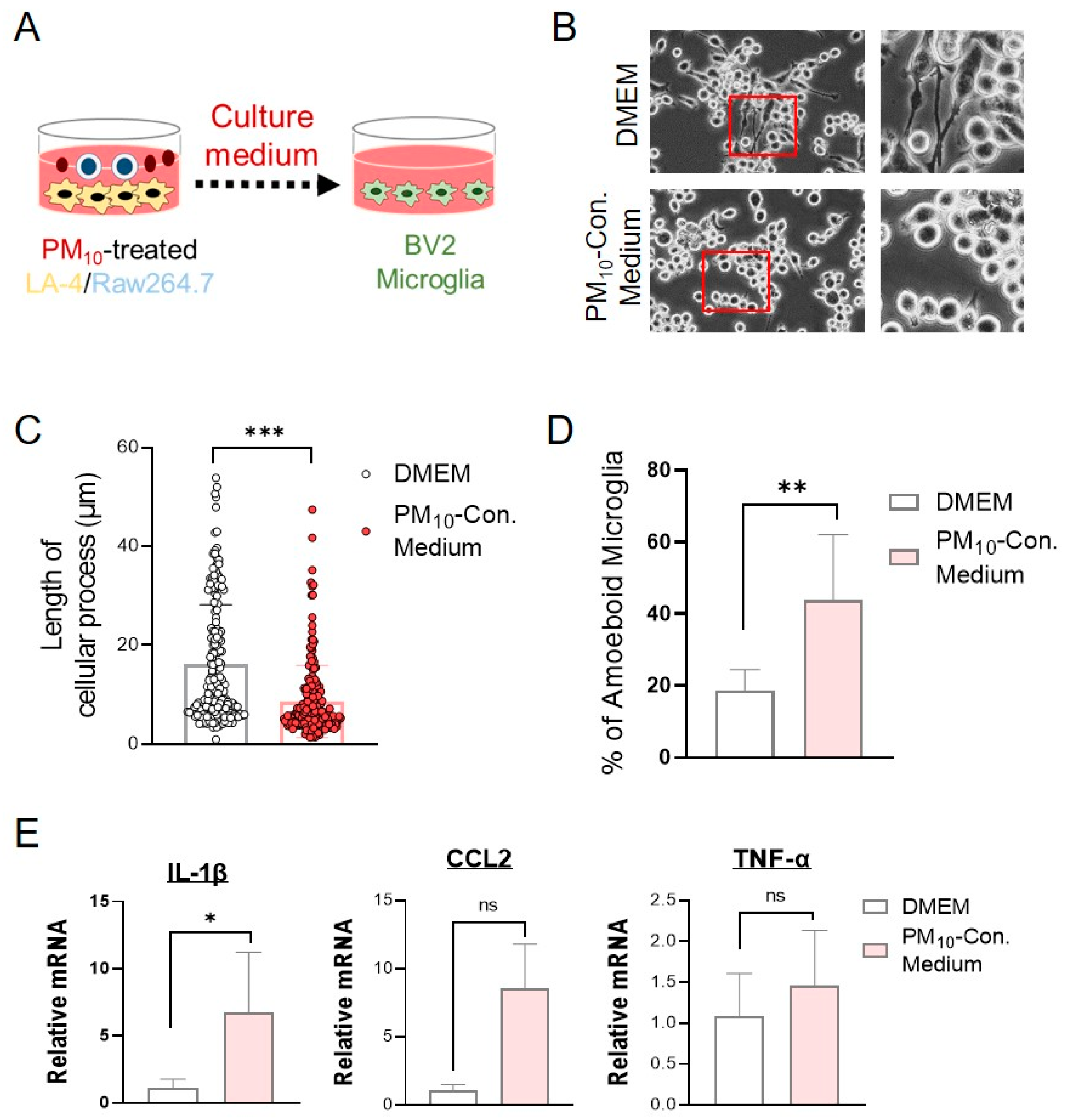
2.3. Short-Term Exposure to PM10 Caused Microglial Activation
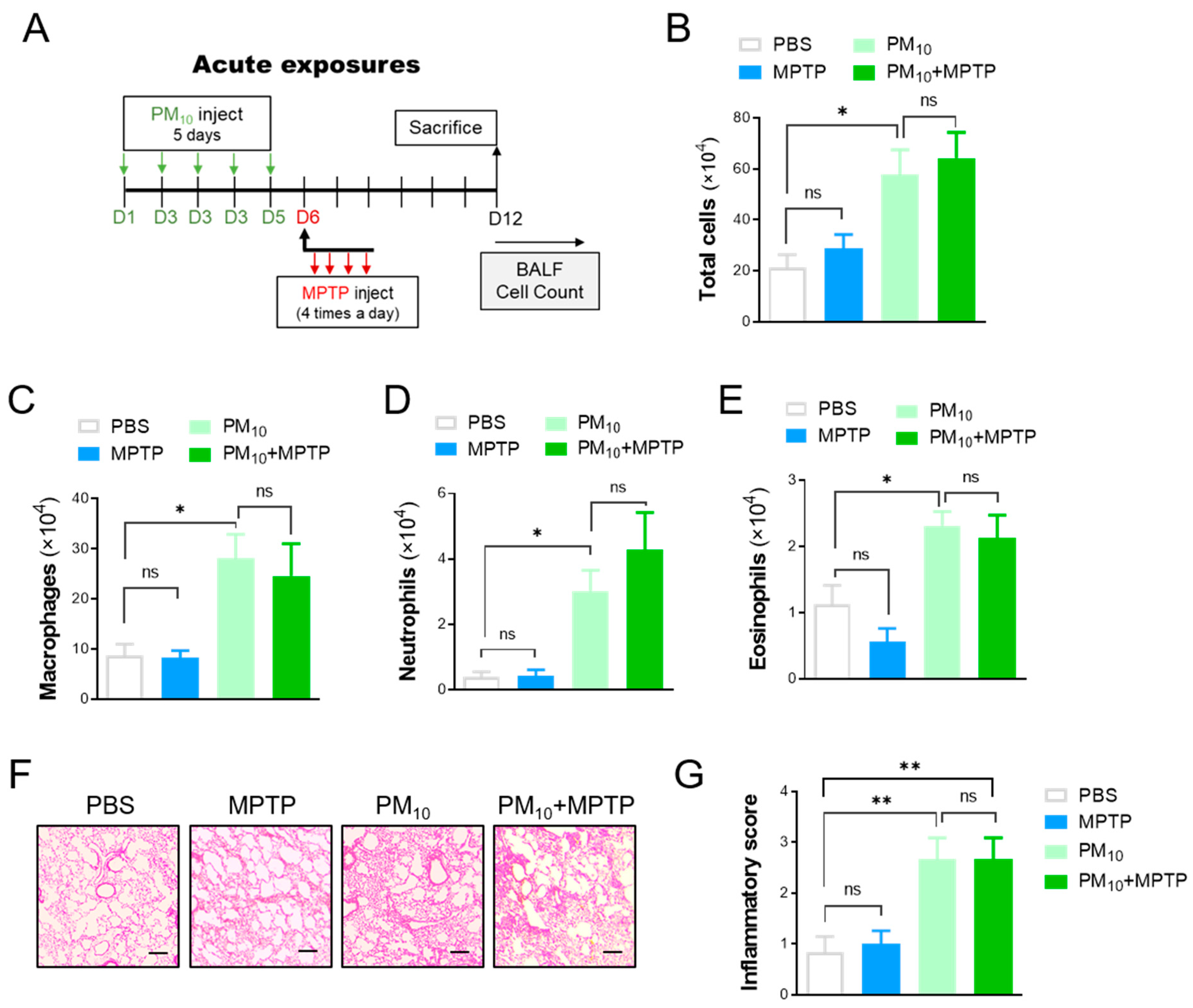
2.4. Short-Term Exposure to PM10 Led to Pulmonary Inflammation
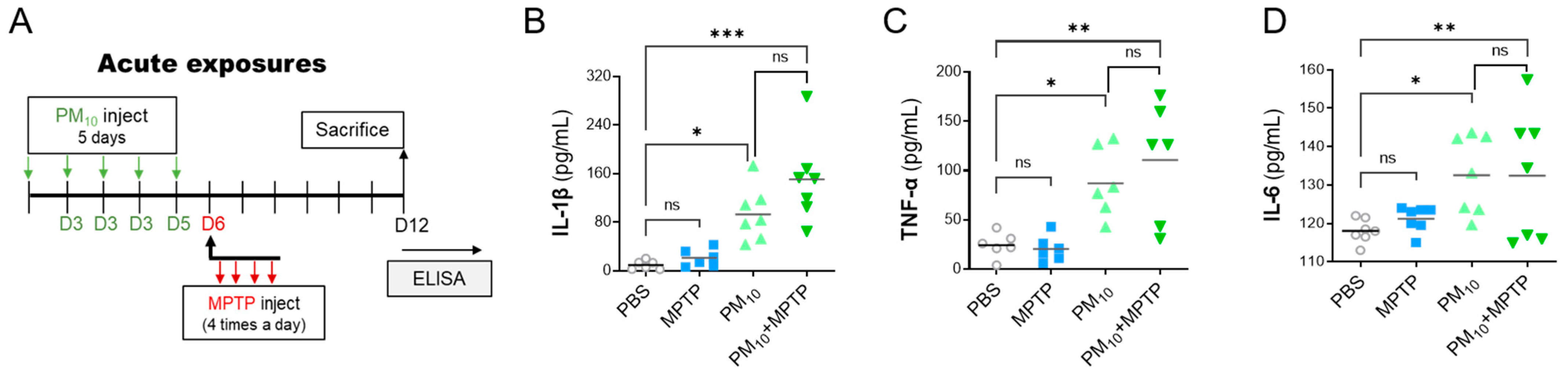
2.5. Short-Term Exposure to PM10 Induced Systemic Inflammation in PD Mice
2.6. PM10 Led to an Inflammatory Response in Cocultured Alveolar Epithelial Cells and Macrophages
2.7. Conditioned Medium from PM10-Stimulated Cocultures of LA-4- and RAW264.7-Activated Microglial Cells
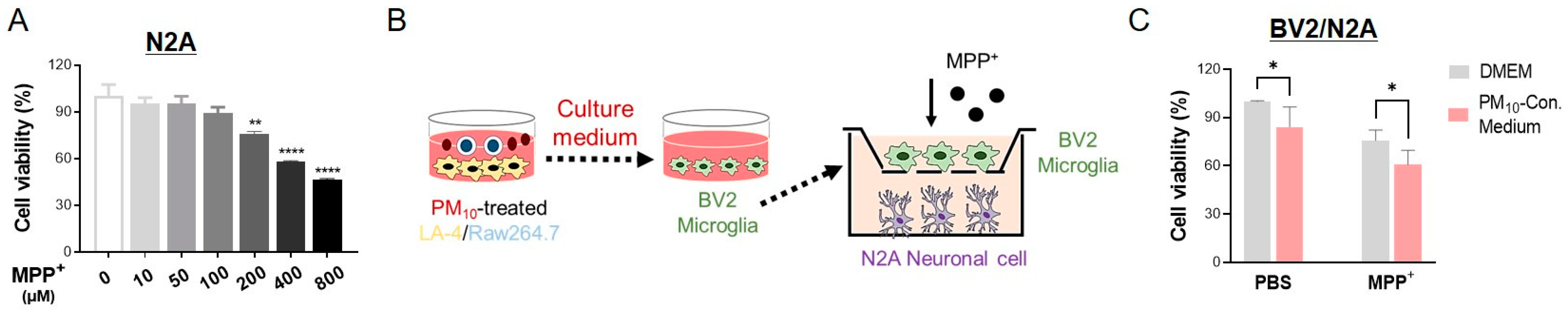
2.8. Activated Microglia Induced by PM10-Conditioned Medium Aggravated Neuronal Toxicity against MPP+
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. MPTP Intoxication
4.3. Intratracheal Instillation of PM10
4.4. Immunohistochemistry
4.5. Unbiased Stereological Estimation
4.6. Analysis of Bronchoalveolar Lavage Fluid
4.7. Locomotor Activity Measurement
4.8. Western Blotting Analysis
4.9. Enzyme-Linked Immunosorbent Assay
4.10. Quantitative Real-Time Polymerase Chain Reaction (Real-Time qPCR)
4.11. Cell Culture and PM10 Stimulus
4.12. Cytotoxicity Assay
4.13. Statistics
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Dockery, D.W.; Pope, C.A., 3rd; Xu, X.; Spengler, J.D.; Ware, J.H.; Fay, M.E.; Ferris, B.G., Jr.; Speizer, F.E. An association between air pollution and mortality in six U.S. cities. N. Engl. J. Med. 1993, 329, 1753–1759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palacios, N. Air pollution and Parkinson’s disease - evidence and future directions. Rev. Environ. Health 2017, 32, 303–313. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Ha, S.U.; Basnet, R. A Review of Epidemiological Research on Adverse Neurological Effects of Exposure to Ambient Air Pollution. Front. Public Health 2016, 4, 157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Block, M.L.; Calderon-Garciduenas, L. Air pollution: Mechanisms of neuroinflammation and CNS disease. Trends Neurosci. 2009, 32, 506–516. [Google Scholar] [CrossRef] [Green Version]
- Calderon-Garciduenas, L.; Reed, W.; Maronpot, R.R.; Henriquez-Roldan, C.; Delgado-Chavez, R.; Calderon-Garciduenas, A.; Dragustinovis, I.; Franco-Lira, M.; Aragon-Flores, M.; Solt, A.C.; et al. Brain inflammation and Alzheimer’s-like pathology in individuals exposed to severe air pollution. Toxicol. Pathol. 2004, 32, 650–658. [Google Scholar] [CrossRef] [PubMed]
- Calderon-Garciduenas, L.; Solt, A.C.; Henriquez-Roldan, C.; Torres-Jardon, R.; Nuse, B.; Herritt, L.; Villarreal-Calderon, R.; Osnaya, N.; Stone, I.; Garcia, R.; et al. Long-term air pollution exposure is associated with neuroinflammation, an altered innate immune response, disruption of the blood-brain barrier, ultrafine particulate deposition, and accumulation of amyloid beta-42 and alpha-synuclein in children and young adults. Toxicol Pathol. 2008, 36, 289–310. [Google Scholar] [CrossRef] [PubMed]
- Lang, A.E.; Lozano, A.M. Parkinson’s disease. First of two parts. N. Engl. J. Med. 1998, 339, 1044–1053. [Google Scholar] [CrossRef]
- Tufekci, K.U.; Meuwissen, R.; Genc, S.; Genc, K. Inflammation in Parkinson’s disease. Adv. Protein Chem. Struct. Biol. 2012, 88, 69–132. [Google Scholar] [CrossRef] [PubMed]
- von Bernhardi, R.; Eugenin-von Bernhardi, L.; Eugenin, J. Microglial cell dysregulation in brain aging and neurodegeneration. Front. Aging Neurosci. 2015, 7, 124. [Google Scholar] [CrossRef] [Green Version]
- Nimmerjahn, A.; Kirchhoff, F.; Helmchen, F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 2005, 308, 1314–1318. [Google Scholar] [CrossRef] [Green Version]
- Xing, Y.F.; Xu, Y.H.; Shi, M.H.; Lian, Y.X. The impact of PM2.5 on the human respiratory system. J. Thorac. Dis. 2016, 8, E69–E74. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Wu, X.; Danesh Yazdi, M.; Braun, D.; Abu Awad, Y.; Wei, Y.; Liu, P.; Di, Q.; Wang, Y.; Schwartz, J.; et al. Long-term effects of PM2.5 on neurological disorders in the American Medicare population: A longitudinal cohort study. Lancet Planet. Health 2020, 4, e557–e565. [Google Scholar] [CrossRef]
- Sama, P.; Long, T.C.; Hester, S.; Tajuba, J.; Parker, J.; Chen, L.C.; Veronesi, B. The cellular and genomic response of an immortalized microglia cell line (BV2) to concentrated ambient particulate matter. Inhal. Toxicol. 2007, 19, 1079–1087. [Google Scholar] [CrossRef] [PubMed]
- Qin, L.; Wu, X.; Block, M.L.; Liu, Y.; Breese, G.R.; Hong, J.S.; Knapp, D.J.; Crews, F.T. Systemic LPS causes chronic neuroinflammation and progressive neurodegeneration. Glia 2007, 55, 453–462. [Google Scholar] [CrossRef] [Green Version]
- Matsuura, K.; Kabuto, H.; Makino, H.; Ogawa, N. Pole test is a useful method for evaluating the mouse movement disorder caused by striatal dopamine depletion. J. Neurosci. Methods 1997, 73, 45–48. [Google Scholar] [CrossRef]
- Jones, B.J.; Roberts, D.J. The quantiative measurement of motor inco-ordination in naive mice using an acelerating rotarod. J. Pharm. Pharmacol. 1968, 20, 302–304. [Google Scholar] [CrossRef] [PubMed]
- Block, M.L.; Wu, X.; Pei, Z.; Li, G.; Wang, T.; Qin, L.; Wilson, B.; Yang, J.; Hong, J.S.; Veronesi, B. Nanometer size diesel exhaust particles are selectively toxic to dopaminergic neurons: The role of microglia, phagocytosis, and NADPH oxidase. FASEB J. 2004, 18, 1618–1620. [Google Scholar] [CrossRef] [Green Version]
- World Health Organization. WHO global Air Quality Guidelines: Particulate Matter (PM2.5 and PM10), Ozone, Nitrogen Dioxide, Sulfur Dioxide and Carbon Monoxide; World Health Organization: Geneva, Switerland, 2021. [Google Scholar]
- Ranft, U.; Schikowski, T.; Sugiri, D.; Krutmann, J.; Kramer, U. Long-term exposure to traffic-related particulate matter impairs cognitive function in the elderly. Environ. Res. 2009, 109, 1004–1011. [Google Scholar] [CrossRef]
- Calderon-Garciduenas, L.; Franco-Lira, M.; Torres-Jardon, R.; Henriquez-Roldan, C.; Barragan-Mejia, G.; Valencia-Salazar, G.; Gonzalez-Maciel, A.; Reynoso-Robles, R.; Villarreal-Calderon, R.; Reed, W. Pediatric respiratory and systemic effects of chronic air pollution exposure: Nose, lung, heart, and brain pathology. Toxicol Pathol 2007, 35, 154–162. [Google Scholar] [CrossRef] [PubMed]
- Becker, S.; Soukup, J.M.; Sioutas, C.; Cassee, F.R. Response of human alveolar macrophages to ultrafine, fine, and coarse urban air pollution particles. Exp. Lung Res. 2003, 29, 29–44. [Google Scholar] [CrossRef] [PubMed]
- Monn, C.; Becker, S. Cytotoxicity and induction of proinflammatory cytokines from human monocytes exposed to fine (PM2.5) and coarse particles (PM10-2.5) in outdoor and indoor air. Toxicol Appl Pharmacol 1999, 155, 245–252. [Google Scholar] [CrossRef]
- Soukup, J.M.; Becker, S. Human alveolar macrophage responses to air pollution particulates are associated with insoluble components of coarse material, including particulate endotoxin. Toxicol. Appl. Pharmacol. 2001, 171, 20–26. [Google Scholar] [CrossRef] [PubMed]
- Ljubimova, J.Y.; Braubach, O.; Patil, R.; Chiechi, A.; Tang, J.; Galstyan, A.; Shatalova, E.S.; Kleinman, M.T.; Black, K.L.; Holler, E. Coarse particulate matter (PM2.5-10) in Los Angeles Basin air induces expression of inflammation and cancer biomarkers in rat brains. Sci. Rep. 2018, 8, 5708. [Google Scholar] [CrossRef] [PubMed]
- Misiukiewicz-Stepien, P.; Paplinska-Goryca, M. Biological effect of PM10 on airway epithelium-focus on obstructive lung diseases. Clin. Immunol. 2021, 227, 108754. [Google Scholar] [CrossRef] [PubMed]
- Vignal, C.; Pichavant, M.; Alleman, L.Y.; Djouina, M.; Dingreville, F.; Perdrix, E.; Waxin, C.; Ouali Alami, A.; Gower-Rousseau, C.; Desreumaux, P.; et al. Effects of urban coarse particles inhalation on oxidative and inflammatory parameters in the mouse lung and colon. Part Fibre Toxicol. 2017, 14, 46. [Google Scholar] [CrossRef] [PubMed]
- Ye, Q.; Wen, Y.; Al-Kuwari, N.; Chen, X. Association Between Parkinson’s Disease and Melanoma: Putting the Pieces Together. Front. Aging Neurosci. 2020, 12, 60. [Google Scholar] [CrossRef]
- Ejma, M.; Madetko, N.; Brzecka, A.; Guranski, K.; Alster, P.; Misiuk-Hojlo, M.; Somasundaram, S.G.; Kirkland, C.E.; Aliev, G. The Links between Parkinson’s Disease and Cancer. Biomedicines 2020, 8, 416. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Gao, X.; Lu, Y.; Chen, H. Meta-analysis of the relationship between Parkinson disease and melanoma. Neurology 2011, 76, 2002–2009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bajaj, A.; Driver, J.A.; Schernhammer, E.S. Parkinson’s disease and cancer risk: A systematic review and meta-analysis. Cancer Causes Control 2010, 21, 697–707. [Google Scholar] [CrossRef] [PubMed]
- Wright, C.Y.; Norval, M.; Kapwata, T.; du Preez, D.D.J.; Wernecke, B.; Tod, B.M.; Visser, W.I. The Incidence of Skin Cancer in Relation to Climate Change in South Africa. Atmosphere 2019, 10, 634. [Google Scholar] [CrossRef] [Green Version]
- Tucci, M.; Passarelli, A.; Mannavola, F.; Felici, C.; Stucci, L.S.; Cives, M.; Silvestris, F. Immune System Evasion as Hallmark of Melanoma Progression: The Role of Dendritic Cells. Front. Oncol. 2019, 9, 1148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGeer, P.L.; Itagaki, S.; Boyes, B.E.; McGeer, E.G. Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson’s and Alzheimer’s disease brains. Neurology 1988, 38, 1285–1291. [Google Scholar] [CrossRef] [PubMed]
- Pajares, M.; Rojo, A.I.; Manda, G.; Bosca, L.; Cuadrado, A. Inflammation in Parkinson’s Disease: Mechanisms and Therapeutic Implications. Cells 2020, 9, 1687. [Google Scholar] [CrossRef]
- Palacios, N.; Fitzgerald, K.; Roberts, A.L.; Hart, J.E.; Weisskopf, M.G.; Schwarzschild, M.A.; Ascherio, A.; Laden, F. A prospective analysis of airborne metal exposures and risk of Parkinson disease in the nurses’ health study cohort. Environ. Health Perspect. 2014, 122, 933–938. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Young, M.T.; Chen, J.C.; Kaufman, J.D.; Chen, H. Ambient Air Pollution Exposures and Risk of Parkinson Disease. Environ. Health Perspect. 2016, 124, 1759–1765. [Google Scholar] [CrossRef]
- Song, S.; Zhou, F.; Chen, W.R. Low-level laser therapy regulates microglial function through Src-mediated signaling pathways: Implications for neurodegenerative diseases. J. Neuroinflammation 2012, 9, 219. [Google Scholar] [CrossRef] [Green Version]
- Zhu, L.; Bi, W.; Lu, D.; Zhang, C.; Shu, X.; Lu, D. Luteolin inhibits SH-SY5Y cell apoptosis through suppression of the nuclear transcription factor-kappaB, mitogen-activated protein kinase and protein kinase B pathways in lipopolysaccharide-stimulated cocultured BV2 cells. Exp. Ther. Med. 2014, 7, 1065–1070. [Google Scholar] [CrossRef] [Green Version]
- Peters, A.; Veronesi, B.; Calderon-Garciduenas, L.; Gehr, P.; Chen, L.C.; Geiser, M.; Reed, W.; Rothen-Rutishauser, B.; Schurch, S.; Schulz, H. Translocation and potential neurological effects of fine and ultrafine particles a critical update. Part Fibre Toxicol. 2006, 3, 13. [Google Scholar] [CrossRef] [Green Version]
- Perry, V.H.; Cunningham, C.; Holmes, C. Systemic infections and inflammation affect chronic neurodegeneration. Nat. Rev. Immunol. 2007, 7, 161–167. [Google Scholar] [CrossRef]
- Ferrari, C.C.; Tarelli, R. Parkinson’s disease and systemic inflammation. Parkinsons Dis. 2011, 2011, 436813. [Google Scholar] [CrossRef] [Green Version]
- Pott Godoy, M.C.; Tarelli, R.; Ferrari, C.C.; Sarchi, M.I.; Pitossi, F.J. Central and systemic IL-1 exacerbates neurodegeneration and motor symptoms in a model of Parkinson’s disease. Brain 2008, 131, 1880–1894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, L.; Yi, J.; Yang, J.; Li, P.; Cheng, X.; Mao, P. Nonsteroidal anti-inflammatory drugs use and risk of Parkinson disease: A dose-response meta-analysis. Medicine 2018, 97, e12172. [Google Scholar] [CrossRef] [PubMed]
- Alster, P.; Madetko, N.; Koziorowski, D.; Friedman, A. Microglial Activation and Inflammation as a Factor in the Pathogenesis of Progressive Supranuclear Palsy (PSP). Front. Neurosci. 2020, 14, 893. [Google Scholar] [CrossRef] [PubMed]
- Mumaw, C.L.; Levesque, S.; McGraw, C.; Robertson, S.; Lucas, S.; Stafflinger, J.E.; Campen, M.J.; Hall, P.; Norenberg, J.P.; Anderson, T.; et al. Microglial priming through the lung-brain axis: The role of air pollution-induced circulating factors. FASEB J. 2016, 30, 1880–1891. [Google Scholar] [CrossRef] [Green Version]
- Calderon-Garciduenas, L.; Kavanaugh, M.; Block, M.; D’Angiulli, A.; Delgado-Chavez, R.; Torres-Jardon, R.; Gonzalez-Maciel, A.; Reynoso-Robles, R.; Osnaya, N.; Villarreal-Calderon, R.; et al. Neuroinflammation, hyperphosphorylated tau, diffuse amyloid plaques, and down-regulation of the cellular prion protein in air pollution exposed children and young adults. J. Alzheimer’s Dis. 2012, 28, 93–107. [Google Scholar] [CrossRef] [Green Version]
- Jackson-Lewis, V.; Przedborski, S. Protocol for the MPTP mouse model of Parkinson’s disease. Nat. Protoc. 2007, 2, 141–151. [Google Scholar] [CrossRef]
- Dijkhoff, I.M.; Drasler, B.; Karakocak, B.B.; Petri-Fink, A.; Valacchi, G.; Eeman, M.; Rothen-Rutishauser, B. Impact of airborne particulate matter on skin: A systematic review from epidemiology to in vitro studies. Part Fibre Toxicol. 2020, 17, 35. [Google Scholar] [CrossRef]
- Ha, J.W.; Song, H.; Hong, S.S.; Boo, Y.C. Marine Alga Ecklonia cava Extract and Dieckol Attenuate Prostaglandin E2 Production in HaCaT Keratinocytes Exposed to Airborne Particulate Matter. Antioxidants 2019, 8, 190. [Google Scholar] [CrossRef] [Green Version]
- Jang, H.S.; Lee, J.E.; Myung, C.H.; Park, J.I.; Jo, C.S.; Hwang, J.S. Particulate Matter-Induced Aryl Hydrocarbon Receptor Regulates Autophagy in Keratinocytes. Biomol. Ther. 2019, 27, 570–576. [Google Scholar] [CrossRef]
- Seok, J.K.; Lee, J.W.; Kim, Y.M.; Boo, Y.C. Punicalagin and (−)-Epigallocatechin-3-Gallate Rescue Cell Viability and Attenuate Inflammatory Responses of Human Epidermal Keratinocytes Exposed to Airborne Particulate Matter PM10. Skin Pharmacol. Physiol. 2018, 31, 134–143. [Google Scholar] [CrossRef]
- Tamagawa, E.; Bai, N.; Morimoto, K.; Gray, C.; Mui, T.; Yatera, K.; Zhang, X.; Xing, L.; Li, Y.; Laher, I.; et al. Particulate matter exposure induces persistent lung inflammation and endothelial dysfunction. Am. J. Physiol. Lung Cell Mol. Physiol. 2008, 295, L79–L85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, K.H.; Lee, S.Y.; Shin, J.; Hwang, J.T.; Jeon, H.N.; Bae, H. Dose-Dependent Neuroprotective Effect of Standardized Bee Venom Phospholipase A2 Against MPTP-Induced Parkinson’s Disease in Mice. Front. Aging Neurosci. 2019, 11, 80, 90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, E.S.; Kim, H.; Lee, G.; Park, S.; Kim, H.; Bae, H. Neuro-protective effects of bee venom by suppression of neuroinflammatory responses in a mouse model of Parkinson’s disease: Role of regulatory T cells. Brain Behav. Immun. 2012, 26, 1322–1330. [Google Scholar] [CrossRef] [PubMed]
- West, M.J.; Slomianka, L.; Gundersen, H.J. Unbiased stereological estimation of the total number of neurons in thesubdivisions of the rat hippocampus using the optical fractionator. Anat. Rec. 1991, 231, 482–497. [Google Scholar] [CrossRef]
- Kim, K.H.; Lee, S.; Lee, H.; Shin, D.; Min, D.; Kim, M.; Ryu, B.; Kim, H.W.; Bae, H. A standardized herbal extract PM014 ameliorates pulmonary fibrosis by suppressing the TGF-beta1 pathway. Sci. Rep. 2018, 8, 16860. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, N.; Hirose, Y.; Ohara, S.; Ono, T.; Watanabe, Y. A simple quantitative bradykinesia test in MPTP-treated mice. Res. Commun. Chem. Pathol. Pharmacol. 1985, 50, 435–441. [Google Scholar]
- Rozas, G.; Lopez-Martin, E.; Guerra, M.J.; Labandeira-Garcia, J.L. The overall rod performance test in the MPTP-treated-mouse model of Parkinsonism. J. Neurosci. Methods 1998, 83, 165–175. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Element | Mass Fraction | |
|---|---|---|
| Certified Value (mg/kg) (1) | Certified Uncertainty (2) (mg/kg) | |
| Arsenic | 7.1 | 0.7 |
| Cadmium | 0.90 | 0.22 |
| Lead | 113 | 17 |
| Nickel | 58 | 7 |
| Gene | Direction | Sequence (5′-> 3′) |
|---|---|---|
| TNF-α | Forward | gcctcttctcattcctgcttg |
| Reverse | ctgatgagagggaggccatt | |
| IL-1β | Forward | cacagcagcacatcaacaag |
| Reverse | gtgctcatgtcctcatcctg | |
| CCL2 | Forward | tgatcccaatgagtaggctggag |
| Reverse | atgtctggacccattccttcttg | |
| B2M | Forward | ctgctacgtaacacagttccaccc |
| Reverse | catgatgcttgatcacatgtctcg |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Choi, D.; Lee, G.; Kim, K.H.; Bae, H. Particulate Matter Exacerbates the Death of Dopaminergic Neurons in Parkinson’s Disease through an Inflammatory Response. Int. J. Mol. Sci. 2022, 23, 6487. https://doi.org/10.3390/ijms23126487
Choi D, Lee G, Kim KH, Bae H. Particulate Matter Exacerbates the Death of Dopaminergic Neurons in Parkinson’s Disease through an Inflammatory Response. International Journal of Molecular Sciences. 2022; 23(12):6487. https://doi.org/10.3390/ijms23126487
Chicago/Turabian StyleChoi, Dabin, Gaheon Lee, Kyung Hwa Kim, and Hyunsu Bae. 2022. "Particulate Matter Exacerbates the Death of Dopaminergic Neurons in Parkinson’s Disease through an Inflammatory Response" International Journal of Molecular Sciences 23, no. 12: 6487. https://doi.org/10.3390/ijms23126487