
EGFR-Mutated Non-Small Cell Lung Cancer and Resistance to Immunotherapy: Role of the Tumor Microenvironment

 ,
, 
 ,
,  and
and 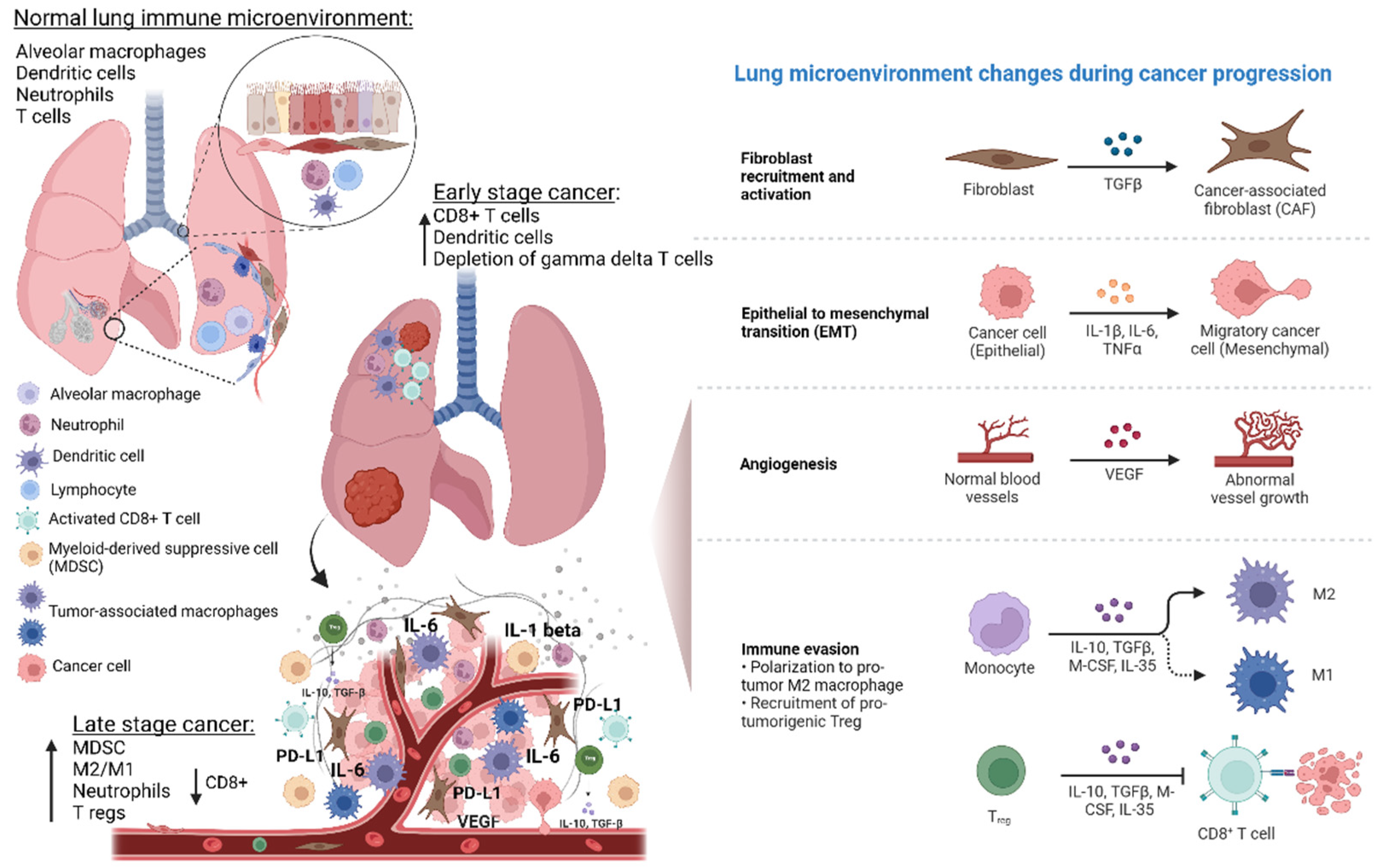{kind=link}
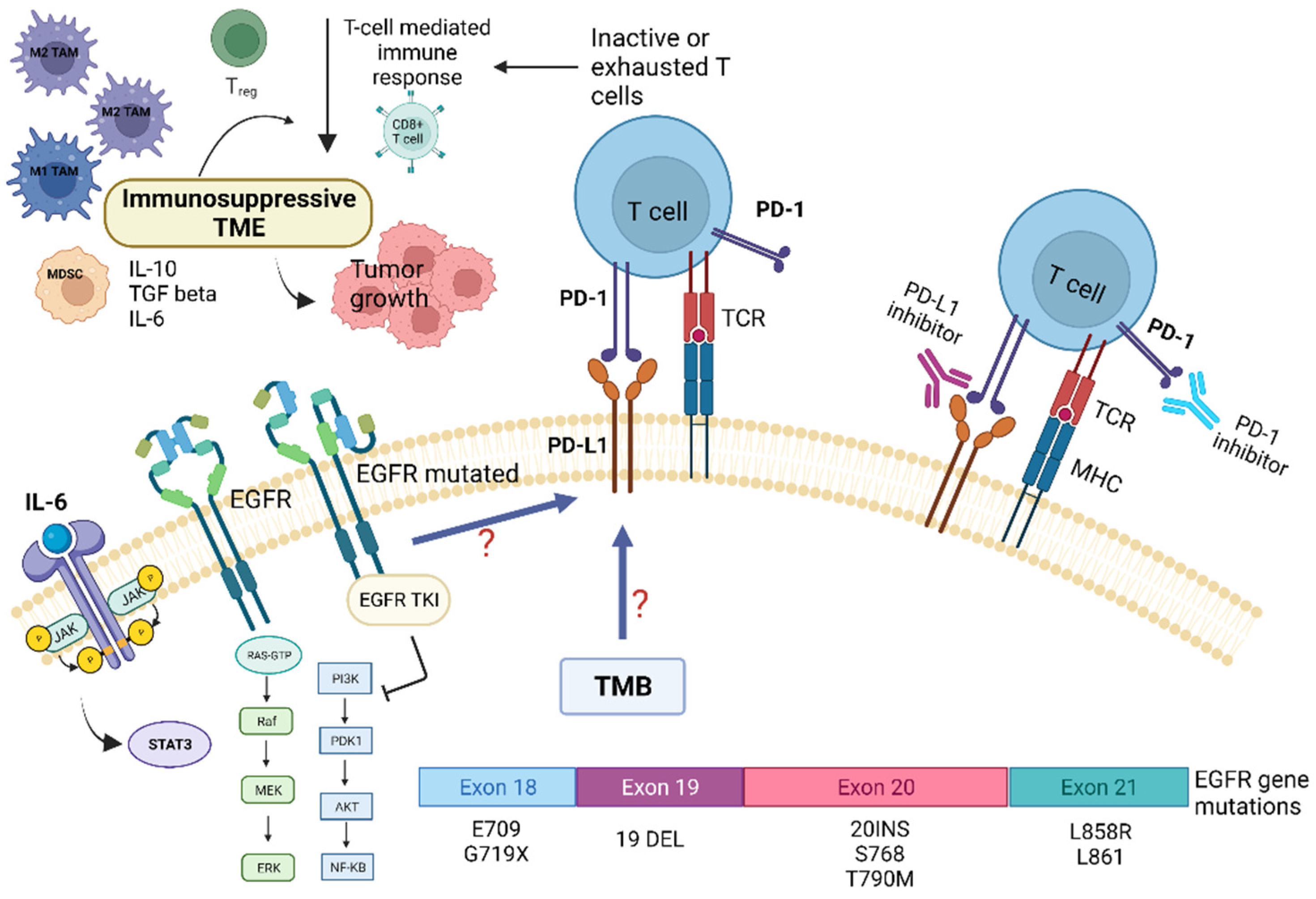{kind=link}
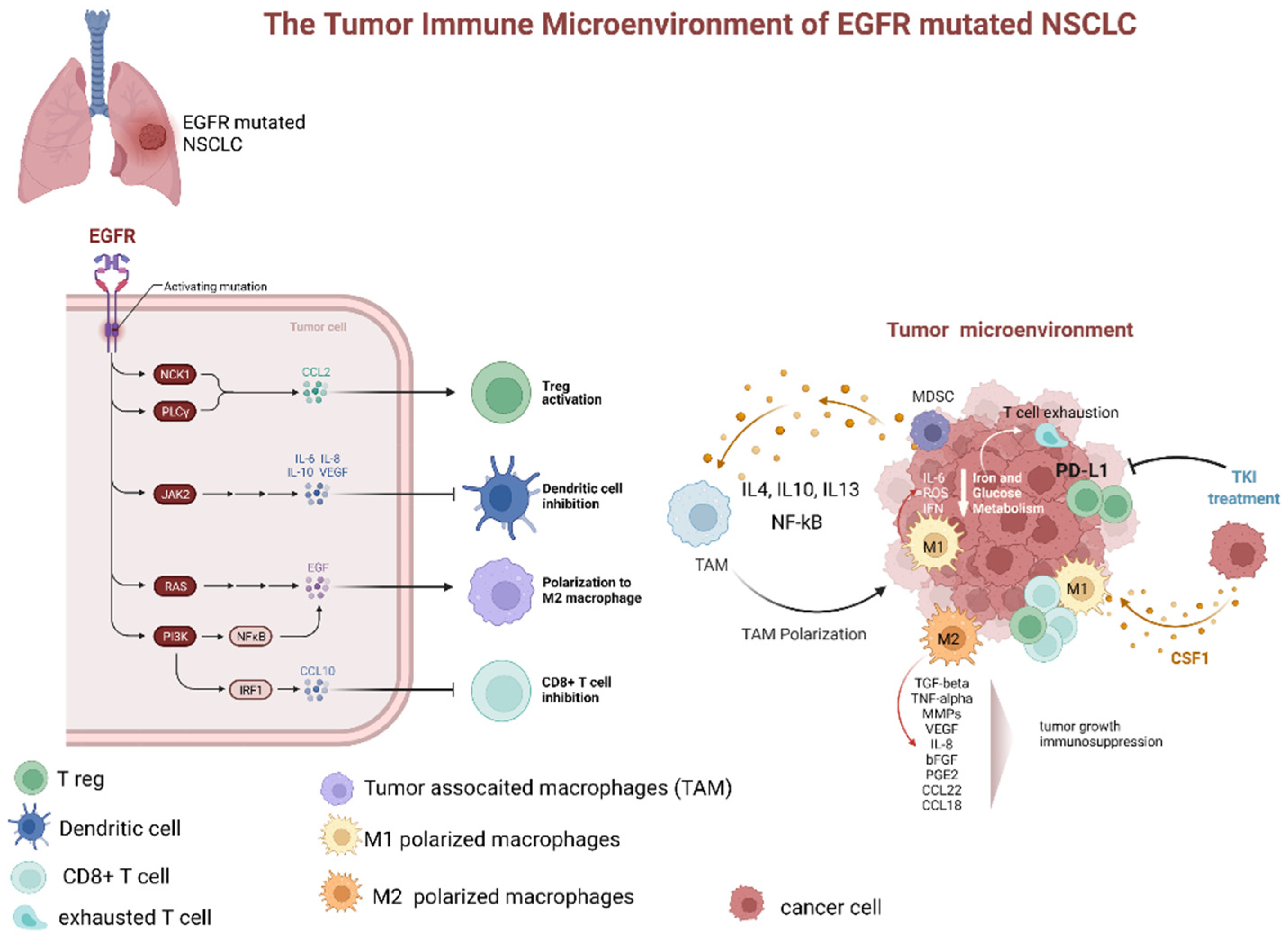{kind=link}
Abstract
:1. Introduction
2. Role of TME in Lung Cancer Evolution and Prognosis
2.1. Subsection
2.1.1. Role of TAMs and Related Inflammation on the Efficacy of Immune Response and Immunotherapy
2.1.2. Role of EGFR Mutations in Influencing TME, TAM Polarization, and Response to Immunotherapy
2.1.3. Dynamic Changes of the TME during TKI Treatment
2.1.4. Rationale for Combining Immunotherapy and TKI Treatment to Overcame Resistance
2.1.5. Overcoming Resistance and Potentiate Response to Immunotherapy in Patients with EGFR-Mutated NSCLC by Targeting TAM Related Inflammation and Cytokine Storm
3. Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Mansuet-Lupo, A.; Alifano, M.; Pécuchet, N.; Biton, J.; Becht, E.; Goc, J.; Germain, C.; Ouakrim, H.; Régnard, J.F.; Cremer, I.; et al. Intratumoral immune cell densities are associated with lung adenocarcinoma gene alterations. Am. J. Respir. Crit. Care Med. 2016, 194, 1403–1412. [Google Scholar] [CrossRef] [PubMed]
- Lynch, T.J.; Bell, D.W.; Sordella, R.; Gurubhagavatula, S.; Okimoto, R.A.; Brannigan, B.W.; Harris, P.L.; Haserlat, S.M.; Supko, J.G.; Haluska, F.G.; et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N. Engl. J. Med. 2004, 350, 2129–2139. [Google Scholar] [CrossRef]
- Wang, D.-H.; Lee, H.-S.; Yoon, D.; Berry, G.; Wheeler, T.M.; Sugarbaker, D.J.; Kheradmand, F.; Engleman, E.; Burt, B.M. Progression of EGFR-Mutant Lung Adenocarcinoma is Driven By Alveolar Macrophages. Clin. Cancer Res. 2017, 23, 778–788. [Google Scholar] [CrossRef] [Green Version]
- Ohashi, K.; Maruvka, Y.E.; Michor, F.; Pao, W. Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitor–Resistant Disease. J. Clin. Oncol. 2013, 31, 1070–1080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruno, D.; Dowlati, A. Immunotherapy in EGFR mutant non-small cell lung cancer: When, who and how? Transl. Lung Cancer Res. 2019, 8, 710–714. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.K.; Man, J.; Lord, S.J.; Links, M.; Gebski, V.; Mok, T.; Yang, J.C.-H. Checkpoint Inhibitors in Metastatic EGFR- Mutated Non–Small Cell Lung Cancer—A Meta-Analysis. J. Thorac. Oncol. 2017, 12, 403–407. [Google Scholar] [CrossRef] [Green Version]
- Osipov, A.; Saung, M.T.; Zheng, L.; Murphy, A.G. Small molecule immunomodulation: The tumor microenvironment and over-coming immune escape. J. Immunother. Cancer. 2019, 7, 224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altorki, N.K.; Markowitz, G.J.; Gao, D.; Port, J.L.; Saxena, A.; Stiles, B.; McGraw, T.; Mittal, V. The lung microenvironment: An important regulator of tumour growth and metastasis. Nat. Rev. Cancer 2019, 19, 9–31. [Google Scholar] [CrossRef] [Green Version]
- Bremnes, R.M.; Busund, L.-T.; Kilvær, T.L.; Andersen, S.; Richardsen, E.; Paulsen, E.E.; Hald, S.; Khanehkenari, M.R.; Cooper, W.A.; Kao, S.C.; et al. The Role of Tumor-Infiltrating Lymphocytes in Development, Progression, and Prognosis of Non–Small Cell Lung Cancer. J. Thorac. Oncol. 2016, 11, 789–800. [Google Scholar] [CrossRef] [Green Version]
- Tuminello, S.; Veluswamy, R.; Lieberman-Cribbin, W.; Gnjatic, S.; Petralia, F.; Wang, P.; Flores, R.; Taioli, E. Prognostic value of immune cells in the tumor microenvironment of early-stage lung cancer: A meta-analysis. Oncotarget 2019, 10, 7142–7155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mantovani, A.; Sozzani, S.; Locati, M.; Allavena, P.; Sica, A. Macrophage polarization: Tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 2002, 23, 549–555. [Google Scholar] [CrossRef]
- Conway, E.M.; Pikor, L.A.; Kung, S.H.Y.; Hamilton, M.J.; Lam, S.; Lam, W.L.; Bennewith, K.L. Macrophages, Inflammation, and Lung Cancer. Am. J. Respir. Crit. Care Med. 2016, 193, 116–130. [Google Scholar] [CrossRef] [PubMed]
- Chung, F.-T.; Lee, K.-Y.; Wang, C.-W.; Heh, C.-C.; Chan, Y.-F.; Chen, H.-W.; Kuo, C.-H.; Feng, P.-H.; Lin, T.-Y.; Wang, C.-H.; et al. Tumor-associated macrophages correlate with response to epidermal growth factor receptor-tyrosine kinase inhibitors in advanced non-small cell lung cancer. Int. J. Cancer 2012, 131, E227–E235. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.; Che, X.; Qiu, X.; Li, Z.; Yang, B.; Wang, S.; Hou, K.; Fan, Y.; Qu, X.; Liu, Y. M2 macrophage infiltration into tumor islets leads to poor prognosis in non-small-cell lung cancer. Cancer Manag. Res. 2019, 11, 6125–6138. [Google Scholar] [CrossRef] [Green Version]
- Son, B.; Lee, S.; Youn, H.; Kim, E.; Kim, W.; Youn, B. The role of tumor microenvironment in therapeutic resistance. Oncotarget 2017, 8, 3933–3945. [Google Scholar] [CrossRef] [Green Version]
- Macciò, A.; Gramignano, G.; Cherchi, M.C.; Tanca, L.; Melis, L.; Madeddu, C. Role of M1-polarized tumor-associated macrophages in the prognosis of advanced ovarian cancer patients. Sci. Rep. 2020, 10, 6096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garrido-Martin, E.M.; Mellows, T.W.P.; Clarke, J.; Ganesan, A.-P.; Wood, O.; Cazaly, A.; Seumois, G.; Chee, S.J.; Alzetani, A.; King, E.V.; et al. M1hot tumor-associated macrophages boost tissue-resident memory T cells infiltration and survival in human lung cancer. J. Immunother. Cancer 2020, 8, e000778. [Google Scholar] [CrossRef]
- Watanabe, H.; Ohashi, K.; Nishii, K.; Seike, K.; Makimoto, G.; Hotta, K.; Maeda, Y.; Kiura, K. A Long-term Response to Nivolumab in a Case of PD-L1-negative Lung Adenocarcinoma with an EGFR Mutation and Surrounding PD-L1-positive Tumor-associated Macrophages. Intern. Med. 2019, 58, 3033–3037. [Google Scholar] [CrossRef] [Green Version]
- Jinushi, M.; Komohara, Y. Tumor-associated macrophages as an emerging target against tumors: Creating a new path from bench to bedside. Biochim. Biophys. Acta 2015, 1855, 123–130. [Google Scholar] [CrossRef]
- Santoni, M.; Massari, F.; Amantini, C.; Nabissi, M.; Maines, F.; Burattini, L.; Berardi, R.; Santoni, G.; Montironi, R.; Tortora, G.; et al. Emerging role of tumor-associated macrophages as therapeutic targets in patients with metastatic renal cell carcinoma. Cancer Immunol. Immunother. 2013, 62, 1757–1768. [Google Scholar] [CrossRef]
- Komohara, Y.; Jinushi, M.; Takeya, M. Clinical significance of macrophage heterogeneity in human malignant tumors. Cancer Sci. 2014, 105, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allavena, P.; Sica, A.; Solinas, G.; Porta, C.; Mantovani, A. The inflammatory micro-environment in tumor progression: The role of tumor-associated macrophages. Crit. Rev. Oncol. Hematol. 2008, 66, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Grohmann, U.; Fallarino, F.; Puccetti, P. Tolerance, DCs and tryptophan: Much ado about IDO. Trends Immunol. 2003, 24, 242–248. [Google Scholar] [CrossRef]
- Macciò, A.; Madeddu, C. Blocking inflammation to improve immunotherapy of advanced cancer. Immunology 2020, 159, 357–364. [Google Scholar] [CrossRef] [Green Version]
- Mascaux, C.; Angelova, M.; Vasaturo, A.; Beane, J.; Hijazi, K.; Anthoine, G.; Buttard, B.; Rothe, F.; Willard-Gallo, K.; Haller, A.; et al. Immune evasion before tumour invasion in early lung squamous carcinogenesis. Nature 2019, 571, 570–575. [Google Scholar] [CrossRef]
- Hori, S.; Nomura, T.; Sakaguchi, S. Control of Regulatory T Cell Development by the Transcription Factor Foxp3. Science 2003, 299, 1057–1061. [Google Scholar] [CrossRef] [Green Version]
- Williams, L.M.; Rudensky, A.Y. Maintenance of the Foxp3-dependent developmental program in mature regulatory T cells requires continued expression of Foxp3. Nat. Immunol. 2007, 8, 277–284. [Google Scholar] [CrossRef]
- Di Pilato, M.; Kim, E.Y.; Cadilha, B.; Prüßmann, J.N.; Nasrallah, M.N.; Seruggia, D.; Usmani, S.; Misale, S.; Zappulli, V.; Carrizosa, E.; et al. Targeting the CBM complex causes Treg cells to prime tumours for immune checkpoint therapy. Nature 2019, 570, 112–116. [Google Scholar] [CrossRef]
- Ivashkiv, L.B. IFNγ: Signalling, epigenetics and roles in immunity, metabolism, disease and cancer immunotherapy. Nat. Rev. Immunol. 2018, 18, 545–558. [Google Scholar] [CrossRef]
- Gallimore, A.; Quezada, S.A.; Roychoudhuri, R. Regulatory T cells in cancer: Where are we now? Immunology 2019, 157, 187–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ayala, M.A.M.; Li, Z.; DuPage, M. Treg programming and therapeutic reprogramming in cancer. Immunology 2019, 157, 198–209. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.O.; Nurieva, R.; Martinez, G.; Kang, H.S.; Chung, Y.; Pappu, B.P.; Shah, B.; Chang, S.H.; Schluns, K.S.; Watowich, S.S.; et al. Molecular Antagonism and Plasticity of Regulatory and Inflammatory T Cell Programs. Immunity 2008, 29, 44–56. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Quiros, J.; Mahuron, K.; Pai, C.-C.; Ranzani, V.; Young, A.; Silveria, S.; Harwin, T.; Abnousian, A.; Pagani, M.; et al. Targeting EZH2 Reprograms Intratumoral Regulatory T Cells to Enhance Cancer Immunity. Cell Rep. 2018, 23, 3262–3274. [Google Scholar] [CrossRef]
- Wan, Y.Y.; Flavell, R.A. Regulatory T-cell functions are subverted and converted owing to attenuated Foxp3 expression. Nature 2007, 445, 766–770. [Google Scholar] [CrossRef]
- Skuljec, J.; Jirmo, A.C.; Habener, A.; Talbot, S.R.; Pul, R.; Grychtol, R.; Aydin, M.; Kleinschnitz, C.; Happle, C.; Hansen, G. Absence of Regulatory T Cells Causes Phenotypic and Functional Switch in Murine Peritoneal Macrophages. Front. Immunol. 2018, 9, 2458. [Google Scholar] [CrossRef] [PubMed]
- Shalapour, S.; Karin, M. Pas de Deux: Control of Anti-tumor Immunity by Cancer-Associated Inflammation. Immunity 2019, 51, 15–26. [Google Scholar] [CrossRef]
- Harris, R.A. Spatial, Temporal, and Functional Aspects of Macrophages during “The Good, the Bad, and the Ugly” Phases of Inflammation. Front. Immunol. 2014, 5, 612. [Google Scholar] [CrossRef] [Green Version]
- Italiani, P.; Boraschi, D.; Italiani, P.; Boraschi, D. From Monocytes to M1/M2 Macrophages: Phenotypical vs. Functional Differentiation. Front. Immunol. 2014, 5, 514. [Google Scholar] [CrossRef] [Green Version]
- Cairo, G.; Recalcati, S.; Mantovani, A.; Locati, M. Iron trafficking and metabolism in macrophages: Contribution to the polarized phenotype. Trends Immunol. 2011, 32, 241–247. [Google Scholar] [CrossRef]
- Wherry, E.J.; Kurachi, M. Molecular and cellular insights into T cell exhaustion. Nat. Rev. Immunol. 2015, 15, 486–499. [Google Scholar] [CrossRef] [PubMed]
- Madeddu, C.; Gramignano, G.; Kotsonis, P.; Coghe, F.; Atzeni, V.; Scartozzi, M.; Maccio, A. Microenvironmental M1 tumor-associated macrophage polarization influences cancer-related anemia in advanced ovarian cancer: Key role of interleukin-6. Haematologica 2018, 103, e388–e391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vargas, A.; Harris, A.J.V.C.C. Biomarker development in the precision medicine era: Lung cancer as a case study. Nat. Cancer 2016, 16, 525–537. [Google Scholar] [CrossRef]
- Hecht, S.S. Cigarette smoking and lung cancer: Chemical mechanisms and approaches to prevention. Lancet Oncol. 2002, 3, 461–469. [Google Scholar] [CrossRef]
- Yeh, H.-H.; Lai, W.-W.; Chen, H.H.W.; Liu, H.-S.; Su, W.-C. Autocrine IL-6-induced Stat3 activation contributes to the pathogenesis of lung adenocarcinoma and malignant pleural effusion. Oncogene 2006, 25, 4300–4309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mondal, A.M.; Horikawa, I.; Pine, S.R.; Fujita, K.; Morgan, K.M.; Vera, E.; Mazur, S.J.; Appella, E.; Vojtesek, B.; Blasco, M.A.; et al. p53 isoforms regulate aging- and tumor-associated replicative senescence in T lymphocytes. J. Clin. Investig. 2013, 123, 5247–5257. [Google Scholar] [CrossRef] [Green Version]
- Maccio, A.; Sanna, E.; Neri, M.; Oppi, S.; Madeddu, C. Cachexia as Evidence of the Mechanisms of Resistance and Tolerance during the Evolution of Cancer Disease. Int. J. Mol. Sci. 2021, 22, 2890. [Google Scholar] [CrossRef]
- Sugiyama, E.; Togashi, Y.; Takeuchi, Y.; Shinya, S.; Tada, Y.; Kataoka, K.; Tane, K.; Sato, E.; Ishii, G.; Goto, K.; et al. Blockade of EGFR improves responsiveness to PD-1 blockade in EGFR-mutated non–small cell lung cancer. Sci. Immunol. 2020, 5, eaav3937. [Google Scholar] [CrossRef]
- Topalian, S.L.; Drake, C.G.; Pardoll, D.M. Immune Checkpoint Blockade: A Common Denominator Approach to Cancer Therapy. Cancer Cell 2015, 27, 450–461. [Google Scholar] [CrossRef] [Green Version]
- Lisberg, A.; Cummings, A.; Goldman, J.; Bornazyan, K.; Reese, N.; Wang, T.; Coluzzi, P.; Ledezma, B.; Mendenhall, M.; Hunt, J.; et al. A Phase II Study of Pembrolizumab in EGFR-Mutant, PD-L1+, Tyrosine Kinase Inhibitor Naïve Patients With Advanced NSCLC. J. Thorac. Oncol. 2018, 13, 1138–1145. [Google Scholar] [CrossRef] [Green Version]
- Pitt, J.M.; Marabelle, A.; Eggermont, A.; Soria, J.-C.; Kroemer, G.; Zitvogel, L. Targeting the tumor microenvironment: Removing obstruction to anticancer immune responses and immunotherapy. Ann. Oncol. 2016, 27, 1482–1492. [Google Scholar] [CrossRef]
- Haratani, K.; Hayashi, H.; Tanaka, T.; Kaneda, H.; Togashi, Y.; Sakai, K.; Hayashi, K.; Tomida, S.; Chiba, Y.; Yonesaka, K.; et al. Tumor immune microenvironment and nivolumab efficacy in EGFR mutation-positive non-small-cell lung cancer based on T790M status after disease progression during EGFR-TKI treatment. Ann. Oncol. 2017, 28, 1532–1539. [Google Scholar] [CrossRef] [PubMed]
- Peranzoni, E.; Lemoine, J.; Vimeux, L.; Feuillet, V.; Barrin, S.; Kantari-Mimoun, C.; Bercovici, N.; Guérin, M.; Biton, J.; Ouakrim, H.; et al. Macrophages impede CD8 T cells from reaching tumor cells and limit the efficacy of anti–PD-1 treatment. Proc. Natl. Acad. Sci. USA 2018, 115, E4041–E4050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teng, F.; Meng, X.; Kong, L.; Yu, J. Progress and challenges of predictive biomarkers of anti PD-1/PD-L1 immunotherapy: A systematic review. Cancer Lett. 2018, 414, 166–173. [Google Scholar] [CrossRef] [PubMed]
- Fehrenbacher, L.; Spira, A.; Ballinger, M.; Kowanetz, M.; Vansteenkiste, J.; Mazieres, J.; Park, K.; Smith, D.; Artal-Cortes, A.; Lewanski, C.; et al. Atezolizumab versus docetaxel for patients with previously treated non-small-cell lung cancer (POPLAR): A multicentre, open-label, phase 2 randomised controlled trial. Lancet 2016, 387, 1837–1846. [Google Scholar] [CrossRef]
- Herbst, R.S.; Soria, J.-C.; Kowanetz, M.; Fine, G.D.; Hamid, O.; Gordon, M.S.; Sosman, J.A.; McDermott, D.F.; Powderly, J.D.; Gettinger, S.N.; et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature 2014, 515, 563–567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsumoto, Y.; Sawa, K.; Fukui, M.; Oyanagi, J.; Izumi, M.; Ogawa, K.; Suzumura, T.; Watanabe, T.; Kaneda, H.; Mitsuoka, S.; et al. Impact of tumor microenvironment on the efficacy of epidermal growth factor receptor-tyrosine kinase inhibitors in patients with EGFR-mutant non-small cell lung cancer. Cancer Sci. 2019, 110, 3244–3254. [Google Scholar] [CrossRef] [Green Version]
- Teng, M.W.L.; Ngiow, S.F.; Ribas, A.; Smyth, M.J. Classifying Cancers Based on T-cell Infiltration and PD-L1. Cancer Res. 2015, 75, 2139–2145. [Google Scholar] [CrossRef] [Green Version]
- Su, S.; Dong, Z.-Y.; Xie, Z.; Yan, L.-X.; Li, Y.-F.; Su, J.; Liu, S.-Y.; Yin, K.; Chen, R.-L.; Huang, S.-M.; et al. Strong Programmed Death Ligand 1 Expression Predicts Poor Response and De Novo Resistance to EGFR Tyrosine Kinase Inhibitors Among NSCLC Patients With EGFR Mutation. J. Thorac. Oncol. 2018, 13, 1668–1675. [Google Scholar] [CrossRef] [Green Version]
- Yoneshima, Y.; Ijichi, K.; Anai, S.; Ota, K.; Otsubo, K.; Iwama, E.; Tanaka, K.; Oda, Y.; Nakanishi, Y.; Okamoto, I. PD-L1 expression in lung adenocarcinoma harboring EGFR mutations or ALK rearrangements. Lung Cancer 2018, 118, 36–40. [Google Scholar] [CrossRef]
- Takashima, Y.; Sakakibara-Konishi, J.; Hatanaka, Y.; Hatanaka, K.C.; Ohhara, Y.; Oizumi, S.; Hida, Y.; Kaga, K.; Kinoshita, I.; Dosaka-Akita, H.; et al. Clinicopathologic Features and Immune Microenvironment of Non–Small-cell Lung Cancer With Primary Resistance to Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitors. Clin. Lung Cancer 2018, 19, 352–359.e1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biswas, S.K.; Lewis, C.E. NF-κB as a central regulator of macrophage function in tumors. J. Leukoc. Biol. 2010, 88, 877–884. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Li, Q.; Yamada, T.; Matsumoto, K.; Matsumoto, I.; Oda, M.; Watanabe, G.; Kayano, Y.; Nishioka, Y.; Sone, S.; et al. Crosstalk to Stromal Fibroblasts Induces Resistance of Lung Cancer to Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitors. Clin. Cancer Res. 2009, 15, 6630–6638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, Y.; Li, X.; Jiang, T.; Zhao, S.; Zhao, C.; Zhang, L.; Liu, X.; Shi, J.; Qiao, M.; Luo, J.; et al. EGFR-targeted therapy alters the tumor microenvironment in EGFR-driven lung tumors: Implications for combination therapies. Int. J. Cancer 2019, 145, 1432–1444. [Google Scholar] [CrossRef] [PubMed]
- Borghaei, H.; Paz-Ares, L.; Horn, L.; Spigel, D.R.; Steins, M.; Ready, N.E.; Chow, L.Q.; Vokes, E.E.; Felip, E.; Holgado, E.; et al. Nivolumab versus Docetaxel in Advanced Nonsquamous Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2015, 373, 1627–1639. [Google Scholar] [CrossRef] [PubMed]
- Herbst, R.S.; Baas, P.; Kim, D.-W.; Felip, E.; Perez-Gracia, J.L.; Han, J.-Y.; Molina, J.; Kim, J.-H.; Arvis, C.D.; Ahn, M.-J.; et al. Pembrolizumab versus docetaxel for previously treated, PD-L1-positive, advanced non-small-cell lung cancer (KEYNOTE-010): A randomised controlled trial. Lancet 2016, 387, 1540–1550. [Google Scholar] [CrossRef]
- Garrido, G.; Rabasa, A.; Garrido, C.; Chao, L.; Garrido, F.; Lora, A.M.G.; Sánchez-Ramírez, B. Upregulation of HLA Class I Expression on Tumor Cells by the Anti-EGFR Antibody Nimotuzumab. Front. Pharmacol. 2017, 8, 595. [Google Scholar] [CrossRef] [Green Version]
- Pollack, B.P.; Sapkota, B.; Cartee, T.V. Epidermal Growth Factor Receptor Inhibition Augments the Expression of MHC Class I and II Genes. Clin. Cancer Res. 2011, 17, 4400–4413. [Google Scholar] [CrossRef] [Green Version]
- Dong, Z.-Y.; Zhang, J.-T.; Liu, S.-Y.; Su, J.; Zhang, C.; Xie, Z.; Zhou, Q.; Tu, H.-Y.; Xu, C.-R.; Yan, L.-X.; et al. EGFR mutation correlates with uninflamed phenotype and weak immunogenicity, causing impaired response to PD-1 blockade in non-small cell lung cancer. Onco. Immunol. 2017, 6, e1356145. [Google Scholar] [CrossRef] [Green Version]
- Li, H.Y.; McSharry, M.; Bullock, B.; Nguyen, T.T.; Kwak, J.; Poczobutt, J.M.; Sippel, T.R.; Heasley, L.E.; Weiser-Evans, M.C.; Clambey, E.T.; et al. The Tumor Microenvironment Regulates Sensitivity of Murine Lung Tumors to PD-1/PD-L1 Antibody Blockade. Cancer Immunol. Res. 2017, 5, 767–777. [Google Scholar] [CrossRef] [Green Version]
- Tariq, M.; Zhang, J.-Q.; Liang, G.-K.; He, Q.-J.; Ding, L.; Yang, B. Gefitinib inhibits M2-like polarization of tumor-associated macrophages in Lewis lung cancer by targeting the STAT6 signaling pathway. Acta Pharmacol. Sin. 2017, 38, 1501–1511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, N.; Fang, W.; Zhan, J.; Hong, S.; Tang, Y.; Kang, S.; Zhang, Y.; He, X.; Zhou, T.; Qin, T.; et al. Upregulation of PD-L1 by EGFR Activation Mediates the Immune Escape in EGFR-Driven NSCLC: Implication for Optional Immune Targeted Therapy for NSCLC Patients with EGFR Mutation. J. Thorac. Oncol. 2015, 10, 910–923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.C.-H.; Gadgeel, S.M.; Sequist, L.V.; Wu, C.-L.; Papadimitrakopoulou, V.A.; Su, W.-C.; Fiore, J.; Saraf, S.; Raftopoulos, H.; Patnaik, A. Pembrolizumab in Combination With Erlotinib or Gefitinib as First-Line Therapy for Advanced NSCLC With Sensitizing EGFR Mutation. J. Thorac. Oncol. 2019, 14, 553–559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- To, K.K.W.; Fong, W.; Cho, W.C.S. Immunotherapy in Treating EGFR-Mutant Lung Cancer: Current Challenges and New Strategies. Front. Oncol. 2021, 11, 635007. [Google Scholar] [CrossRef]
- Chan, D.W.-K.; Choi, H.C.-W.; Lee, V.H.-F. Treatment-Related Adverse Events of Combination EGFR Tyrosine Kinase Inhibitor and Immune Checkpoint Inhibitor in EGFR-Mutant Advanced Non-Small Cell Lung Cancer: A Systematic Review and Meta-Analysis. Cancers 2022, 14, 2157. [Google Scholar] [CrossRef]
- Oxnard, G.R.; Yang, J.C.-H.; Yu, H.; Kim, S.-W.; Saka, H.; Horn, L.; Goto, K.; Ohe, Y.; Mann, H.; Thress, K.S.; et al. TATTON: A multi-arm, phase Ib trial of osimertinib combined with selumetinib, savolitinib, or durvalumab in EGFR-mutant lung cancer. Ann. Oncol. 2020, 31, 507–516. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.C.-H.; Shepherd, F.A.; Kim, D.-W.; Lee, G.-W.; Lee, J.S.; Chang, G.-C.; Lee, S.S.; Wei, Y.-F.; Lee, Y.G.; Laus, G.; et al. Osimertinib Plus Durvalumab versus Osimertinib Monotherapy in EGFR T790M–Positive NSCLC following Previous EGFR TKI Therapy: CAURAL Brief Report. J. Thorac. Oncol. 2019, 14, 933–939. [Google Scholar] [CrossRef] [Green Version]
- Creelan, B.C.; Yeh, T.C.; Kim, S.-W.; Nogami, N.; Kim, D.-W.; Chow, L.Q.M.; Kanda, S.; Taylor, R.; Tang, W.; Tang, M.; et al. A Phase 1 study of gefitinib combined with durvalumab in EGFR TKI-naive patients with EGFR mutation-positive locally advanced/metastatic non-small-cell lung cancer. Br. J. Cancer 2020, 124, 383–390. [Google Scholar] [CrossRef]
- Gettinger, S.; Hellmann, M.D.; Chow, L.Q.; Borghaei, H.; Antonia, S.; Brahmer, J.R.; Goldman, J.W.; Gerber, D.; Juergens, R.A.; Shepherd, F.A.; et al. Nivolumab Plus Erlotinib in Patients With EGFR-Mutant Advanced NSCLC. J. Thorac. Oncol. 2018, 13, 1363–1372. [Google Scholar] [CrossRef] [Green Version]
- Ayeni, D.; Miller, B.; Kuhlmann, A.; Ho, P.-C.; Robles-Oteiza, C.; Gaefele, M.; Levy, S.; de Miguel, F.J.; Perry, C.; Guan, T.; et al. Tumor regression mediated by oncogene withdrawal or erlotinib stimulates infiltration of inflammatory immune cells in EGFR mutant lung tumors. J. Immunother. Cancer 2019, 7, 172. [Google Scholar] [CrossRef] [Green Version]
- Lin, K.; Cheng, J.; Yang, T.; Li, Y.; Zhu, B. EGFR-TKI down-regulates PD-L1 in EGFR mutant NSCLC through inhibiting NF-κB. Biochem. Biophys. Res. Commun. 2015, 463, 95–101. [Google Scholar] [CrossRef] [PubMed]
- Luo, Q.; Gu, Y.; Zheng, W.; Wu, X.; Gong, F.; Gu, L.; Sun, Y.; Xu, Q. Erlotinib inhibits T-cell-mediated immune response via down-regulation of the c-Raf/ERK cascade and Akt signaling pathway. Toxicol. Appl. Pharmacol. 2011, 251, 130–136. [Google Scholar] [CrossRef] [PubMed]
- Venugopalan, A.; Lee, M.-J.; Niu, G.; Medina-Echeverz, J.; Tomita, Y.; Lizak, M.J.; Cultraro, C.M.; Simpson, R.M.; Chen, X.; Trepel, J.B.; et al. EGFR-targeted therapy results in dramatic early lung tumor regression accompanied by imaging response and immune infiltration in EGFR mutant transgenic mouse models. Oncotarget 2016, 7, 54137–54156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tu, E.; McGlinchey, K.; Wang, J.; Martin, P.; Ching, S.L.; Floc’H, N.; Kurasawa, J.; Starrett, J.H.; Lazdun, Y.; Wetzel, L.; et al. Anti–PD-L1 and anti-CD73 combination therapy promotes T cell response to EGFR-mutated NSCLC. JCI Insight 2022, 7, e142843. [Google Scholar] [CrossRef] [PubMed]
- Le, X.; Negrao, M.V.; Reuben, A.; Federico, L.; Diao, L.; McGrail, D.; Nilsson, M.; Robichaux, J.; Munoz, I.G.; Patel, S.; et al. Characterization of the Immune Landscape of EGFR-Mutant NSCLC Identifies CD73/Adenosine Pathway as a Potential Therapeutic Target. J. Thorac. Oncol. 2020, 16, 583–600. [Google Scholar] [CrossRef]
- Passarelli, A.; Aieta, M.; Sgambato, A.; Gridelli, C. Targeting Immunometabolism Mediated by CD73 Pathway in EGFR-Mutated Non-small Cell Lung Cancer: A New Hope for Overcoming Immune Resistance. Front. Immunol. 2020, 11, 1479. [Google Scholar] [CrossRef]
- Soo, R.A.; Lim, S.M.; Syn, N.; Teng, R.; Soong, R.; Mok, T.; Cho, B.C. Immune checkpoint inhibitors in epidermal growth factor receptor mutant non-small cell lung cancer: Current controversies and future directions. Lung Cancer 2018, 115, 12–20. [Google Scholar] [CrossRef]
- Cassetta, L.; Kitamura, T. Targeting Tumor-Associated Macrophages as a Potential Strategy to Enhance the Response to Immune Checkpoint Inhibitors. Front. Cell Dev. Biol. 2018, 6, 38. [Google Scholar] [CrossRef]
- Lin, A.; Wei, T.; Meng, H.; Luo, P.; Zhang, J. Role of the dynamic tumor microenvironment in controversies regarding immune checkpoint inhibitors for the treatment of non-small cell lung cancer (NSCLC) with EGFR mutations. Mol. Cancer 2019, 18, 139. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Gao, A.; Zhang, F.; Yang, Z.; Wang, S.; Fang, Y.; Li, J.; Wang, J.; Shi, W.; Wang, L.; et al. ILT4 inhibition prevents TAM- and dysfunctional T cell-mediated immunosuppression and enhances the efficacy of anti-PD-L1 therapy in NSCLC with EGFR activation. Theranostics 2021, 11, 3392–3416. [Google Scholar] [CrossRef]
- Shibahara, D.; Tanaka, K.; Iwama, E.; Kubo, N.; Ota, K.; Azuma, K.; Harada, T.; Fujita, J.; Nakanishi, Y.; Okamoto, I. Intrinsic and Extrinsic Regulation of PD-L2 Expression in Oncogene-Driven Non–Small Cell Lung Cancer. J. Thorac. Oncol. 2018, 13, 926–937. [Google Scholar] [CrossRef] [Green Version]
- Wu, Z.; Man, S.; Sun, R.; Li, Z.; Wu, Y.; Zuo, D. Recent advances and challenges of immune checkpoint inhibitors in immunotherapy of non-small cell lung cancer. Int. Immunopharmacol. 2020, 85, 106613. [Google Scholar] [CrossRef]
- Garassino, M.C.; Cho, B.-C.; Kim, J.-H.; Mazières, J.; Vansteenkiste, J.; Lena, H.; Jaime, J.C.; Gray, J.E.; Powderly, J.; Chouaid, C.; et al. Durvalumab as third-line or later treatment for advanced non-small-cell lung cancer (ATLANTIC): An open-label, single-arm, phase 2 study. Lancet Oncol. 2018, 19, 521–536. [Google Scholar] [CrossRef]
- Akbay, E.A.; Koyama, S.; Carretero, J.; Altabef, A.; Tchaicha, J.H.; Christensen, C.L.; Mikse, O.R.; Cherniack, A.D.; Beauchamp, E.M.; Pugh, T.J.; et al. Activation of the PD-1 Pathway Contributes to Immune Escape in EGFR-Driven Lung Tumors. Cancer Discov. 2013, 3, 1355–1363. [Google Scholar] [CrossRef] [Green Version]
- Azuma, K.; Ota, K.; Kawahara, A.; Hattori, S.; Iwama, E.; Harada, T.; Matsumoto, K.; Takayama, K.; Takamori, S.; Kage, M.; et al. Association of PD-L1 overexpression with activating EGFR mutations in surgically resected nonsmall-cell lung cancer. Ann. Oncol. 2014, 25, 1935–1940. [Google Scholar] [CrossRef]
- Mazieres, J.; Drilon, A.; Lusque, A.B.; Mhanna, L.; Cortot, A.; Mezquita, L.; Thai, A.A.; Mascaux, C.; Couraud, S.; Veillon, R.; et al. Immune checkpoint inhibitors for patients with advanced lung cancer and oncogenic driver alterations: Results from the IMMUNOTARGET registry. Ann. Oncol. 2019, 30, 1321–1328. [Google Scholar] [CrossRef]
- Yang, L.; He, Y.T.; Dong, S.; Wei, X.W.; Chen, Z.H.; Zhang, B.; Chen, W.D.; Yang, X.R.; Wang, F.; Shang, X.M.; et al. Single-cell transcriptome analysis revealed a suppressive tumor immune microenvironment in EGFR mutant lung adenocarcinoma. J. Immunother. Cancer 2022, 10, e003534. [Google Scholar] [CrossRef]
- Li, T.; Pang, X.; Wang, J.; Wang, S.; Guo, Y.; He, N.; Xing, P.; Li, J. Exploration of the Tumor-Suppressive Immune Microenvironment by Integrated Analysis in EGFR-Mutant Lung Adenocarcinoma. Front. Oncol. 2021, 11, 591922. [Google Scholar] [CrossRef]
- Mantovani, G.; Maccio, A.; Madeddu, C.; Massa, E.; Mudu, M.C.; Mulas, C.; Gramignano, G.; Massidda, S.; Murgia, V.; Lusso, M.R.; et al. Immunotherapy (recombinant interleukin 2), hormone therapy (medroxyprogesterone acetate) and antioxidant agents as combined maintenance treatment of responders to previous chemotherapy. Int. J. Oncol. 2001, 18, 383–391. [Google Scholar] [CrossRef]
- Mantovani, G.; Macciò, A.; Lai, P.; Massa, E.; Massa, D.; Mulas, C.; Succu, G.; Mudu, M.C.; Manca, G.; Versace, R.; et al. Results of a Dose-Intense Phase 1 Study of a Combination Chemotherapy Regimen With Cisplatin and Epidoxorubicin Including Medroxyprogesterone Acetate and Recombinant Interleukin-2 in Patients With Inoperable Primary Lung Cancer. J. Immunother. 2000, 23, 267–274. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Madeddu, C.; Donisi, C.; Liscia, N.; Lai, E.; Scartozzi, M.; Macciò, A. EGFR-Mutated Non-Small Cell Lung Cancer and Resistance to Immunotherapy: Role of the Tumor Microenvironment. Int. J. Mol. Sci. 2022, 23, 6489. https://doi.org/10.3390/ijms23126489
Madeddu C, Donisi C, Liscia N, Lai E, Scartozzi M, Macciò A. EGFR-Mutated Non-Small Cell Lung Cancer and Resistance to Immunotherapy: Role of the Tumor Microenvironment. International Journal of Molecular Sciences. 2022; 23(12):6489. https://doi.org/10.3390/ijms23126489
Chicago/Turabian StyleMadeddu, Clelia, Clelia Donisi, Nicole Liscia, Eleonora Lai, Mario Scartozzi, and Antonio Macciò. 2022. "EGFR-Mutated Non-Small Cell Lung Cancer and Resistance to Immunotherapy: Role of the Tumor Microenvironment" International Journal of Molecular Sciences 23, no. 12: 6489. https://doi.org/10.3390/ijms23126489
APA StyleMadeddu, C., Donisi, C., Liscia, N., Lai, E., Scartozzi, M., & Macciò, A. (2022). EGFR-Mutated Non-Small Cell Lung Cancer and Resistance to Immunotherapy: Role of the Tumor Microenvironment. International Journal of Molecular Sciences, 23(12), 6489. https://doi.org/10.3390/ijms23126489