
Targeting Cell Cycle Progression in HER2+ Breast Cancer: An Emerging Treatment Opportunity


Abstract
:Layman summary
Abstract
1. Introduction
2. Cell Cycle and Cell Cycle Inhibitors
3. Approved CDKi
4. Palbociclib
5. Ribociclib
6. Amebaciclib
7. CDKi in HER2+/ER+ BC
8. CDKi in HER2+/ER- BC
9. Towards a Common Goal of a Chemotherapy-Sparing Treatment Plan
Clinical Trials Evaluating the Efficacy of Cell Cycle Inhibition in Combination with HER2-Directed Therapies
10. Future Directions
11. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Momenimovahed, Z.; Salehiniya, H. Epidemiological Characteristics of and Risk Factors for Breast Cancer in the World. Breast Cancer Targets Ther. 2019, 11, 151–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeSantis, C.E.; Ma, J.; Goding Sauer, A.; Newman, L.A.; Jemal, A. Breast Cancer Statistics, 2017, Racial Disparity in Mortality by State: Breast Cancer Statistics, 2017. CA Cancer J. Clin. 2017, 67, 439–448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, D.; Koirala, N.; Li, X.; Khan, N.; Dong, F.; Zhang, W.; Mulay, P.; Shrikhande, G.; Puskas, J.; Drazba, J.; et al. Screening of Polymer-Based Drug Delivery Vehicles Targeting Folate Receptors in Triple-Negative Breast Cancer. J. Vasc. Interv. Radiol. 2020, 31, 1866–1873.e2. [Google Scholar] [CrossRef]
- Schnitt, S.J. Classification and Prognosis of Invasive Breast Cancer: From Morphology to Molecular Taxonomy. Mod. Pathol. 2010, 23, S60–S64. [Google Scholar] [CrossRef] [Green Version]
- Godoy-Ortiz, A.; Sanchez-Muñoz, A.; Chica Parrado, M.R.; Álvarez, M.; Ribelles, N.; Rueda Dominguez, A.; Alba, E. Deciphering HER2 Breast Cancer Disease: Biological and Clinical Implications. Front. Oncol. 2019, 9, 1124. [Google Scholar] [CrossRef]
- Wieduwilt, M.J.; Moasser, M.M. The Epidermal Growth Factor Receptor Family: Biology Driving Targeted Therapeutics. Cell. Mol. Life Sci. 2008, 65, 1566–1584. [Google Scholar] [CrossRef] [Green Version]
- Roskoski, R. The ErbB/HER Family of Protein-Tyrosine Kinases and Cancer. Pharmacol. Res. 2014, 79, 34–74. [Google Scholar] [CrossRef]
- Hayes, D.F. HER2 and Breast Cancer—A Phenomenal Success Story. N. Engl. J. Med. 2019, 381, 1284–1286. [Google Scholar] [CrossRef]
- Bae, S.Y.; La Choi, Y.; Kim, S.; Kim, M.; Kim, J.; Jung, S.P.; Choi, M.-Y.; Lee, S.K.; Kil, W.H.; Lee, J.E.; et al. HER3 Status by Immunohistochemistry Is Correlated with Poor Prognosis in Hormone Receptor-Negative Breast Cancer Patients. Breast Cancer Res. Treat. 2013, 139, 741–750. [Google Scholar] [CrossRef]
- Hellyer, N.J.; Cheng, K.; Koland, J.G. ErbB3 (HER3) Interaction with the P85 Regulatory Subunit of Phosphoinositide 3-Kinase. Biochem. J. 1998, 333, 757–763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ménard, S.; Fortis, S.; Castiglioni, F.; Agresti, R.; Balsari, A. HER2 as a Prognostic Factor in Breast Cancer. Oncology 2001, 61, 67–72. [Google Scholar] [CrossRef] [PubMed]
- Dey, N.; Williams, C.; Leyland-Jones, B.; De, P. A Critical Role for HER3 in HER2-Amplified and Non-Amplified Breast Cancers: Function of a Kinase-Dead RTK. Am. J. Transl. Res. 2015, 7, 733–750. [Google Scholar] [PubMed]
- Soltoff, S.P.; Carraway, K.L.; Prigent, S.A.; Gullick, W.G.; Cantley, L.C. ErbB3 Is Involved in Activation of Phosphatidylinositol 3-Kinase by Epidermal Growth Factor. Mol. Cell. Biol. 1994, 14, 3550–3558. [Google Scholar] [CrossRef] [PubMed]
- Dey, N.; Sun, Y.; Carlson, J.H.; Wu, H.; Lin, X.; Leyland-Jones, B.; De, P. Anti-Tumor Efficacy of BEZ235 Is Complemented by Its Anti-Angiogenic Effects via Downregulation of PI3K-MTOR-HIF1alpha Signaling in HER2-Defined Breast Cancers. Am. J. Cancer Res. 2016, 6, 714–746. [Google Scholar]
- Muthuswamy, S.K.; Gilman, M.; Brugge, J.S. Controlled Dimerization of ErbB Receptors Provides Evidence for Differential Signaling by Homo- and Heterodimers. Mol. Cell. Biol. 1999, 19, 6845–6857. [Google Scholar] [CrossRef] [Green Version]
- Schmit, F.; Utermark, T.; Zhang, S.; Wang, Q.; Von, T.; Roberts, T.M.; Zhao, J.J. PI3K Isoform Dependence of PTEN-Deficient Tumors Can Be Altered by the Genetic Context. Proc. Natl. Acad. Sci. USA 2014, 111, 6395–6400. [Google Scholar] [CrossRef] [Green Version]
- Dey, N.; Aske, J.; Lin, X.; Sun, Y.; Leyland-Jones, B.; Friedman, L.; De, P. A Tipping-Point for Apoptosis Following Dual Inhibition of HER2 Signaling Network by T-DM1 plus GDC-0980 Maximizes Anti-Tumor Efficacy. Am. J. Cancer Res. 2021, 11, 2867–2892. [Google Scholar]
- Kreutzfeldt, J.; Rozeboom, B.; Dey, N.; De, P. The Trastuzumab Era: Current and Upcoming Targeted HER2+ Breast Cancer Therapies. Am. J. Cancer Res. 2020, 10, 1045–1067. [Google Scholar]
- McDonald, C.B.; Bhat, V.; Mikles, D.C.; Deegan, B.J.; Seldeen, K.L.; Farooq, A. Bivalent Binding Drives the Formation of the Grb2-Gab1 Signaling Complex in a Noncooperative Manner. FEBS J. 2012, 279, 2156–2173. [Google Scholar] [CrossRef] [Green Version]
- Song, X.; Fan, P.-D.; Bantikassegn, A.; Guha, U.; Threadgill, D.W.; Varmus, H.; Politi, K. ERBB3-Independent Activation of the PI3K Pathway in EGFR-Mutant Lung Adenocarcinomas. Cancer Res. 2015, 75, 1035–1045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartman, Z.; Zhao, H.; Agazie, Y.M. HER2 Stabilizes EGFR and Itself by Altering Autophosphorylation Patterns in a Manner That Overcomes Regulatory Mechanisms and Promotes Proliferative and Transformation Signaling. Oncogene 2013, 32, 4169–4180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, A.; Unni, N.; Peng, Y. The Changing Paradigm for the Treatment of HER2-Positive Breast Cancer. Cancers 2020, 12, 2081. [Google Scholar] [CrossRef]
- Urso, L.; Vernaci, G.; Carlet, J.; Lo Mele, M.; Fassan, M.; Zulato, E.; Faggioni, G.; Menichetti, A.; Di Liso, E.; Griguolo, G.; et al. ESR1 Gene Mutation in Hormone Receptor-Positive HER2-Negative Metastatic Breast Cancer Patients: Concordance Between Tumor Tissue and Circulating Tumor DNA Analysis. Front. Oncol. 2021, 11, 625636. [Google Scholar] [CrossRef] [PubMed]
- Giuliano, M.; Hu, H.; Wang, Y.-C.; Fu, X.; Nardone, A.; Herrera, S.; Mao, S.; Contreras, A.; Gutierrez, C.; Wang, T.; et al. Upregulation of ER Signaling as an Adaptive Mechanism of Cell Survival in HER2-Positive Breast Tumors Treated with Anti-HER2 Therapy. Clin. Cancer Res. 2015, 21, 3995–4003. [Google Scholar] [CrossRef] [Green Version]
- Nahta, R.; O’Regan, R.M. Therapeutic Implications of Estrogen Receptor Signaling in HER2-Positive Breast Cancers. Breast Cancer Res. Treat. 2012, 135, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Eggersmann, T.K.; Degenhardt, T.; Gluz, O.; Wuerstlein, R.; Harbeck, N. CDK4/6 Inhibitors Expand the Therapeutic Options in Breast Cancer: Palbociclib, Ribociclib and Abemaciclib. BioDrugs 2019, 33, 125–135. [Google Scholar] [CrossRef]
- Abukhdeir, A.M.; Park, B.H. P21 and P27: Roles in Carcinogenesis and Drug Resistance. Expert Rev. Mol. Med. 2008, 10, e19. [Google Scholar] [CrossRef] [Green Version]
- Kong, T.; Liu, M.; Ji, B.; Bai, B.; Cheng, B.; Wang, C. Role of the Extracellular Signal-Regulated Kinase 1/2 Signaling Pathway in Ischemia-Reperfusion Injury. Front. Physiol. 2019, 10, 1038. [Google Scholar] [CrossRef] [Green Version]
- Finn, R.S.; Dering, J.; Conklin, D.; Kalous, O.; Cohen, D.J.; Desai, A.J.; Ginther, C.; Atefi, M.; Chen, I.; Fowst, C.; et al. PD 0332991, a Selective Cyclin D Kinase 4/6 Inhibitor, Preferentially Inhibits Proliferation of Luminal Estrogen Receptor-Positive Human Breast Cancer Cell Lines in Vitro. Breast Cancer Res. 2009, 11, R77. [Google Scholar] [CrossRef] [Green Version]
- Goel, S.; Wang, Q.; Watt, A.C.; Tolaney, S.M.; Dillon, D.A.; Li, W.; Ramm, S.; Palmer, A.C.; Yuzugullu, H.; Varadan, V.; et al. Overcoming Therapeutic Resistance in HER2-Positive Breast Cancers with CDK4/6 Inhibitors. Cancer Cell 2016, 29, 255–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asghar, U.; Witkiewicz, A.K.; Turner, N.C.; Knudsen, E.S. The History and Future of Targeting Cyclin-Dependent Kinases in Cancer Therapy. Nat. Rev. Drug Discov. 2015, 14, 130–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, N.U.; Amiri-Kordestani, L.; Palmieri, D.; Liewehr, D.J.; Steeg, P.S. CNS Metastases in Breast Cancer: Old Challenge, New Frontiers. Clin. Cancer Res. 2013, 19, 6404–6418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mounsey, L.A.; Deal, A.M.; Keith, K.C.; Benbow, J.M.; Shachar, S.S.; Zagar, T.; Dees, E.C.; Carey, L.A.; Ewend, M.G.; Anders, C.K. Changing Natural History of HER2–Positive Breast Cancer Metastatic to the Brain in the Era of New Targeted Therapies. Clin. Breast Cancer 2018, 18, 29–37. [Google Scholar] [CrossRef] [Green Version]
- Murthy, R.K.; Loi, S.; Okines, A.; Paplomata, E.; Hamilton, E.; Hurvitz, S.A.; Lin, N.U.; Borges, V.; Abramson, V.; Anders, C.; et al. Tucatinib, Trastuzumab, and Capecitabine for HER2-Positive Metastatic Breast Cancer. N. Engl. J. Med. 2020, 382, 597–609. [Google Scholar] [CrossRef]
- Gelbert, L.M.; Cai, S.; Lin, X.; Sanchez-Martinez, C.; del Prado, M.; Lallena, M.J.; Torres, R.; Ajamie, R.T.; Wishart, G.N.; Flack, R.S.; et al. Preclinical Characterization of the CDK4/6 Inhibitor LY2835219: In-Vivo Cell Cycle-Dependent/Independent Anti-Tumor Activities Alone/in Combination with Gemcitabine. Investig. New Drugs 2014, 32, 825–837. [Google Scholar] [CrossRef] [Green Version]
- Michaud, K.; Solomon, D.A.; Oermann, E.; Kim, J.-S.; Zhong, W.-Z.; Prados, M.D.; Ozawa, T.; James, C.D.; Waldman, T. Pharmacologic Inhibition of Cyclin-Dependent Kinases 4 and 6 Arrests the Growth of Glioblastoma Multiforme Intracranial Xenografts. Cancer Res. 2010, 70, 3228–3238. [Google Scholar] [CrossRef] [Green Version]
- Raub, T.J.; Wishart, G.N.; Kulanthaivel, P.; Staton, B.A.; Ajamie, R.T.; Sawada, G.A.; Gelbert, L.M.; Shannon, H.E.; Sanchez-Martinez, C.; De Dios, A. Brain Exposure of Two Selective Dual CDK4 and CDK6 Inhibitors and the Antitumor Activity of CDK4 and CDK6 Inhibition in Combination with Temozolomide in an Intracranial Glioblastoma Xenograft. Drug Metab. Dispos. 2015, 43, 1360–1371. [Google Scholar] [CrossRef] [Green Version]
- Tien, A.-C.; Li, J.; Bao, X.; Derogatis, A.; Kim, S.; Mehta, S.; Sanai, N. A Phase 0 Trial of Ribociclib in Recurrent Glioblastoma Patients Incorporating a Tumor Pharmacodynamic- and Pharmacokinetic-Guided Expansion Cohort. Clin. Cancer Res. 2019, 25, 5777–5786. [Google Scholar] [CrossRef] [Green Version]
- Tolaney, S.M.; Sahebjam, S.; Le Rhun, E.; Bachelot, T.; Kabos, P.; Awada, A.; Yardley, D.; Chan, A.; Conte, P.; Diéras, V.; et al. A Phase II Study of Abemaciclib in Patients with Brain Metastases Secondary to Hormone Receptor–Positive Breast Cancer. Clin. Cancer Res. 2020, 26, 5310–5319. [Google Scholar] [CrossRef]
- Cobleigh, M.A.; Vogel, C.L.; Tripathy, D.; Robert, N.J.; Scholl, S.; Fehrenbacher, L.; Wolter, J.M.; Paton, V.; Shak, S.; Lieberman, G.; et al. Multinational Study of the Efficacy and Safety of Humanized Anti-HER2 Monoclonal Antibody in Women Who Have HER2-Overexpressing Metastatic Breast Cancer That Has Progressed after Chemotherapy for Metastatic Disease. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 1999, 17, 2639–2648. [Google Scholar] [CrossRef] [PubMed]
- Slamon, D.J.; Leyland-Jones, B.; Shak, S.; Fuchs, H.; Paton, V.; Bajamonde, A.; Fleming, T.; Eiermann, W.; Wolter, J.; Pegram, M.; et al. Use of Chemotherapy plus a Monoclonal Antibody against HER2 for Metastatic Breast Cancer That Overexpresses HER2. N. Engl. J. Med. 2001, 344, 783–792. [Google Scholar] [CrossRef] [PubMed]
- Romond, E.H.; Perez, E.A.; Bryant, J.; Suman, V.J.; Geyer, C.E.; Davidson, N.E.; Tan-Chiu, E.; Martino, S.; Paik, S.; Kaufman, P.A.; et al. Trastuzumab plus Adjuvant Chemotherapy for Operable HER2-Positive Breast Cancer. N. Engl. J. Med. 2005, 353, 1673–1684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swain, S.M.; Baselga, J.; Kim, S.-B.; Ro, J.; Semiglazov, V.; Campone, M.; Ciruelos, E.; Ferrero, J.-M.; Schneeweiss, A.; Heeson, S.; et al. Pertuzumab, Trastuzumab, and Docetaxel in HER2-Positive Metastatic Breast Cancer. N. Engl. J. Med. 2015, 372, 724–734. [Google Scholar] [CrossRef] [Green Version]
- Gianni, L.; Pienkowski, T.; Im, Y.-H.; Roman, L.; Tseng, L.-M.; Liu, M.-C.; Lluch, A.; Staroslawska, E.; de la Haba-Rodriguez, J.; Im, S.-A.; et al. Efficacy and Safety of Neoadjuvant Pertuzumab and Trastuzumab in Women with Locally Advanced, Inflammatory, or Early HER2-Positive Breast Cancer (NeoSphere): A Randomised Multicentre, Open-Label, Phase 2 Trial. Lancet Oncol. 2012, 13, 25–32. [Google Scholar] [CrossRef]
- Von Minckwitz, G.; Procter, M.; de Azambuja, E.; Zardavas, D.; Benyunes, M.; Viale, G.; Suter, T.; Arahmani, A.; Rouchet, N.; Clark, E.; et al. Adjuvant Pertuzumab and Trastuzumab in Early HER2-Positive Breast Cancer. N. Engl. J. Med. 2017, 377, 122–131. [Google Scholar] [CrossRef]
- Rugo, H.S.; Im, S.-A.; Cardoso, F.; Cortés, J.; Curigliano, G.; Musolino, A.; Pegram, M.D.; Wright, G.S.; Saura, C.; Escrivá-de-Romaní, S.; et al. Efficacy of Margetuximab vs Trastuzumab in Patients With Pretreated ERBB2-Positive Advanced Breast Cancer: A Phase 3 Randomized Clinical Trial. JAMA Oncol. 2021, 7, 573–584. [Google Scholar] [CrossRef]
- Verma, S.; Miles, D.; Gianni, L.; Krop, I.E.; Welslau, M.; Baselga, J.; Pegram, M.; Oh, D.-Y.; Diéras, V.; Guardino, E.; et al. Trastuzumab Emtansine for HER2-Positive Advanced Breast Cancer. N. Engl. J. Med. 2012, 367, 1783–1791. [Google Scholar] [CrossRef] [Green Version]
- Krop, I.E.; Kim, S.-B.; González-Martín, A.; LoRusso, P.M.; Ferrero, J.-M.; Smitt, M.; Yu, R.; Leung, A.C.F.; Wildiers, H. TH3RESA study collaborators Trastuzumab Emtansine versus Treatment of Physician’s Choice for Pretreated HER2-Positive Advanced Breast Cancer (TH3RESA): A Randomised, Open-Label, Phase 3 Trial. Lancet Oncol. 2014, 15, 689–699. [Google Scholar] [CrossRef]
- Von Minckwitz, G.; Huang, C.-S.; Mano, M.S.; Loibl, S.; Mamounas, E.P.; Untch, M.; Wolmark, N.; Rastogi, P.; Schneeweiss, A.; Redondo, A.; et al. Trastuzumab Emtansine for Residual Invasive HER2-Positive Breast Cancer. N. Engl. J. Med. 2019, 380, 617–628. [Google Scholar] [CrossRef]
- Modi, S.; Saura, C.; Yamashita, T.; Park, Y.H.; Kim, S.-B.; Tamura, K.; Andre, F.; Iwata, H.; Ito, Y.; Tsurutani, J.; et al. Trastuzumab Deruxtecan in Previously Treated HER2-Positive Breast Cancer. N. Engl. J. Med. 2020, 382, 610–621. [Google Scholar] [CrossRef] [PubMed]
- Cortés, J.; Kim, S.-B.; Chung, W.-P.; Im, S.-A.; Park, Y.H.; Hegg, R.; Kim, M.H.; Tseng, L.-M.; Petry, V.; Chung, C.-F.; et al. Trastuzumab Deruxtecan versus Trastuzumab Emtansine for Breast Cancer. N. Engl. J. Med. 2022, 386, 1143–1154. [Google Scholar] [CrossRef] [PubMed]
- Geyer, C.E.; Forster, J.; Lindquist, D.; Chan, S.; Romieu, C.G.; Pienkowski, T.; Jagiello-Gruszfeld, A.; Crown, J.; Chan, A.; Kaufman, B.; et al. Lapatinib plus Capecitabine for HER2-Positive Advanced Breast Cancer. N. Engl. J. Med. 2006, 355, 2733–2743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, A.; Delaloge, S.; Holmes, F.A.; Moy, B.; Iwata, H.; Harvey, V.J.; Robert, N.J.; Silovski, T.; Gokmen, E.; von Minckwitz, G.; et al. Neratinib after Trastuzumab-Based Adjuvant Therapy in Patients with HER2-Positive Breast Cancer (ExteNET): A Multicentre, Randomised, Double-Blind, Placebo-Controlled, Phase 3 Trial. Lancet Oncol. 2016, 17, 367–377. [Google Scholar] [CrossRef]
- Saura, C.; Oliveira, M.; Feng, Y.-H.; Dai, M.-S.; Chen, S.-W.; Hurvitz, S.A.; Kim, S.-B.; Moy, B.; Delaloge, S.; Gradishar, W.; et al. Neratinib Plus Capecitabine Versus Lapatinib Plus Capecitabine in HER2-Positive Metastatic Breast Cancer Previously Treated with ≥ 2 HER2-Directed Regimens: Phase III NALA Trial. J. Clin. Oncol. 2020, 38, 3138–3149. [Google Scholar] [CrossRef]
- Bonelli, P.; Tuccillo, F.M.; Borrelli, A.; Schiattarella, A.; Buonaguro, F.M. CDK/CCN and CDKI Alterations for Cancer Prognosis and Therapeutic Predictivity. BioMed Res. Int. 2014, 2014, 361020. [Google Scholar] [CrossRef]
- Halachmi, S.; Marden, E.; Martin, G.; MacKay, H.; Abbondanza, C.; Brown, M. Estrogen Receptor-Associated Proteins: Possible Mediators of Hormone-Induced Transcription. Science 1994, 264, 1455–1458. [Google Scholar] [CrossRef]
- Pavithran, H.; Kumavath, R. Emerging Role of Pioneer Transcription Factors in Targeted ERα Positive Breast Cancer. Explor. Target. Anti-Tumor Ther. 2021, 2, 26–35. [Google Scholar] [CrossRef]
- Giacinti, C.; Giordano, A. RB and Cell Cycle Progression. Oncogene 2006, 25, 5220–5227. [Google Scholar] [CrossRef] [Green Version]
- Harbour, J.W.; Dean, D.C. The Rb/E2F Pathway: Expanding Roles and Emerging Paradigms. Genes Dev. 2000, 14, 2393–2409. [Google Scholar] [CrossRef] [Green Version]
- Cooper, G.M.; Hausman, R.E. The eukaryotic cell cycle. In The Cell: A Molecular Approach; Sinauer Associates: Sunderland, MA, USA, 2013; pp. 641–647. ISBN 978-0-87893-964-0. [Google Scholar]
- The Cancer Genome Atlas Network. Comprehensive Molecular Portraits of Human Breast Tumours. Nature 2012, 490, 61–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landis, M.W.; Pawlyk, B.S.; Li, T.; Sicinski, P.; Hinds, P.W. Cyclin D1-Dependent Kinase Activity in Murine Development and Mammary Tumorigenesis. Cancer Cell 2006, 9, 13–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corona, S.P.; Ravelli, A.; Cretella, D.; Cappelletti, M.R.; Zanotti, L.; Dester, M.; Gobbi, A.; Petronini, P.G.; Generali, D. CDK4/6 Inhibitors in HER2-Positive Breast Cancer. Crit. Rev. Oncol. Hematol. 2017, 112, 208–214. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.S.; Crown, J.P.; Lang, I.; Boer, K.; Bondarenko, I.M.; Kulyk, S.O.; Ettl, J.; Patel, R.; Pinter, T.; Schmidt, M.; et al. The Cyclin-Dependent Kinase 4/6 Inhibitor Palbociclib in Combination with Letrozole versus Letrozole Alone as First-Line Treatment of Oestrogen Receptor-Positive, HER2-Negative, Advanced Breast Cancer (PALOMA-1/TRIO-18): A Randomised Phase 2 Study. Lancet Oncol. 2015, 16, 25–35. [Google Scholar] [CrossRef]
- Hortobagyi, G.N.; Stemmer, S.M.; Burris, H.A.; Yap, Y.-S.; Sonke, G.S.; Paluch-Shimon, S.; Campone, M.; Blackwell, K.L.; André, F.; Winer, E.P.; et al. Ribociclib as First-Line Therapy for HR-Positive, Advanced Breast Cancer. N. Engl. J. Med. 2016, 375, 1738–1748. [Google Scholar] [CrossRef]
- Sledge, G.W.; Toi, M.; Neven, P.; Sohn, J.; Inoue, K.; Pivot, X.; Burdaeva, O.; Okera, M.; Masuda, N.; Kaufman, P.A.; et al. MONARCH 2: Abemaciclib in Combination With Fulvestrant in Women With HR+/HER2− Advanced Breast Cancer Who Had Progressed While Receiving Endocrine Therapy. J. Clin. Oncol. 2017, 35, 2875–2884. [Google Scholar] [CrossRef]
- Marra, A.; Curigliano, G. Are All Cyclin-Dependent Kinases 4/6 Inhibitors Created Equal? NPJ Breast Cancer 2019, 5, 27. [Google Scholar] [CrossRef] [Green Version]
- Finn, R.S.; Martin, M.; Rugo, H.S.; Jones, S.; Im, S.-A.; Gelmon, K.; Harbeck, N.; Lipatov, O.N.; Walshe, J.M.; Moulder, S.; et al. Palbociclib and Letrozole in Advanced Breast Cancer. N. Engl. J. Med. 2016, 375, 1925–1936. [Google Scholar] [CrossRef]
- Kim, S.; Tiedt, R.; Loo, A.; Horn, T.; Delach, S.; Kovats, S.; Haas, K.; Engstler, B.S.; Cao, A.; Pinzon-Ortiz, M.; et al. The Potent and Selective Cyclin-Dependent Kinases 4 and 6 Inhibitor Ribociclib (LEE011) Is a Versatile Combination Partner in Preclinical Cancer Models. Oncotarget 2018, 9, 35226–35240. [Google Scholar] [CrossRef] [Green Version]
- Chen, P.; Lee, N.V.; Hu, W.; Xu, M.; Ferre, R.A.; Lam, H.; Bergqvist, S.; Solowiej, J.; Diehl, W.; He, Y.-A.; et al. Spectrum and Degree of CDK Drug Interactions Predicts Clinical Performance. Mol. Cancer Ther. 2016, 15, 2273–2281. [Google Scholar] [CrossRef] [Green Version]
- Jingwen, B.; Yaochen, L.; Guojun, Z. Cell Cycle Regulation and Anticancer Drug Discovery. Cancer Biol. Med. 2017, 14, 348. [Google Scholar] [CrossRef] [PubMed]
- Tripathy, D.; Bardia, A.; Sellers, W.R. Ribociclib (LEE011): Mechanism of Action and Clinical Impact of This Selective Cyclin-Dependent Kinase 4/6 Inhibitor in Various Solid Tumors. Clin. Cancer Res. 2017, 23, 3251–3262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goetz, M.P.; Toi, M.; Campone, M.; Sohn, J.; Paluch-Shimon, S.; Huober, J.; Park, I.H.; Trédan, O.; Chen, S.-C.; Manso, L.; et al. MONARCH 3: Abemaciclib As Initial Therapy for Advanced Breast Cancer. J. Clin. Oncol. 2017, 35, 3638–3646. [Google Scholar] [CrossRef] [PubMed]
- Dean, J.L.; McClendon, A.K.; Hickey, T.E.; Butler, L.M.; Tilley, W.D.; Witkiewicz, A.K.; Knudsen, E.S. Therapeutic Response to CDK4/6 Inhibition in Breast Cancer Defined by Ex Vivo Analyses of Human Tumors. Cell Cycle 2012, 11, 2756–2761. [Google Scholar] [CrossRef]
- ElChaarani, B.; Stires, H.; Pohlmann, P.R.; Riggins, R. Pre-Clinical Analysis of the CDK4/6 Inhibitor Palbociclib in HER2-Positive Breast Cancer. J. Clin. Oncol. 2017, 35, e12520. [Google Scholar] [CrossRef]
- Croessmann, S.; Formisano, L.; Kinch, L.N.; Gonzalez-Ericsson, P.I.; Sudhan, D.R.; Nagy, R.J.; Mathew, A.; Bernicker, E.H.; Cristofanilli, M.; He, J.; et al. Combined Blockade of Activating ERBB2 Mutations and ER Results in Synthetic Lethality of ER+/HER2 Mutant Breast Cancer. Clin. Cancer Res. 2019, 25, 277–289. [Google Scholar] [CrossRef] [Green Version]
- Lenferink, A.E.; Busse, D.; Flanagan, W.M.; Yakes, F.M.; Arteaga, C.L. ErbB2/Neu Kinase Modulates Cellular P27(Kip1) and Cyclin D1 through Multiple Signaling Pathways. Cancer Res. 2001, 61, 6583–6591. [Google Scholar]
- Nikolai, B.C.; Lanz, R.B.; York, B.; Dasgupta, S.; Mitsiades, N.; Creighton, C.J.; Tsimelzon, A.; Hilsenbeck, S.G.; Lonard, D.M.; Smith, C.L.; et al. HER2 Signaling Drives DNA Anabolism and Proliferation through SRC-3 Phosphorylation and E2F1-Regulated Genes. Cancer Res. 2016, 76, 1463–1475. [Google Scholar] [CrossRef] [Green Version]
- Goel, S.; Pernas, S.; Tan-Wasielewski, Z.; Barry, W.T.; Bardia, A.; Rees, R.; Andrews, C.; Tahara, R.K.; Trippa, L.; Mayer, E.L.; et al. Ribociclib Plus Trastuzumab in Advanced HER2-Positive Breast Cancer: Results of a Phase 1b/2 Trial. Clin. Breast Cancer 2019, 19, 399–404. [Google Scholar] [CrossRef]
- Biganzoli, L.; Brain, E.; Malorni, L.; Risi, E.; Regan, M.M. Phase II Randomized Trial of Neoadjuvant Trastuzumab and Pertuzumab (TP) with Either Palbociclib + Letrozole (Pal+L) or Paclitaxel (Pac) for Elderly Patients with Estrogen Receptor & HER2 Positive (ER+/HER2+) Breast Cancer (BC) (International Breast Cancer Study Group IBCSG 55-17, TOUCH). Ann. Oncol. 2019, 30, v96. [Google Scholar] [CrossRef]
- Loibl, S.; Metzger, O.; Mandrekar, S.J.; Mundhenke, C.; Seiler, S.; Valagussa, P.; DeMichele, A.; Lim, E.; Tripathy, D.; Winer, E.P.; et al. PATINA: A Randomized, Open Label, Phase III Trial to Evaluate the Efficacy and Safety of Palbociclib + Anti-HER2 Therapy + Endocrine Therapy (ET) vs. Anti-HER2 Therapy + ET after Induction Treatment for Hormone Receptor Positive (HR+)/HER2-Positive Metastatic Breast Cancer (MBC). Ann. Oncol. 2018, 29, VIII121. [Google Scholar] [CrossRef]
- Shagisultanova, E.; Chalasani, P.; Brown-Glaberman, U.A.; Gradishar, W.J.; Brenner, A.J.; Stopeck, A.; Gao, D.; McSpadden, T.; Kabos, P.; Borges, V.F.; et al. Tucatinib, Palbociclib, and Letrozole in HR+/HER2+ Metastatic Breast Cancer: Report of Phase IB Safety Cohort. J. Clin. Oncol. 2019, 37, 1029. [Google Scholar] [CrossRef]
- Villagrasa, P.; Prat, A.; Oliveira, M.; De La Pena, L.; Gonzalez, X.; Cortes, J.; Pernas Simon, S.; Rios, J.; Canes, J.; Ciruelos, E. SOLTI-1303 PATRICIA: A Phase II Study of Palbociclib and Trastuzumab (HR+ with or without Letrozole) in Trastuzumab-pretreated, Postmenopausal Patients with HER2-positive Metastatic Breast Cancer. J. Clin. Oncol. 2018, 36, TPS1101. [Google Scholar] [CrossRef]
- Zhang, J.; Meng, Y.; Wang, B.; Wang, L.; Cao, J.; Tao, Z.; Li, T.; Yao, W.; Hu, X. Dalpiciclib Combined With Pyrotinib and Letrozole in Women With HER2-Positive, Hormone Receptor-Positive Metastatic Breast Cancer (LORDSHIPS): A Phase Ib Study. Front. Oncol. 2022, 12, 775081. [Google Scholar] [CrossRef]
- Thu, K.; Soria-Bretones, I.; Mak, T.; Cescon, D. Targeting the Cell Cycle in Breast Cancer: Towards the next Phase. Cell Cycle 2018, 17, 1871–1885. [Google Scholar] [CrossRef] [Green Version]
- Du, Q.; Guo, X.; Wang, M.; Li, Y.; Sun, X.; Li, Q. The Application and Prospect of CDK4/6 Inhibitors in Malignant Solid Tumors. J. Hematol. Oncol. 2020, 13, 41. [Google Scholar] [CrossRef]
- Chang, F.; Lee, J.T.; Navolanic, P.M.; Steelman, L.S.; Shelton, J.G.; Blalock, W.L.; Franklin, R.A.; McCubrey, J.A. Involvement of PI3K/Akt Pathway in Cell Cycle Progression, Apoptosis, and Neoplastic Transformation: A Target for Cancer Chemotherapy. Leukemia 2003, 17, 590–603. [Google Scholar] [CrossRef] [Green Version]
- Wee, P.; Wang, Z. Epidermal Growth Factor Receptor Cell Proliferation Signaling Pathways. Cancers 2017, 9, 52. [Google Scholar] [CrossRef] [Green Version]
- Liang, J.; Zubovitz, J.; Petrocelli, T.; Kotchetkov, R.; Connor, M.K.; Han, K.; Lee, J.-H.; Ciarallo, S.; Catzavelos, C.; Beniston, R.; et al. PKB/Akt Phosphorylates P27, Impairs Nuclear Import of P27 and Opposes P27-Mediated G1 Arrest. Nat. Med. 2002, 8, 1153–1160. [Google Scholar] [CrossRef]
- Larionov, A.A. Current Therapies for Human Epidermal Growth Factor Receptor 2-Positive Metastatic Breast Cancer Patients. Front. Oncol. 2018, 8, 89. [Google Scholar] [CrossRef]
- González-Ruiz, L.; González-Moles, M.Á.; González-Ruiz, I.; Ruiz-Ávila, I.; Ayén, Á.; Ramos-García, P. An Update on the Implications of Cyclin D1 in Melanomas. Pigment. Cell Melanoma Res. 2020, 33, 788–805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciriello, G.; Gatza, M.L.; Beck, A.H.; Wilkerson, M.D.; Rhie, S.K.; Pastore, A.; Zhang, H.; McLellan, M.; Yau, C.; Kandoth, C.; et al. Comprehensive Molecular Portraits of Invasive Lobular Breast Cancer. Cell 2015, 163, 506–519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- METABRIC Group; Curtis, C.; Shah, S.P.; Chin, S.-F.; Turashvili, G.; Rueda, O.M.; Dunning, M.J.; Speed, D.; Lynch, A.G.; Samarajiwa, S.; et al. The Genomic and Transcriptomic Architecture of 2,000 Breast Tumours Reveals Novel Subgroups. Nature 2012, 486, 346–352. [Google Scholar] [CrossRef] [PubMed]
- Pereira, B.; Chin, S.-F.; Rueda, O.M.; Vollan, H.-K.M.; Provenzano, E.; Bardwell, H.A.; Pugh, M.; Jones, L.; Russell, R.; Sammut, S.-J.; et al. The Somatic Mutation Profiles of 2,433 Breast Cancers Refine Their Genomic and Transcriptomic Landscapes. Nat. Commun. 2016, 7, 11479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Razavi, P.; Chang, M.T.; Xu, G.; Bandlamudi, C.; Ross, D.S.; Vasan, N.; Cai, Y.; Bielski, C.M.; Donoghue, M.T.A.; Jonsson, P.; et al. The Genomic Landscape of Endocrine-Resistant Advanced Breast Cancers. Cancer Cell 2018, 34, 427–438.e6. [Google Scholar] [CrossRef] [Green Version]
- Rueda, O.M.; Sammut, S.-J.; Seoane, J.A.; Chin, S.-F.; Caswell-Jin, J.L.; Callari, M.; Batra, R.; Pereira, B.; Bruna, A.; Ali, H.R.; et al. Dynamics of Breast-Cancer Relapse Reveal Late-Recurring ER-Positive Genomic Subgroups. Nature 2019, 567, 399–404. [Google Scholar] [CrossRef]
- Kato, S.; Adashek, J.J.; Shaya, J.; Okamura, R.; Jimenez, R.E.; Lee, S.; Sicklick, J.K.; Kurzrock, R. Concomitant MEK and Cyclin Gene Alterations: Implications for Response to Targeted Therapeutics. Clin. Cancer Res. 2021, 27, 2792–2797. [Google Scholar] [CrossRef]
- Kato, S.; Okamura, R.; Adashek, J.J.; Khalid, N.; Lee, S.; Nguyen, V.; Sicklick, J.K.; Kurzrock, R. Targeting G1/S Phase Cell-Cycle Genomic Alterations and Accompanying Co-Alterations with Individualized CDK4/6 Inhibitor–Based Regimens. JCI Insight 2021, 6, e142547. [Google Scholar] [CrossRef]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The CBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data: Figure 1. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [Green Version]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative Analysis of Complex Cancer Genomics and Clinical Profiles Using the CBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [Green Version]
- Witkiewicz, A.K.; Cox, D.; Knudsen, E.S. CDK4/6 Inhibition Provides a Potent Adjunct to Her2-Targeted Therapies in Preclinical Breast Cancer Models. Genes Cancer 2014, 5, 261–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, M.; Scott, S.; Evans, K.W.; Yuca, E.; Saridogan, T.; Zheng, X.; Wang, H.; Korkut, A.; Cruz Pico, C.X.; Demirhan, M.; et al. Combining Neratinib with CDK4/6, MTOR, and MEK Inhibitors in Models of HER2-Positive Cancer. Clin. Cancer Res. 2021, 27, 1681–1694. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, N.; Conklin, D.; Beckmann, R.; Luo, T.; Chau, K.; Thomas, J.; Mc Nulty, A.; Marchal, C.; Kalous, O.; von Euw, E.; et al. Preclinical Activity of Abemaciclib Alone or in Combination with Antimitotic and Targeted Therapies in Breast Cancer. Mol. Cancer Ther. 2018, 17, 897–907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tolaney, S.M.; Wardley, A.M.; Zambelli, S.; Hilton, J.F.; Troso-Sandoval, T.A.; Ricci, F.; Im, S.-A.; Kim, S.-B.; Johnston, S.R.; Chan, A.; et al. Abemaciclib plus Trastuzumab with or without Fulvestrant versus Trastuzumab plus Standard-of-Care Chemotherapy in Women with Hormone Receptor-Positive, HER2-Positive Advanced Breast Cancer (MonarcHER): A Randomised, Open-Label, Phase 2 Trial. Lancet Oncol. 2020, 21, 763–775. [Google Scholar] [CrossRef]
- Gianni, L.; Bisagni, G.; Colleoni, M.; Del Mastro, L.; Zamagni, C.; Mansutti, M.; Zambetti, M.; Frassoldati, A.; De Fato, R.; Valagussa, P.; et al. Neoadjuvant Treatment with Trastuzumab and Pertuzumab plus Palbociclib and Fulvestrant in HER2-Positive, ER-Positive Breast Cancer (NA-PHER2): An Exploratory, Open-Label, Phase 2 Study. Lancet Oncol. 2018, 19, 249–256. [Google Scholar] [CrossRef]
- Ciruelos, E.; Villagrasa, P.; Pascual, T.; Oliveira, M.; Pernas, S.; Paré, L.; Escrivá-de-Romaní, S.; Manso, L.; Adamo, B.; Martínez, E.; et al. Palbociclib and Trastuzumab in HER2-Positive Advanced Breast Cancer: Results from the Phase II SOLTI-1303 PATRICIA Trial. Clin. Cancer Res. 2020, 26, 5820–5829. [Google Scholar] [CrossRef] [PubMed]
- Ademuyiwa, F.O.; Northfelt, D.; O’Connor, T.; Levine, E.; Luo, J.; Tao, Y.; Hoog, J.; Laury, M.; Summa, T.; Hammerschmidt, T.; et al. Abstract P2-13-01: Phase 2 Study of Neoadjuvant Palbociclib, Letrozole, and Trastuzumab in Patients with ER+ HER2+ Breast Cancer (PALTAN). Cancer Res. 2022, 82, P2-13-01. [Google Scholar] [CrossRef]
- Metzger, O.; Mandrekar, S.; Loibl, S.; Mundhenke, C.; Seiler, S.; Valagussa, P.; Lim, E.; Tripathy, D.; Winer, E.; Huang, C.; et al. Abstract OT3-02-07: PATINA: A Randomized, Open Label, Phase III Trial to Evaluate the Efficacy and Safety of Palbociclib + Anti-HER2 Therapy + Endocrine Therapy (ET) vs. Anti-HER2 Therapy + ET after Induction Treatment for Hormone Receptor Positive (HR+)/HER2-Positive Metastatic Breast Cancer (MBC). Cancer Res. 2019, 79, OT3-02-07. [Google Scholar]
- Spring, L.M.; Clark, S.L.; Li, T.; Goel, S.; Tayob, N.; Viscosi, E.; Abraham, E.; Juric, D.; Isakoff, S.J.; Mayer, E.; et al. Phase 1b Clinical Trial of Ado-Trastuzumab Emtansine and Ribociclib for HER2-Positive Metastatic Breast Cancer. NPJ Breast Cancer 2021, 7, 103. [Google Scholar] [CrossRef]
- Roberts, P.J.; Bisi, J.E.; Strum, J.C.; Combest, A.J.; Darr, D.B.; Usary, J.E.; Zamboni, W.C.; Wong, K.-K.; Perou, C.M.; Sharpless, N.E. Multiple Roles of Cyclin-Dependent Kinase 4/6 Inhibitors in Cancer Therapy. JNCI J. Natl. Cancer Inst. 2012, 104, 476–487. [Google Scholar] [CrossRef]
- LoRusso, P.M.; Weiss, D.; Guardino, E.; Girish, S.; Sliwkowski, M.X. Trastuzumab Emtansine: A Unique Antibody-Drug Conjugate in Development for Human Epidermal Growth Factor Receptor 2–Positive Cancer. Clin. Cancer Res. 2011, 17, 6437–6447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saura, C.; Matito, J.; Oliveira, M.; Wildiers, H.; Brufksy, A.M.; Waters, S.H.; Hurvitz, S.A.; Moy, B.; Kim, S.-B.; Gradishar, W.J.; et al. Biomarker Analysis of the Phase III NALA Study of Neratinib + Capecitabine versus Lapatinib + Capecitabine in Patients with Previously Treated Metastatic Breast Cancer. Clin. Cancer Res. 2021, 27, 5818–5827. [Google Scholar] [CrossRef] [PubMed]
- Giordano, S.H.; Franzoi, M.A.B.; Temin, S.; Anders, C.K.; Chandarlapaty, S.; Crews, J.R.; Kirshner, J.J.; Krop, I.E.; Lin, N.U.; Morikawa, A.; et al. Systemic Therapy for Advanced Human Epidermal Growth Factor Receptor 2-Positive Breast Cancer: ASCO Guideline Update. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2022, 36, JCO2200519. [Google Scholar] [CrossRef] [PubMed]
- Esposito, V. Analysis of Cell Cycle Regulator Proteins in Non-Small Cell Lung Cancer. J. Clin. Pathol. 2004, 57, 58–63. [Google Scholar] [CrossRef] [Green Version]
- Patnaik, A.; Rosen, L.S.; Tolaney, S.M.; Tolcher, A.W.; Goldman, J.W.; Gandhi, L.; Papadopoulos, K.P.; Beeram, M.; Rasco, D.W.; Hilton, J.F.; et al. Efficacy and Safety of Abemaciclib, an Inhibitor of CDK4 and CDK6, for Patients with Breast Cancer, Non–Small Cell Lung Cancer, and Other Solid Tumors. Cancer Discov. 2016, 6, 740–753. [Google Scholar] [CrossRef] [Green Version]
- Skowron, M.A.; Vermeulen, M.; Winkelhausen, A.; Becker, T.K.; Bremmer, F.; Petzsch, P.; Schönberger, S.; Calaminus, G.; Köhrer, K.; Albers, P.; et al. CDK4/6 Inhibition Presents as a Therapeutic Option for Paediatric and Adult Germ Cell Tumours and Induces Cell Cycle Arrest and Apoptosis via Canonical and Non-Canonical Mechanisms. Br. J. Cancer 2020, 123, 378–391. [Google Scholar] [CrossRef]
- D’Andrilli, G.; Giordano, A.; Bovicelli, A. Epithelial Ovarian Cancer: The Role of Cell Cycle Genes in the Different Histotypes. Open Clin. Cancer J. 2008, 2, 7–12. [Google Scholar] [CrossRef]
- Giannone, G.; Tuninetti, V.; Ghisoni, E.; Genta, S.; Scotto, G.; Mittica, G.; Valabrega, G. Role of Cyclin-Dependent Kinase Inhibitors in Endometrial Cancer. Int. J. Mol. Sci. 2019, 20, 2353. [Google Scholar] [CrossRef] [Green Version]
- D’Andrilli, G.; Kumar, C.; Scambia, G.; Giordano, A. Cell Cycle Genes in Ovarian Cancer: Steps Toward Earlier Diagnosis and Novel Therapies. Clin. Cancer Res. 2004, 10, 8132–8141. [Google Scholar] [CrossRef] [Green Version]
- Otto, T.; Sicinski, P. Cell Cycle Proteins as Promising Targets in Cancer Therapy. Nat. Rev. Cancer 2017, 17, 93–115. [Google Scholar] [CrossRef] [Green Version]
- Gianni, L.; Colleoni, M.; Bisagni, G.; Mansutti, M.; Zamagni, C.; Del Mastro, L.; Zambelli, S.; Bianchini, G.; Frassoldati, A.; Maffeis, I.; et al. Effects of Neoadjuvant Trastuzumab, Pertuzumab and Palbociclib on Ki67 in HER2 and ER-Positive Breast Cancer. NPJ Breast Cancer 2022, 8, 1. [Google Scholar] [CrossRef] [PubMed]
- Tolaney, S.M.; Toi, M.; Neven, P.; Sohn, J.; Grischke, E.-M.; Llombart-Cussac, A.; Soliman, H.; Wang, H.; Wijayawardana, S.; Jansen, V.M.; et al. Clinical Significance of PIK3CA and ESR1 Mutations in Circulating Tumor DNA: Analysis from the MONARCH 2 Study of Abemaciclib plus Fulvestrant. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2022, 28, 1500–1506. [Google Scholar] [CrossRef] [PubMed]
- Bhinder, B.; Gilvary, C.; Madhukar, N.S.; Elemento, O. Artificial Intelligence in Cancer Research and Precision Medicine. Cancer Discov. 2021, 11, 900–915. [Google Scholar] [CrossRef] [PubMed]

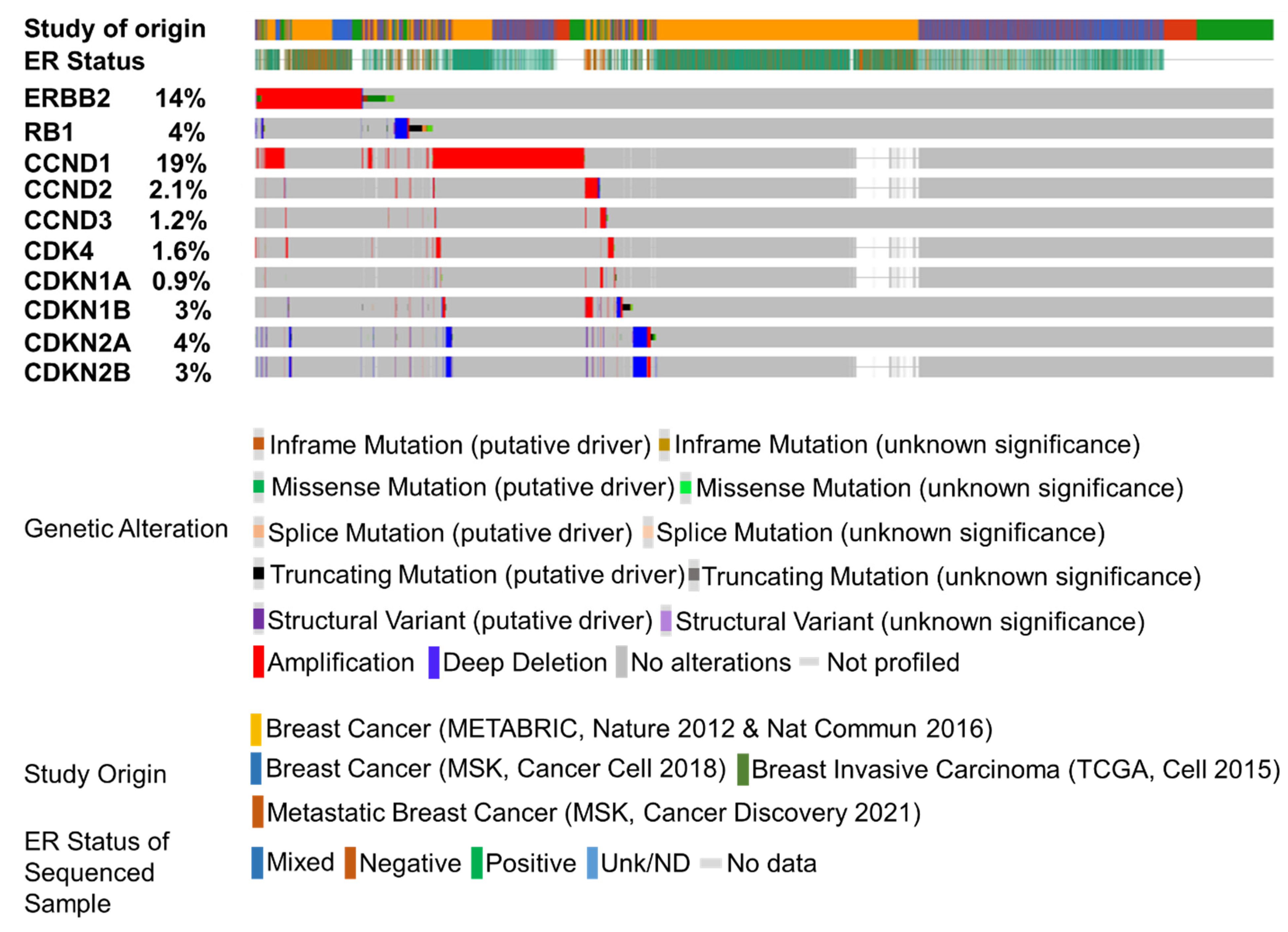

{kind=link}
{kind=link}
| Agent | Molecular Target | Key Trial | Indication (Approval Date) |
|---|---|---|---|
| Monoclonal antibody | |||
| Trastuzumab | HER2 (Subdomain IV) | Phase II [41] and phase III Herceptin trials [42] NSABP B-31 (NCT00004067) NCCTG N9831 (NCT00005970) [43] | 1st line: HER2+ MBC with paclitaxel (1998). 2nd/3rd line: Monotherapy for HER2+ MBC with ≥1 chemotherapy regimens for metastatic disease (1998). Adjuvant: In combination with doxorubicin, cyclophosphamide, and paclitaxel for the treatment of node-positive HER2+ BC (2006). |
| Pertuzumab | HER2 (Subdomain II) | Metastatic: CLEOPATRA (NCT00567190) [44] Neoadjuvant: NeoSphere (NCT00545688) [45] Adjuvant: APHINITY (NCT01358877) [46] | 1st line: HER2+ BC (metastatic (2012) or neoadjuvant (2013)) with trastuzumab and docetaxel (triplet therapy). Adjuvant treatment of HER2+ early BC (EBC) in combination with trastuzumab and chemotherapy (2017). |
| Margetuximab | HER2 (with Fc-engineered region) | SOPHIA (NCT02492711) [47] | 3rd line: Metastatic HER2+ BC with ≥2 anti-HER2 regimens with at least one for metastatic disease (2020). |
| Antibody-drug conjugate (ADC) | |||
| Ado-trastuzumab emtansine (T-DM1) | HER2 (with microtubule inhibitor) | EMILIA (NCT00829166) [48] TH3RESA (NCT01419197) [49] KATHERINE (NCT01772472) [50] | 2nd line: (Monotherapy) MBC was previously treated with trastuzumab and a taxane (2013). (Monotherapy) Adjuvant treatment for HER2+ EBC with the residual invasive disease after neoadjuvant taxane and trastuzumab-based treatment (2019). |
| Trastuzumab deruxtecan (DS-8201) | HER2 (with topoisomerase I inhibitor) | DESTINY-Breast01 (NCT03248492) [51] DESTINY-Breast03 (NCT03529110) [52] | 3rd line: (Monotherapy) Metastatic HER2+ BC who have received two or more prior anti-HER2-based regimens in the metastatic setting (2019). 2nd line: (Monotherapy) Metastatic HER2+ BC previously treated with anti-HER2 antibodies and a taxane in the metastatic setting or (neo) adjuvant setting (2022). |
| Small molecule inhibitors (Tyrosine kinase inhibitors [TKI]) | |||
| Lapatinib | EGFR/HER1, HER2 | Phase III Lapatinib trial (NCT00078572) [53] | 2nd line: HER2+ MBC who have received prior therapy including an anthracycline, taxane, and trastuzumab (2007). |
| Neratinib | Pan-HER (EGFR/HER1, HER2, HER4) | ExteNET (NCT00878709) [54] NALA (NCT01808573) [55] | Extended adjuvant treatment of early-stage HER2+ BC (2017). 3rd line: Metastatic HER2+ BC who have received two or more prior anti-HER2-based regimens for their metastatic disease (2020). |
| Tucatinib | HER2 | HER2CLIMB (NCT02614794) [35] | 3rd line: Metastatic HER2-positive BC, including patients with brain metastases, with one or more prior anti-HER2 regimens in the metastatic setting (2020). |
| Trial (NCT ID) | Arms | Phase, Expected Enrolment & Site | Primary Outcome | Setting |
|---|---|---|---|---|
| CDKi, ribociclib in combination with trastuzumab Or T-DM1 for advanced/metastatic HER2-positive breast cancer (NCT02657343) [80] |
| Phase Ib/II N = 25 USA | Maximum Tolerated Dose (MTD) and/or recommended Phase2 Dose (RP2D) Clinical Benefit Rate (CBR) | Metastatic |
| Ribociclib with trastuzumab plus letrozole in postmenopausal HR+, HER2-positive advanced breast cancer patients (NCT03913234) |
| Phase Ib/II N = 95 South Korea | Progression-free survival | Metastatic |
| T-DM1 and Palbociclib for Metastatic HER2 Breast Cancer (NCT03530696) |
| Phase II N = 46 USA | Progression-free survival | Metastatic |
| Neoadjuvant treatment with palbociclib and exemestane plus trastuzumab and pyrotinib in ER-positive, HER2-positive breast cancer (neoPEHP) (NCT04858516) |
| Phase II N=57 China | Pathological complete response | Neoadjuvant |
| To reduce the use of chemotherapy in postmenopausal patients with ER-positive and HER2-positive breast cancer (TOUCH) (NCT03644186) [81] |
| Phase II N = 144 Belgium, France, Italy, Switzerland | Pathological complete response | Neoadjuvant |
| Palbociclib, trastuzumab, lapatinib, and fulvestrant treatment in patients with brain metastasis for ER-positive, HER2-positive breast cancer (NCT04334330) |
| Phase II N = 34 China | Objective response rate in the CNS | Metastatic (Brain) |
| Clinical Study of the Targeted Therapy, Palbociclib, to Treat Metastatic Breast Cancer (PATINA) (NCT02947685) [82] |
| Phase III N = 496 Multiple centers worldwide | Progression-free survival (PFS) | Metastatic |
| Tucatinib, Palbociclib, and Letrozole in Metastatic Hormone Receptor-Positive and HER2-positive Breast Cancer (NCT03054363) [83] |
| Phase Ib/II N = 42 USA | Phase 1b adverse events (AE) Progression-free survival (PFS) | Metastatic |
| Palbociclib and Trastuzumab With Endocrine Therapy in HER2-positive Metastatic Breast Cancer (PATRICIA II) (NCT02448420) [84] |
| Phase II N = 102 Spain | Progression-Free Survival (PFS) | Metastatic |
| Anastrozole, Palbociclib, Trastuzumab and Pertuzumab in HER2-positive, HER2-positive Metastatic Breast (NCT03304080) |
| Phase I/II N = 36 USA | Dose-Limiting Toxicity (DLT) Maximum Tolerated Dose (MTD) Clinical Benefit Rate (CBR) | Metastatic |
| T-DM1 With or Without Abemaciclib for the Treatment of HER2-Positive Metastatic Breast Cancer (NCT04351230) |
| Phase II N = 0 (Withdrawn) USA | Progression-free survival (PFS) | Metastatic |
| Pyrotinib, Letrozole and SHR6390 in ER+/HER2+ Advanced Breast Cancer (PLEASURABLE) (NCT03772353) [85] |
| Phase I/II N = 79 China | Phase 1b adverse events (AE) Progression-free survival (PFS) | Metastatic |
| Pyrotinib with CDKi SHR6390 for Trastuzumab-treated Advanced HER2-Positive Breast Cancer (INPHASE) (NCT04095390) |
| Phase II N = 60 China | Objective Overall Response Rate (ORR) | Metastatic |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Koirala, N.; Dey, N.; Aske, J.; De, P. Targeting Cell Cycle Progression in HER2+ Breast Cancer: An Emerging Treatment Opportunity. Int. J. Mol. Sci. 2022, 23, 6547. https://doi.org/10.3390/ijms23126547
Koirala N, Dey N, Aske J, De P. Targeting Cell Cycle Progression in HER2+ Breast Cancer: An Emerging Treatment Opportunity. International Journal of Molecular Sciences. 2022; 23(12):6547. https://doi.org/10.3390/ijms23126547
Chicago/Turabian StyleKoirala, Nischal, Nandini Dey, Jennifer Aske, and Pradip De. 2022. "Targeting Cell Cycle Progression in HER2+ Breast Cancer: An Emerging Treatment Opportunity" International Journal of Molecular Sciences 23, no. 12: 6547. https://doi.org/10.3390/ijms23126547
APA StyleKoirala, N., Dey, N., Aske, J., & De, P. (2022). Targeting Cell Cycle Progression in HER2+ Breast Cancer: An Emerging Treatment Opportunity. International Journal of Molecular Sciences, 23(12), 6547. https://doi.org/10.3390/ijms23126547