
Twelve-Nitrogen-Atom Cyclic Structure Stabilized by 3d-Element Atoms: Quantum Chemical Modeling



Abstract
:1. Introduction


2. Method
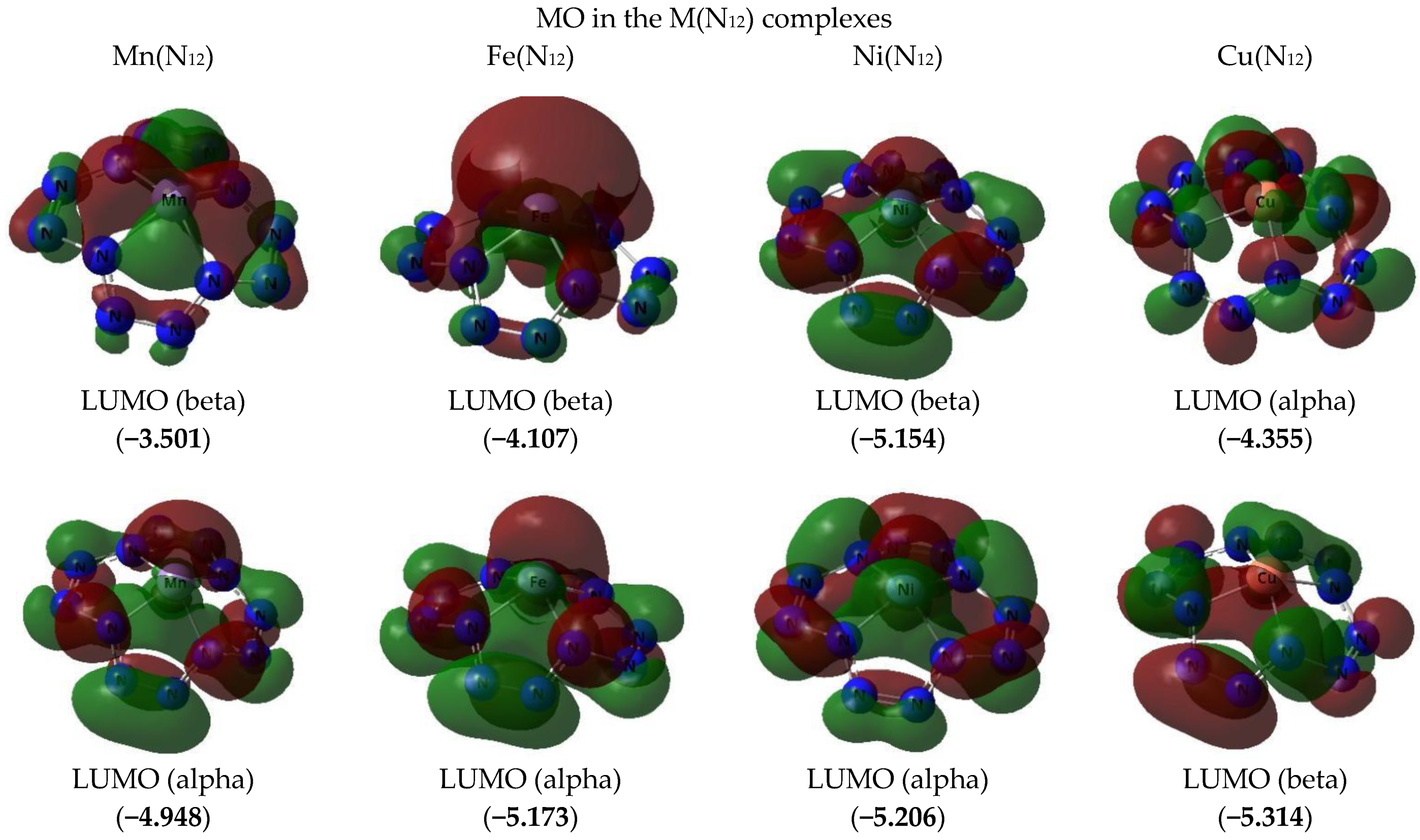
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chachkov, D.V.; Mikhailov, O.V. New Chemical Compound with Unusual Ratio of Number of Carbon and Nitrogen Atoms—C(N12): Quantum-Chemical Modelling. RSC Adv. 2021, 11, 35974–35981. [Google Scholar] [CrossRef]
- Klapötke, T.M.; Harcourt, R.D. The interconversion of N12 to N8 and two equivalents of N2. J. Mol. Struct. 2001, 541, 237–242. [Google Scholar] [CrossRef]
- Olah, G.A.; Prakash, G.K.S.; Rasul, G. N62+ and N42+ Dications and Their N12 and N10 Azido Derivatives: DFT/GIAO-MP2 Theoretical Studies. J. Am. Chem. Soc. 2001, 123, 3308–3310. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.S.; Zhao, J.F. Theoretical Study of Potential Energy Surfaces for N12 Clusters. J. Phys. Chem. A 2002, 106, 5367–5372. [Google Scholar] [CrossRef]
- Bruney, L.Y.; Bledson, T.M.; Strout, D.L. What Makes an N12 Cage Stable? Inorg. Chem. 2003, 42, 8117–8120. [Google Scholar] [CrossRef] [PubMed]
- Samartzis, P.C.; Woodtke, A.M. All-nitrogen chemistry: How far are we from N60? Int. Rev. Phys. Chem. 2006, 25, 1952–2005. [Google Scholar] [CrossRef]
- Greschner, M.J.; Zhang, M.; Majumdar, A.; Liu, H.; Peng, F.; Tse, J.S.; Yao, Y. A New Allotrope of Nitrogen as High-Energy Density Material. J. Phys. Chem. A 2016, 120, 2920–2925. [Google Scholar] [CrossRef]
- Mikhailov, O.V.; Chachkov, D.V. Molecular structures and thermodynamics of stable N4, N6 and N8 neutral polynitrogens according to data of QCISD(T)/TZVP method. Chem. Phys. Lett. 2020, 753, 137594. [Google Scholar] [CrossRef]
- Chachkov, D.V.; Mikhailov, O.V. Tetra-, hexa-, and octanitrogen molecules: A quantum chemical design and thermodynamic properties. Russ. Chem. Bull. 2020, 69, 2067–2072. [Google Scholar] [CrossRef]
- Mikhailov, O.V. Molecular and Electronic Structures of Neutral Polynitrogens: Review on the Theory and Experiment in 21st Century. Int. J. Mol. Sci. 2022, 23, 2841. [Google Scholar] [CrossRef]
- Straka, M. N6 ring as a planar hexagonal ligand in novel M(η6-N6) species. Chem. Phys. Lett. 2002, 358, 531–536. [Google Scholar] [CrossRef]
- Lee, E.P.F.; Dyke, J.M.; Claridge, R.P. Ab Initio Calculations on Al2N4 and AlNn (n = 4 to 7): Potential Precursors of High Energy Density Materials. J. Phys. Chem. A 2002, 106, 8680–8695. [Google Scholar] [CrossRef]
- Choi, C.; Yoo, H.-W.; Goh, E.M.; Cho, S.G.; Jung, Y.S. Ti(N5)4 as a Potential Nitrogen-Rich Stable High-Energy Density Material. J. Phys. Chem. A 2016, 120, 4249–4255. [Google Scholar] [CrossRef] [PubMed]
- Brathwaite, A.D.; Abbott-Lyon, H.L.; Duncan, M.A. Distinctive Coordination of CO vs N2 to Rhodium Cations: An Infrared and Computational Study. J. Phys. Chem. A 2016, 120, 7659–7670. [Google Scholar] [CrossRef]
- Ding, K.; Xu, H.; Yang, Y.; Li, T.; Chen, Z.; Ge, Z.; Zhu, W.; Zheng, W. Mass Spectrometry and Theoretical Investigation of VNn+ (n = 8, 9, and 10) Clusters. J. Phys. Chem. A 2018, 122, 4687–4695. [Google Scholar] [CrossRef]
- Bykov, M.; Bykova, E.; Koemets, E.; Fedotenko, T.; Aprilis, G.; Glazyrin, K.; Liermann, H.-P.; Ponomareva, A.V.; Tidholm, J.; Tasnadi, F.; et al. High-pressure synthesis of a nitrogen-rich inclusion compound ReN8 xN2 with conjugated polymeric nitrogen chains. Angew. Chem. Int. Ed. 2018, 57, 9048–9053. [Google Scholar] [CrossRef] [Green Version]
- Ge, Z.; Ding, K.; Li, Y.; Xu, H.; Chen, Z.; Ma, Y.; Li, T.; Zhu, W.; Zheng, W. Structural evolution of LiNn+ (n = 2, 4, 6, 8, and 10) clusters: Mass spectrometry and theoretical calculations. RSC Adv. 2019, 9, 6762–6769. [Google Scholar] [CrossRef] [Green Version]
- Ding, K.; Li, T.; Xu, H.; Li, Y.; Ge, Z.; Zhu, W.; Zheng, W. Mass spectrometry detection of LiN12+ cluster and theoretical investigation of its structures and stability. Chem. Phys. Lett. 2020, 747, 137310. [Google Scholar] [CrossRef]
- Ding, K.; Chen, H.; Xu, H.; Yang, B.; Ge, Z.; Lu, C.; Zheng, W. Identification of octahedral coordinated ZrN12+ cationic clusters by mass spectrometry and structure searches. Dalton Trans. 2021, 50, 10187–10192. [Google Scholar] [CrossRef]
- Yuan, J.N.; Xia, K.; Wu, J.F.; Sun, J. High-energy-density pentazolate salts: CaN10 and BaN10. Sci. China Phys. Mech. Astron. 2021, 64, 218211. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar] [CrossRef] [Green Version]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P.; Burke, K.; Wang, Y. Generalized gradient approximation for the exchange-correlation hole of a many-electron system. Phys. Rev. B 1996, 54, 16533–16539. [Google Scholar] [CrossRef] [Green Version]
- Medvedev, M.G.; Bushmarinov, I.S.; Sun, J.; Perdew, J.P.; Lyssenko, K.A. Density functional theory is straying from the path toward the exact functional. Science 2017, 355, 49–52. [Google Scholar] [CrossRef] [PubMed]
- Mikhailov, O.V.; Chachkov, D.V. DFT Quantum-Chemical Modeling Molecular Structures of Cobalt Macrocyclic Complexes with Porphyrazine or Its Benzo-Derivatives and Two Oxygen Acido Ligands. Int. J. Mol. Sci. 2020, 21, 9085. [Google Scholar] [CrossRef]
- Hoe, W.M.; Cohen, A.; Handy, N.C. Assessment of a new local exchange functional OPTX. Chem. Phys. Lett. 2001, 341, 319–328. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [Green Version]
- Paulsen, H.; Duelund, L.; Winkler, H.; Toftlund, H.; Trautwein, A.X. Free Energy of Spin-Crossover Complexes Calculated with Density Functional Methods. Inorg. Chem. 2001, 40, 2201–2203. [Google Scholar] [CrossRef]
- Swart, M.; Groenhof, A.R.; Ehlers, A.W.; Lammertsma, K. Validation of Exchange−Correlation Functionals for Spin States of Iron Complexes. J. Phys. Chem. A 2004, 108, 5479–5483. [Google Scholar] [CrossRef]
- Swart, M.; Ehlers, A.W.; Lammertsma, K. Performance of the OPBE exchange-correlation functional. Mol. Phys. 2004, 102, 2467–2474. [Google Scholar] [CrossRef]
- Swart, M. Metal–ligand bonding in metallocenes: Differentiation between spin state, electrostatic and covalent bonding. Inorg. Chim. Acta 2007, 360, 179–189. [Google Scholar] [CrossRef]
- Møller, C.; Plesset, M.S. Note on an approximation treatment for many-electron systems. Phys. Rev. 1934, 46, 618–622. [Google Scholar] [CrossRef] [Green Version]
- Head-Gordon, M.; Pople, J.A.; Frisch, M.J. MP2 energy evaluation by direct methods. Chem. Phys. Lett. 1988, 153, 503–506. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. (Eds.) Gaussian 09, Revision A.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Weinhold, F.; Landis, C.R.; Glendening, E.D. What is NBO analysis and how is it useful? Int. Rev. Phys. Chem. 2016, 35, 399–440. [Google Scholar] [CrossRef]
- Ochterski, J.W. Thermochemistry in Gaussian; Gaussian, Inc.: Wallingford, CT, USA, 2000. [Google Scholar]

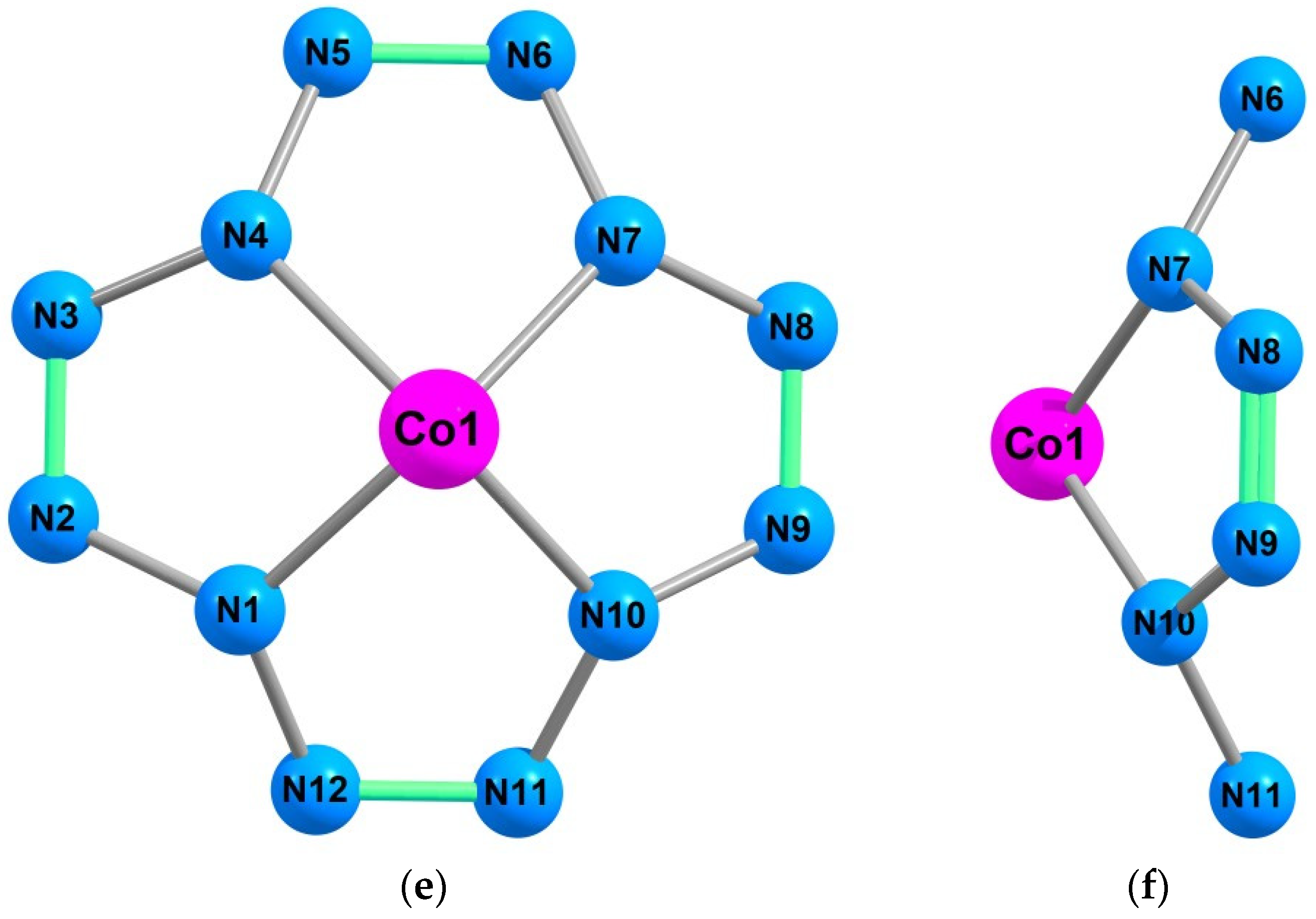


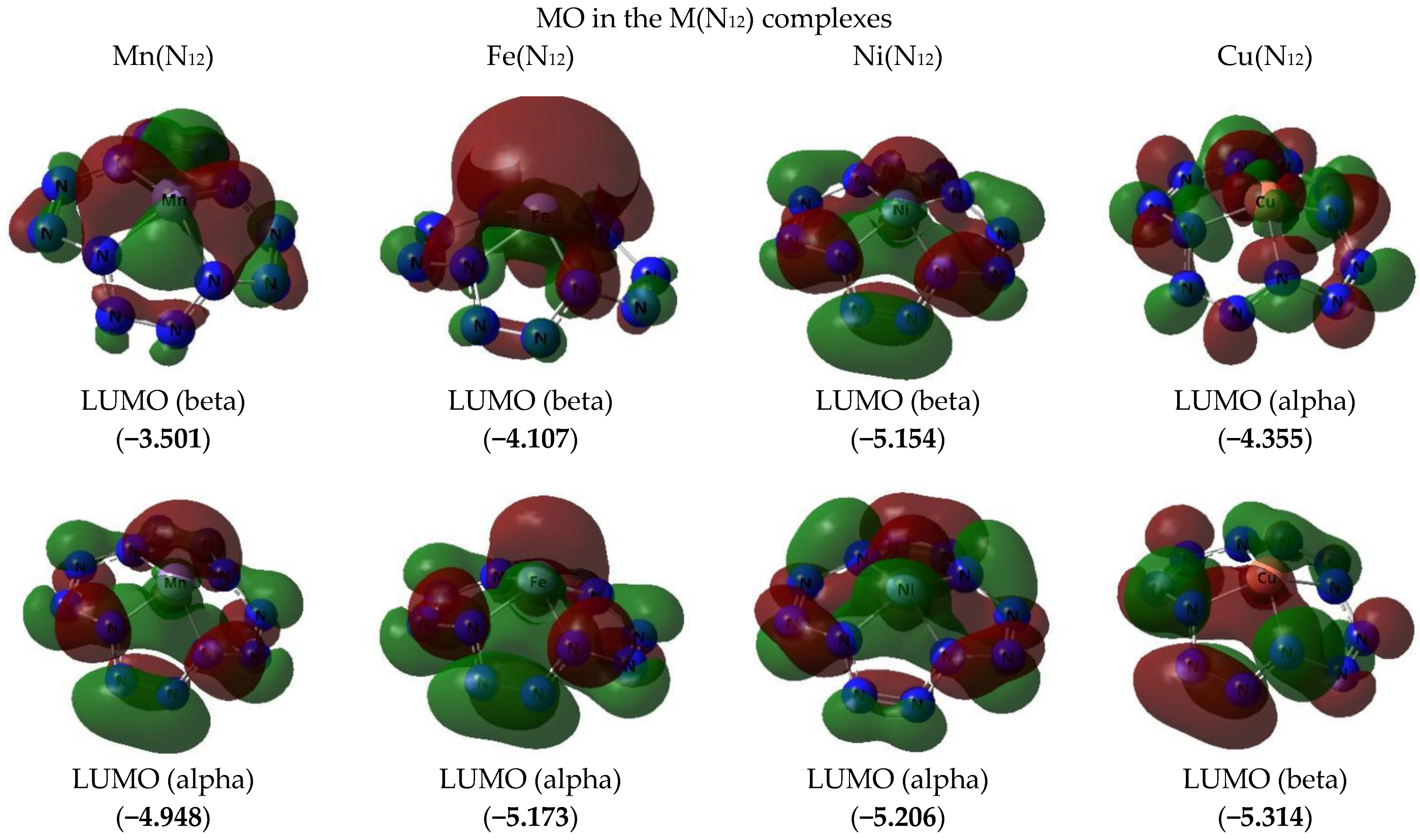

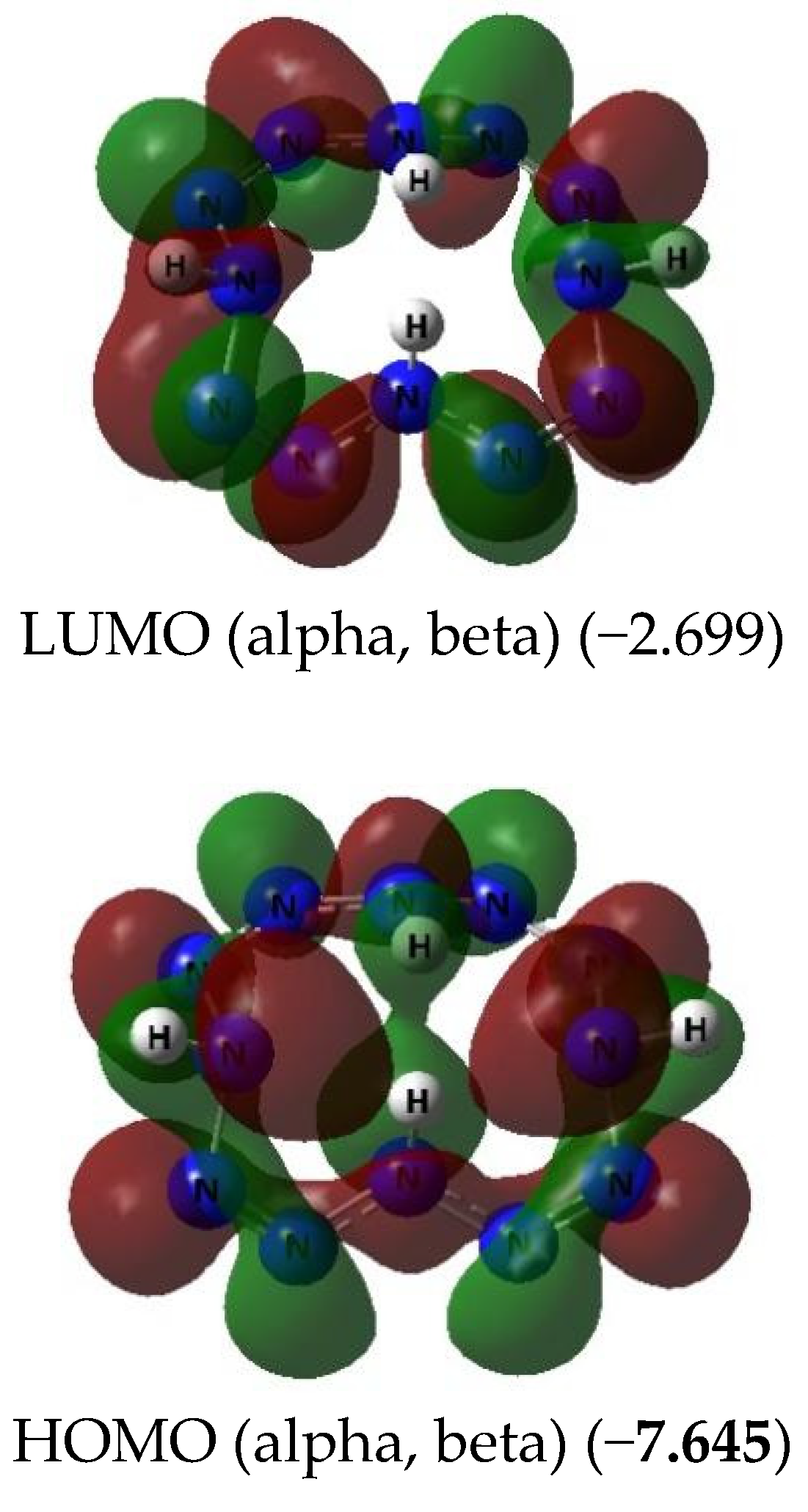


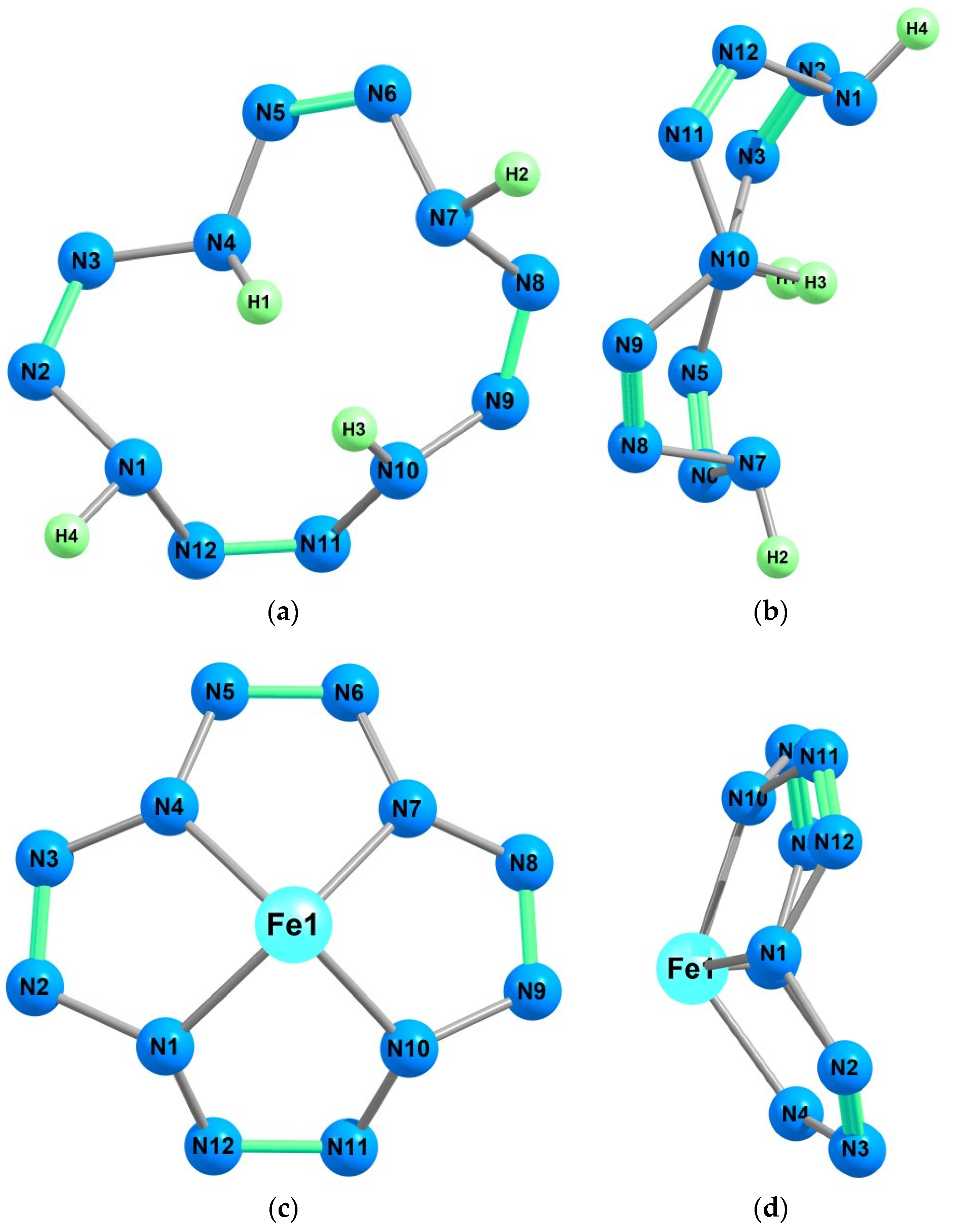{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 3d-Element (M) | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Structural Parameter | Ti | V | Cr | Mn | Fe | Co | Ni | Cu | (H4N12) |
| M–N bond lengths in the MN4 chelate node, pm | |||||||||
| M1N1 | 193.5 | 188.2 | 186.3 | 191.1 | 183.7 | 181.2 | 179.6 | 186.0 | - |
| M1N4 | 193.5 | 188.2 | 186.3 | 191.1 | 182.6 | 181.2 | 179.9 | 184.2 | - |
| M1N7 | 193.5 | 188.2 | 186.3 | 190.0 | 182.6 | 181.2 | 179.6 | 184.2 | - |
| M1N10 | 193.5 | 188.2 | 186.3 | 190.0 | 183.7 | 181.2 | 179.5 | 186.0 | - |
| Nitrogen–nitrogen bond lengths in macrocycle, pm | |||||||||
| N1N2 | 138.0 | 137.9 | 139.1 | 131.7 | 145.3 | 137.4 | 140.6 | 146.3 | 148.1 |
| N2N3 | 125.2 | 125.0 | 123.8 | 129.6 | 123.4 | 124.8 | 121.3 | 123.2 | 123.4 |
| N3N4 | 138.0 | 137.9 | 139.1 | 131.7 | 137.2 | 137.4 | 151.2 | 134.5 | 135.1 |
| N4N5 | 138.0 | 137.9 | 139.1 | 145.3 | 138.5 | 137.4 | 130.2 | 139.9 | 135.1 |
| N5N6 | 125.2 | 125.0 | 123.8 | 123.7 | 124.3 | 124.8 | 131.4 | 122.5 | 123.4 |
| N6N7 | 138.0 | 137.9 | 139.1 | 136.0 | 138.5 | 137.4 | 130.2 | 139.9 | 148.1 |
| N7N8 | 138.0 | 137.9 | 139.1 | 138.8 | 137.2 | 137.4 | 151.2 | 134.5 | 149.5 |
| N8N9 | 125.2 | 125.0 | 123.8 | 124.2 | 123.4 | 124.8 | 121.3 | 123.2 | 122.8 |
| N9N10 | 138.0 | 137.9 | 139.1 | 138.7 | 145.3 | 137.4 | 140.6 | 146.3 | 136.1 |
| N10N11 | 138.0 | 137.9 | 139.1 | 136.1 | 131.5 | 137.4 | 136.1 | 131.1 | 136.0 |
| N11N12 | 125.2 | 125.0 | 123.8 | 123.7 | 130.0 | 124.8 | 125.6 | 129.8 | 122.8 |
| N12N1 | 138.0 | 137.9 | 139.1 | 145.2 | 131.5 | 137.4 | 136.1 | 131.1 | 149.5 |
| Distance from the center of the M atom to the plane formed by donor nitrogen atoms in the MN4 chelate node. pm | |||||||||
| 104.4 | 97.5 | 89.9 | 90.9 | 83.2 | 78.5 | 67.4 | 70.3 | - | |
| Bond angles in the MN4 chelate node, deg | |||||||||
| N1M1N4 | 73.1 | 74.4 | 76.6 | 77.9 | 77.8 | 79.2 | 81.9 | 81.8 | - |
| N4M1N7 | 73.1 | 74.4 | 76.6 | 75.9 | 77.6 | 79.2 | 82.3 | 81.0 | - |
| N7M1N10 | 73.1 | 74.4 | 76.6 | 75.5 | 77.8 | 79.2 | 81.9 | 81.8 | - |
| N10M1N1 | 73.1 | 74.4 | 76.6 | 75.9 | 79.3 | 79.2 | 81.6 | 82.7 | - |
| Bond angles sum (BAS), deg | 292.4 | 297.6 | 306.4 | 305.2 | 312.5 | 316.8 | 327.7 | 327.3 | - |
| Deviation from coplanarity, deg | 67.6 | 62.4 | 53.6 | 54.8 | 47.5 | 43.2 | 32.3 | 32.7 | - |
| Non-bond angles in the MN4 chelate node, deg | |||||||||
| N1N4N7 | 90.0 | 90.0 | 90.0 | 89.1 | 90.7 | 90.0 | 89.7 | 90.8 | 103.9 |
| N4N7N10 | 90.0 | 90.0 | 90.0 | 90.9 | 90.7 | 90.0 | 89.7 | 90.8 | 68.1 |
| N7N10N1 | 90.0 | 90.0 | 90.0 | 90.9 | 89.3 | 90.0 | 90.3 | 89.2 | 96.9 |
| N10N1N4 | 90.0 | 90.0 | 90.0 | 89.1 | 89.3 | 90.0 | 90.3 | 89.2 | 68.1 |
| Non-bond angles sum (NBAS), deg | 360.0 | 360.0 | 360.0 | 360.0 | 360.0 | 360.0 | 360.0 | 360.0 | 337.0 |
| Deviation from coplanarity, deg | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 23.0 |
| Bond angles in 5-membered cycles, deg | |||||||||
| M1N1N2 | 119.7 | 119.8 | 118.6 | 116.1 | 117.0 | 117.4 | 116.5 | 113.0 | - |
| N1N2N3 | 112.4 | 111.9 | 112.6 | 114.8 | 109.9 | 112.7 | 116.0 | 111.1 | 114.9 |
| N2N3N4 | 112.4 | 111.9 | 112.6 | 114.8 | 114.4 | 112.7 | 110.1 | 119.2 | 116.7 |
| N3N4M1 | 119.7 | 119.8 | 118.6 | 116.1 | 118.9 | 117.4 | 114.6 | 114.6 | - |
| M1N4N5 | 119.7 | 119.8 | 118.6 | 116.6 | 118.6 | 117.4 | 113.4 | 114.9 | - |
| N4N5N6 | 112.4 | 111.9 | 112.6 | 110.8 | 112.2 | 112.7 | 113.9 | 114.6 | 116.8 |
| N5N6N7 | 112.4 | 111.9 | 112.6 | 115.5 | 112.2 | 112.7 | 113.9 | 114.6 | 114.9 |
| N6N7M1 | 119.7 | 119.8 | 118.6 | 118.9 | 118.6 | 117.4 | 113.3 | 114.9 | - |
| M1N7N8 | 119.7 | 119.8 | 118.6 | 118.5 | 118.9 | 117.4 | 114.6 | 114.6 | - |
| N7N8N9 | 112.4 | 111.9 | 112.6 | 113.0 | 114.4 | 112.7 | 110.1 | 119.2 | 116.4 |
| N8N9N10 | 112.4 | 111.9 | 112.6 | 113.0 | 109.9 | 112.7 | 116.0 | 111.0 | 120.9 |
| N9N10M1 | 119.7 | 119.8 | 118.6 | 118.5 | 117.0 | 117.4 | 116.6 | 113.0 | - |
| M1N10N11 | 119.7 | 119.8 | 118.6 | 118.9 | 116.8 | 117.4 | 115.3 | 111.3 | - |
| N10N11N12 | 112.4 | 111.9 | 112.6 | 115.4 | 113.4 | 112.7 | 113.6 | 116.3 | 120.9 |
| N11N12N1 | 112.4 | 111.9 | 112.6 | 110.8 | 113.4 | 112.7 | 113.6 | 116.3 | 116.4 |
| N12N1M1 | 119.7 | 119.8 | 118.6 | 116.6 | 116.8 | 117.4 | 115.3 | 111.3 | - |
| Deviation of macrocycle (N12) from coplanarity, deg | 80.4 | 76.4 | 85.2 | 97.8 | 74.0 | 75.6 | 93.6 | 103.0 | 137.7 |
| The difference between macrocycle deviation (N12) in (H4N12) and in M(N12), deg | 57.3 | 61.3 | 52.5 | 39.9 | 63.7 | 52.1 | 44.1 | 34.7 | - |
| M | Ti | V | Cr | Mn | Fe | Co | Ni | Cu | (H4) |
|---|---|---|---|---|---|---|---|---|---|
| μ | 8.43 | 7.30 | 4.81 | 5.40 | 4.65 | 4.00 | 2.92 | 3.11 | 4.75 |
| Effective Charge on Atom, in Units of Electron Charge (ē) | <S**2> | |||||||
|---|---|---|---|---|---|---|---|---|
| M | M1 | N1 (N4) | N2 (N6) | N3 (N5) | N7 (N10) | N8 (N12) | N9 (N11) | |
| Ti | 1.1781 | −0.3028 | 0.0042 | 0.0042 | −0.3028 | 0.0041 | 0.0042 | 0 |
| (−0.3028) | −0.0042 | −0.0042 | (−0.3028) | −0.0041 | −0.0042 | |||
| V | 0.7623 | −0.2071 | 0.0083 | 0.0083 | −0.2071 | 0.0083 | 0.0083 | 0.7532 |
| (−0.2071) | −0.0083 | −0.0083 | (−0.2071) | −0.0083 | −0.0083 | |||
| Cr | 0.7069 | −0.2275 | 0.0253 | 0.0254 | −0.2275 | 0.0255 | 0.0254 | 2.2481 |
| (−0.2275) | −0.0253 | −0.0254 | (−0.2275) | −0.0255 | −0.0254 | |||
| Mn | 0.8408 | −0.1900 | 0.0002 | 0.0002 | −0.2975 | 0.0118 | 0.0121 | 3.9244 |
| (−0.1899) | −0.0629 | (−0.0081) | (−0.2975) | (−0.0078) | −0.0629 | |||
| Fe | 0.5588 | −0.1263 | 0 | 0.0636 | −0.2315 | 0.0636 | 0 | 2.0654 |
| (−0.2316) | −0.0102 | −0.0101 | (−0.1263) | −0.0045 | −0.0045 | |||
| Co | 0.5406 | −0.1783 | 0.0216 | 0.0216 | −0.1783 | 0.0216 | 0.0217 | 3.7832 |
| (−0.1783) | −0.0216 | −0.0216 | (−0.1783) | −0.0216 | −0.0217 | |||
| Ni | 0.4074 | −0.1928 | 0.0748 | 0.0427 | −0.1190 | 0.0427 | 0.0748 | 0.0364 |
| (−0.1190) | (−0.0096) | (−0.0095) | (−0.1928) | −0.0002 | −0.0002 | |||
| Cu | 0.6256 | −0.1576 | −0.0131 | 0.0729 | −0.2579 | 0.073 | −0.0131 | 0.7751 |
| (−0.2581) | −0.0361 | −0.0361 | (−0.1577) | −0.0068 | −0.0729 | |||
| (H4) | - | −0.4932 | −0.0262 | 0.0543 | −0.4934 | −0.0316 | 0.058 | 0 |
| (−0.3478) | (−0.0263) | −0.0544 | (−0.3109) | (−0.0319) | −0.058 | |||
| M | Standard Thermodynamic Parameters of Formation | ||
|---|---|---|---|
| H0f,298, kJ/mol | S0f,298, J/mol∙K | G0f,298, kJ/mol | |
| Ti | 1562.3 | 398.3 | 1792.8 |
| V | 1662.2 | 399.1 | 1890.4 |
| Cr | 1833.8 | 403.2 | 2061.7 |
| Mn | 1571.7 | 404.1 | 1801.0 |
| Fe | 1744.8 | 403.4 | 1972.9 |
| Co | 1823.6 | 404.2 | 2052.2 |
| Ni | 1933.7 | 412.6 | 2159.7 |
| Cu | 2033.9 | 405.0 | 2263.3 |
| H4 | 1705.9 | 399.3 | 2004.8 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mikhailov, O.V.; Chachkov, D.V. Twelve-Nitrogen-Atom Cyclic Structure Stabilized by 3d-Element Atoms: Quantum Chemical Modeling. Int. J. Mol. Sci. 2022, 23, 6560. https://doi.org/10.3390/ijms23126560
Mikhailov OV, Chachkov DV. Twelve-Nitrogen-Atom Cyclic Structure Stabilized by 3d-Element Atoms: Quantum Chemical Modeling. International Journal of Molecular Sciences. 2022; 23(12):6560. https://doi.org/10.3390/ijms23126560
Chicago/Turabian StyleMikhailov, Oleg V., and Denis V. Chachkov. 2022. "Twelve-Nitrogen-Atom Cyclic Structure Stabilized by 3d-Element Atoms: Quantum Chemical Modeling" International Journal of Molecular Sciences 23, no. 12: 6560. https://doi.org/10.3390/ijms23126560
APA StyleMikhailov, O. V., & Chachkov, D. V. (2022). Twelve-Nitrogen-Atom Cyclic Structure Stabilized by 3d-Element Atoms: Quantum Chemical Modeling. International Journal of Molecular Sciences, 23(12), 6560. https://doi.org/10.3390/ijms23126560