Abstract
Inherited retinal diseases (IRD) are a group of heterogeneous disorders, most of which lead to blindness with limited therapeutic options. Pathogenic variants in RBP4, coding for a major blood carrier of retinol, retinol-binding protein 4, are responsible for a peculiar form of IRD. The aim of this study was to investigate if retinal function of an RBP4-related IRD patient can be improved by retinol administration. Our patient presented a peculiar white-dot retinopathy, reminiscent of vitamin A deficient retinopathy. Using a customized next generation sequencing (NGS) IRD panel we discovered a novel loss-of-function homozygous pathogenic variant in RBP4: c.255G >A, p.(Trp85*). Western blotting revealed the absence of RBP4 protein in the patient’s serum. Blood retinol levels were undetectable. The patient was put on a high-dose oral retinol regimen (50,000 UI twice a week). Subjective symptoms and retinal function markedly and sustainably improved at 5-months and 1-year follow-up. Here we show that this novel IRD case can be treated by oral retinol administration.
1. Introduction
Inherited retinal diseases are a heterogeneous group of disorders, the most common of which—retinitis pigmentosa—progresses towards blindness with limited therapeutic options. Some rare forms of IRD, linked with inborn defects of small molecule metabolism can benefit from metabolic and/or dietary treatment [1,2]. For instance, vitamin A and E intake improves vision in patients with abetalipoproteinemia (OMIM# 200100), a hereditary defect of fat-soluble vitamin absorption and transport [3]. Dietary arginine restriction and vitamin B6 intake can reduce blood ornithine level and slow retinal degeneration in patients with gyrate atrophy (OMIM#258870) [4,5]. Low phytanic acid diet may slow retinal degeneration progression rates in adult Refsum disease (OMIM#266500) [6,7].
Retinol-binding protein 4 (RBP4, Uniprot#Q5VY30) is a major blood transporter of retinol from hepatocyte to target organs. Pathogenic variants in RBP4 are associated with both ocular developmental abnormalities and retinal degeneration (OMIM#615147). Ocular developmental abnormalities range from a mild iris coloboma to micro- and anophthalmia. Retinal degeneration consists of a rod–cone dystrophy, also known as retinitis pigmentosa.
We report here that high-dose vitamin A is able to improve visual function in a new case of RBP4-associated retinopathy.
2. Results
2.1. Detailed Case Description
A male patient of Algerian ancestry was initially assessed at age 12 years. He had decreased visual acuity and progressive night blindness since early childhood. Best corrected visual acuity was 20/32 for both eyes with spectacle correction −0.50(−1.0)5° in the right eye (RE) and −0.75(−0.25)170° in the left eye (LE).
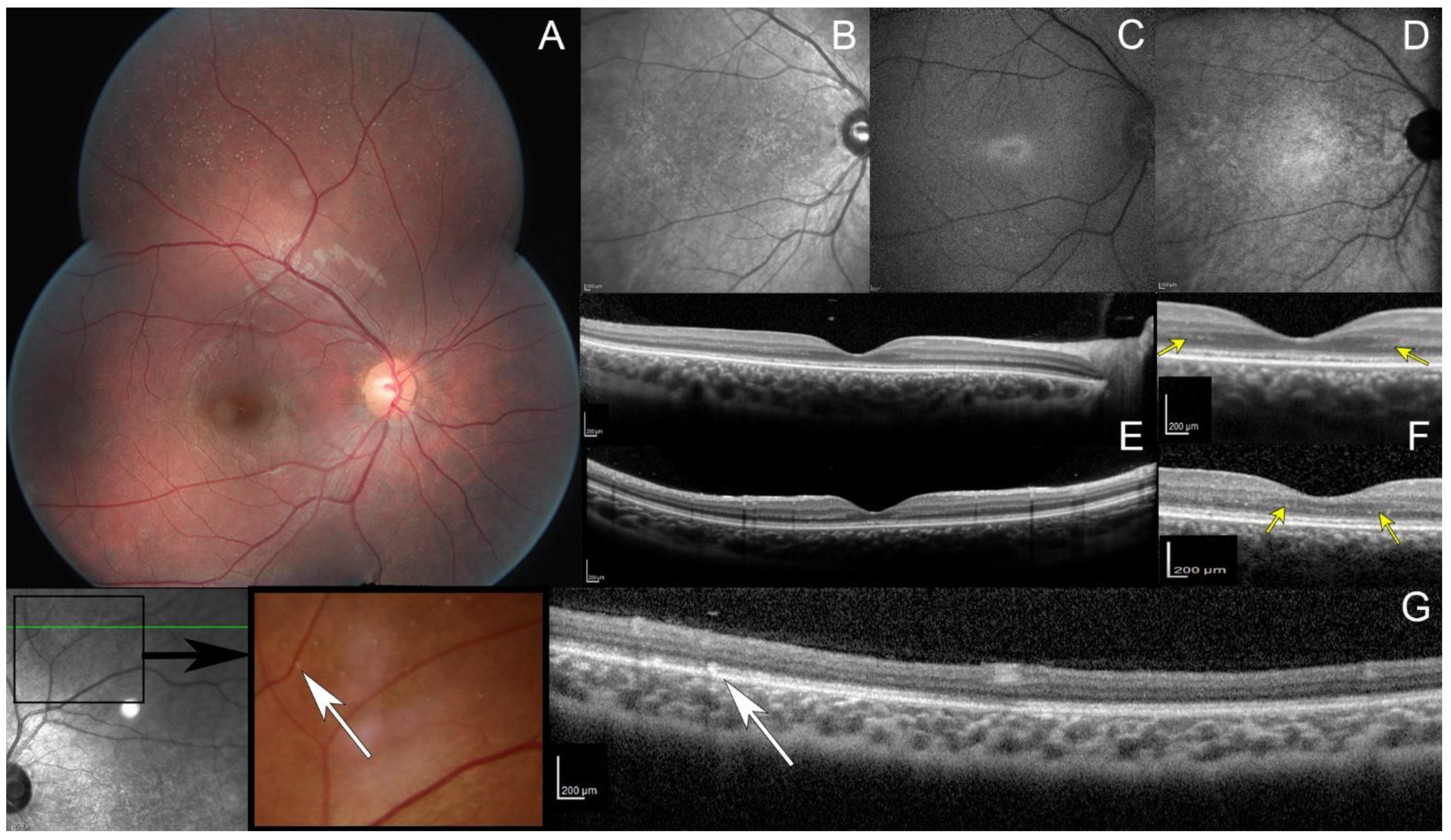
Goldman kinetic visual field tested on V4e, V1e, and V1e targets was normal while the III1e and smaller targets were not perceived by the patient (Figure S1 in Supplementary Data). Automated static perimetry showed a pericentral ring scotoma. Dark-adapted responses (DA0.01) of ISCEV standard full-field electroretinogram (ffERG) were undetectable while DA3.0 and DA10.0 revealed severely reduced and delayed responses; light-adapted responses (LA3.0 and LA3.0 flicker) were severely reduced with implicit time shift in keeping with generalized rod–cone dysfunction (Figure S2 in Supplementary Data). Full Stimulus Threshold (FST) revealed severely reduced threshold for the white stimulus (Figure S3 in Supplementary Data). Fundus examination revealed numerous white dots scattered over the mid and far periphery with no pigmentary or atrophic retinal changes (Figure 1A and Figure S4 in Supplementary Data). On infrared reflectance (IRR) imaging, both maculae were granular (Figure 1B). Increased image averaging on short-wave fundus autofluorescence (SWAF) revealed an ellipsoid-shaped ring of increased autofluorescence around the fovea on an overall hypoautofluorescent background (Figure 1C). Near-infrared fundus autofluorescence (NIRAF) imaging also showed a reduced signal (Figure 1D). Spectral domain optical coherence tomography (SD-OCT) centered on the fovea revealed an hyporeflective irregular ellipsoid zone (EZ) (Figure 1E). The outer nuclear layer (ONL) thickness was preserved with an unusual hyper reflective band on both sides of the fovea (Figure 1F, yellow arrows). SD-OCT performed through the peripheral white dots revealed hyperreflective dots above the retinal pigment epithelium (RPE), interrupting the EZ (Figure 1G, white arrows).
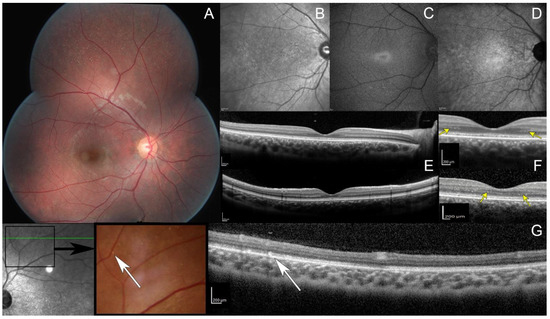
Figure 1.
Multimodal retinal imaging. (A) Fundus photo, multiple white dots scattered over the midperipheral retina. Note the absence of intraretinal pigment migration and the lack of retinal vessel attenuation. (B) Infrared reflectance image, macular granularity. (C) Short-wavelength fundus autofluorescence imaging obtained with significant averaging due to the generalized reduced autofluorescence, small peri-foveal hyperautofluorescent ring with indistinct borders. (D) Near infrared fundus autofluorescence, small hypoautofluorescent dots. (E,F) spectral domain optic coherence tomography (SD-OCT, top: horizontal scan; and bottom: vertical scan), hypo reflective and fragmented ellipsoid zone with no interdigitation zone; preserved outer nuclear layer thickness with an unusual hyper reflective band on both sides of the fovea (yellow arrows). (G) OCT (scan passing through white dots, white arrows), hyperreflective dots above the retinal pigment epithelium with a focal interruption of the ellipsoid zone.
A general examination was performed to detect any vitamin A deficiency-related alterations. The only skin issue was subtle acne vulgaris on the forehead. Past medical history was unremarkable and the patient declined any dietary restrictions.
2.2. Genetic and Functional Studies
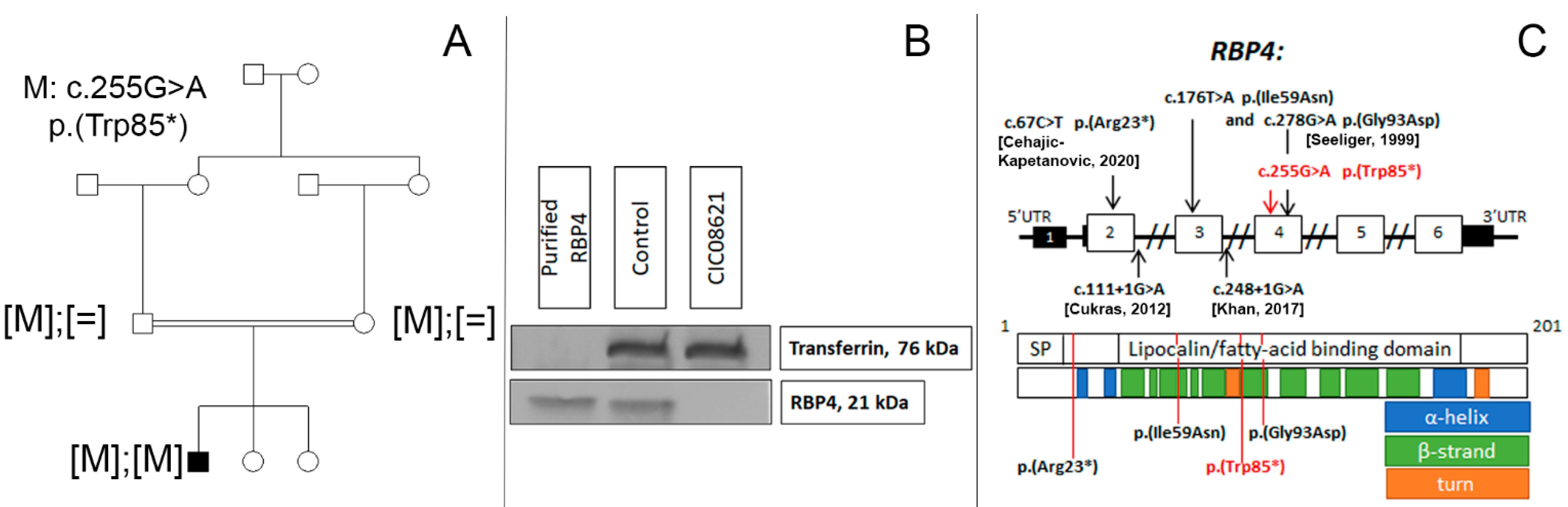
The family consisted of unaffected first-degree-cousin parents of Algerian descent and two unaffected sisters (Figure 2A). Targeted NGS identified a novel homozygous nonsense variant c.255G >A, p.(Trp85*) in RBP4 which co-segregated with disease in this family. This variant was classified as pathogenic Ia (PVS1, PP1-S, PM2) in accordance with ACMG standards [8]. Western blot analysis revealed the absence of the RBP4 protein band in the serum of the patient (Figure 2B). Blood retinol level was also undetectable.
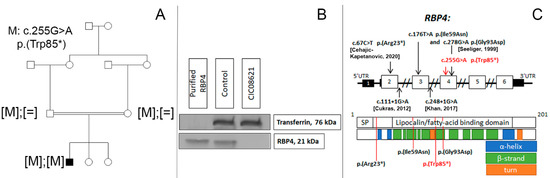
Figure 2.
(A) Novel biallelic RBP4 variant co-segregating with the disease. (B) Western blot analysis showing the absence of RBP4 in peripheral blood of our patient (CIC08621) carrying the homozygous nonsense variant in RBP4, compared to an unaffected control and recombinant RPB4. Transferrin is used as serum loading control. (C) RBP4 gene and protein structure. SP—signal peptide domain. Disulfide bonds (22-178, 88-192, 138-147) are not shown. Previously reported and novel (in red) variants linked with inherited retinal degeneration [9,10].
2.3. Treatment and Follow-Up
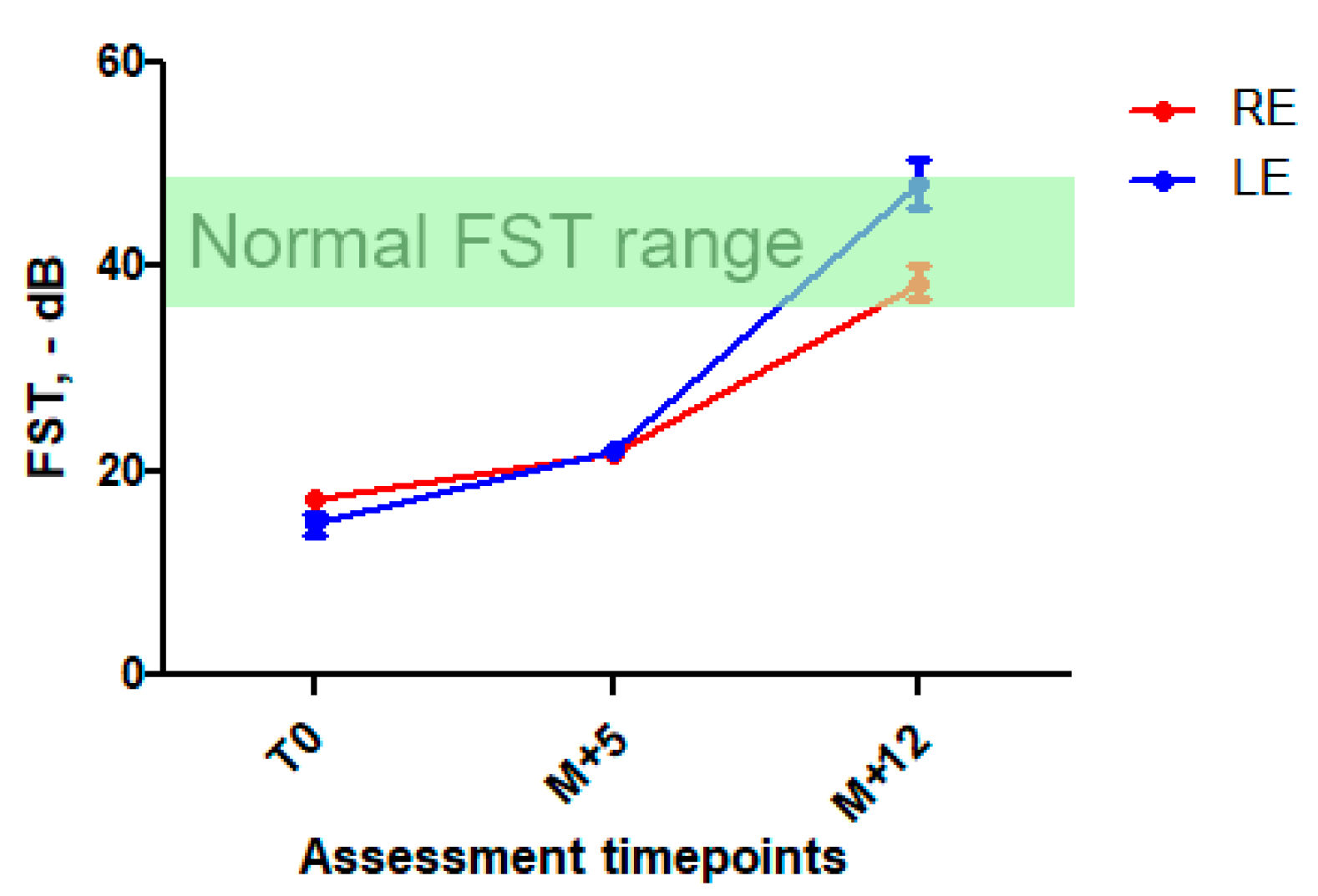
After discussion with the patient and his parents and their informed consent, high-dose oral vitamin A was initiated with retinol palmitate, 50,000 UI twice a week. Liver function was monitored. Five months after initiation of the treatment, the patient reported a significant subjective night vision improvement and a better tolerance of light-to-dark transitions. There was an improvement in FST at this time point (+25% in RE and 40% in LE, respectively) sustained at 1-year follow-up (Figure 3 and Figure S3 in Supplementary Data), while visual acuity, ERG, and retinal imaging (more specifically, white dots, ONL and EZ aspects) remained unchanged. We also observed a marked visual field improvement: III1e and II1e targets became perceived by the patient (Figure S1 in Supplementary Data). There were no signs of general retinol toxicity and liver function remained normal.
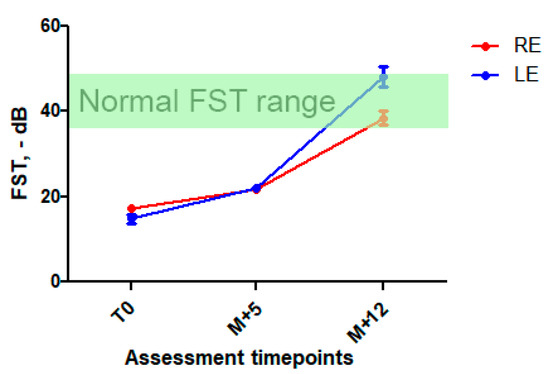
Figure 3.
Full stimulus threshold before and after high-dose vitamin A intake. Mean FST ± SD at first assessment, at M + 5 and M + 12 follow-up. A significant improvement in FST was observed at M + 5 (25 and 40% decrease) and was sustained at M + 12 (123 et 215% in RE and LE, respectively). In green, normal FST range [11,12].
3. Discussion
The visual system is highly dependent upon retinol supply. Retinoids are essential for normal ocular development as a whole and for the maintenance of a normal tissue structure. Abnormalities in nutritional supply, storage, transport, delivery, or metabolism of retinoids are linked with a broad spectrum of developmental and degenerative diseases of the eye [13].
RBP4 is the major plasma carrier of retinol and retinoids. It forms a heterohexameric complex with transthyretin in the blood, a characteristic which protects small molecules of RBP4 (21k Da) from glomerular filtration [14]. There are two different pathways for retinoids to reach their target tissues. The main pathway depends upon RBP4, which is complexed to retinol (holo-RBP4) and then recognized and bond to a membrane retinoid receptor STRA6, located at the basal side of the RPE. STRA6 allows the internalization of retinol [15]. The minor pathway is under the control of the scavenger class B type I receptor (Sr-BI) which enables the absorption of the protein-free fraction of retinoids transported within circulating lipoprotein complexes and chylomicrons. In the absence of RBP4, this second low-rate route could be sufficient to provide retinoids to all other tissues except for the RPE, leading to a retinal disease [16,17,18].
Pathogenic variants in RBP4 are responsible for rod–cone dystrophy associated with various degrees of microphthalmia, coloboma, and comedogenic acne. Only few patients harboring RBP4 defects have been reported to date [9,10,19,20] (Figure 2C). Our patient presents a mild phenotype unlike patients harboring RBP4 gene defects reported previously which could be explained in part by his young age (12 years) compared to the late adult cases with advanced retinal degeneration reported in the literature. The observed white dot retinopathy in our patient could be an early feature of progressive inherited retinal degeneration.
In contrast, our patient had a very unusual phenotype for RBP4 deficiency. His ocular findings were reminiscent of fundus albipunctatus (FA, OMIM#136880) due to the presence of night blindness, numerous peripheral white dots, and no signs of retinal degeneration. FA clinical phenotype is typically associated with pathogenic variants in RDH5 (OMIM# 601617). However, retinal white dots in our patient were less numerous and clearly not organized in a network pattern as in RDH5-retinopathy. The ERG was also different showing a generalized severe rod–cone dysfunction distinct from the Riggs-type ERG [21] classically associated with RDH5-retinopathy including normal cone function and cone-dominated dark-adapted responses. Retinol-dehydrogenase 5 is a visual cycle enzyme oxidizing 11-cis-retinol into 11-cis-retinal. The lack of enzyme activity leads to 11-cis and 13-cis retinyl esters accumulation which are thought to be the origin of the white dots in FA [22,23].
Another retinal disease very close to our patient’s presentation is retinitis punctata albescens (RPA, OMIM#136880 or Bothnia dystrophy, OMIM#607475). White dots are usually present in the early stages of the disease and are due to an accumulation of all-trans-retinyl esters in the retinal pigment epithelium secondary to an impairment of the cellular retinaldehyde–binding protein 1 (CRABP1, Uniprot#P29762), slowing down the isomerization of all-trans-retinyl esters in 11-cis-retinol. Pathogenic variants in RLBP1, encoding CRABP1, are responsible for this phenotype. As its name implies, RPA is a progressive retinal degeneration. Unlike in our patient, RPA is characterized by moderate narrowing of the retinal vasculature, optic disc pallor, pigmentary changes, and peripheral scalloped areas of chorioretinal atrophy [24,25]. ERG responses in RPA are, however, closer to those of our patient, with severely affected rod responses and more preserved cone responses. A limited recovery of ERG responses after prolonged dark adaptation is also reported in RPA [26] as in RDH5-retinopathy. This was unfortunately not tested in our patient. White dots have also been described in RHO (OMIM#180380) [27], PRPH2 (OMIM#179605) [28], LRAT (OMIM#604863) [29,30], and RPE65 (OMIM#180069) [31,32] gene defects, but the phenotype in these cases is more severe and progressive; thus, delineating it from our patient’s clinical picture.
Functional and morphological retinal changes in our patient were close to those reported in vitamin A deficient retinopathy (VAD). Functionally, generalized photoreceptor dysfunction with rod responses being more altered than cone responses is also characteristic for VAD [33,34]. White retinal dots can be a feature in VAD [33,34], but they are somewhat different in shape (indistinct borders) and hypoautofluorescent on SWAF [35]. Our case also presented intriguing finding with an additional hyperreflective band in the parafoveal region on OCT (Figure 2F). We hypothesize that this alteration could be a partial duplication of the outer plexiform layer and may be related to the important role of retinoids in retinal development [36].
Another inherited form of retinal degeneration linked with an altered absorption and biodistribution of vitamin A is abetalipoproteinemia (or Bassen–Kornzweig syndrome). Biallelic gene defects in MTTP (OMIM#157147), encoding Mitochondrial Triglyceride Transfer Protein (Uniprot#P55157) lead to impaired assembly and secretion of plasma lipoproteins that contain apolipoprotein B (very low- and low-density lipoproteins and chylomicrons). Lipoproteins facilitate absorption and carry a free fraction of fat-soluble vitamins (A, D, E, K). Upon reduced MTTP activity, there is a reduced retinol and tocopherol absorption, transport, and delivery to the target organs, including the eye, which results in retinal degeneration. Early treatment with high-dose vitamins A and E in patients with abetalipoproteinemia resulted in improvement in their retinal function and a slower retinal degeneration progression rate [3,37,38].
Oral administration of high-dose retinol palmitate has already shown to raise the level of free plasma retinol and retinyl esters in RBP4-deficient patients [39] but no visual outcome had been reported so far. Nevertheless, experimental studies report that RBP-/- mice are able to use alternative RBP-independent pathways for retinol supply to the retina with phenotypic rescue provided by a retinol-sufficient diet [17]. In order to compensate for the lack of RBP4-related retinol transport and attempt to enhance the free fraction of retinoids delivered via the slow RBP4-independent pathway, we decided to prescribe high doses of retinol to our patient. After five months of high-dose oral vitamin A, the patient reported subjective improvement in dimly lighted environments which was supported by FST and kinetic perimetry changes from baseline. This effect further improved after one year follow-up.
Adverse and toxic effects of long-term high retinol intake are numerous including bone toxicity (hypercalcemia and osteoporosis), neurotoxicity (intracranial hypertension), and liver toxicity (liver enlargement, cirrhosis) [40]. Saturation of the RBP4-independent pathway of retinol delivery could also be detrimental for xanthophyll pigment uptake by the retina, as this slow pathway is competitive between retinol and lutein/zeaxanthin transport [41]. However, toxicity did not occur in patients with abetalipoproteinemia on prolonged high-dose retinol treatment, as the overall blood levels remained low [3]. Our patient did not experience any adverse effect after 1 year of retinol intake. Liver enzymes and blood calcium were normal. Long-term follow-up with close monitoring for vitamin A tolerability will determine whether high doses of retinol are able to prevent retinal degeneration.
4. Materials and Methods
4.1. Clinical Studies
The patient was clinically investigated at the national reference center for rare ocular diseases REFERET of the Centre Hospitalier National d’Ophtalmologie des Quinze-Vingts as previously described [42]. Briefly, best-corrected visual acuity (BCVA), refractive error, slit-lamp biomicroscopy, static and kinetic visual fields, full-field electroretinogram according to the standards of International Society for Clinical Electrophysiology of Vision [43] (Espion, Diagnosys LLC, Lowell, MA), fundus photography, spectral domain optical coherence tomography (Spectralis OCT, Heidelberg Engineering, Inc., Heidelberg, Germany), infrared and short-wavelength autofluorescence (Heidelberg Retinal Tomograph, Heidelberg Engineering, Inc.) were performed. Full stimulus threshold was assessed using achromatic full-field stimuli (Diagnosys Espion system, Diagnosys LLC, Lowell, MA, USA) as previously described [11,12].
4.2. Genetic Analysis
Blood samples from the index case and from his parents were collected for genetic research and genomic DNA was extracted as previously reported [44]. These DNA samples were stored and obtained from the NeuroSensCol DNA bank, for research in neuroscience (PI: JA Sahel, co-PI I Audo, partner with CHNO des Quinze-Vingts, Inserm and CNRS, certified NFS96-900). Targeted next generation sequencing (NGS) was performed in collaboration with an external company (IntegraGen, Evry, France) [45]. The RBP4 (MIM#180250) variant selected after NGS was validated in the index case and relatives by Sanger sequencing (refseq: NM_001323517.1, primer sequences and conditions available on demand).
4.3. Western Blot
Western blot identification of RBP4 was performed as described in a previously published protocol [46]. Loading control was performed using anti-transferrin monoclonal antibody (Abcam, Discovery Drive, Cambridge Biomedical Campus, Cambridge, UK, ab109503).
5. Conclusions
We report here the first case of RBP4-related retinopathy manifesting as a white-dot retinopathy whose visual function improved after high-dose oral vitamin A intake. Such treatment should be considered in early stages of RBP4-related retinopathy. Future research will determine whether high level of vitamin A intake is able to reverse the phenotype sustainably.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/ijms23126590/s1.
Author Contributions
Conceptualization, C.Z., J.-A.S. and I.A.; methodology, B.W., M.N., C.C., A.A., C.A., C.D. and S.S.; validation, C.Z., J.-A.S., I.A., B.W., M.N., C.C., A.A., C.A., C.D. and S.S.; formal analysis, V.M.S., C.Z., J.-A.S., I.A., B.W., M.N., C.C., A.A., C.A., C.D. and S.S.; investigation, V.M.S., C.Z., I.A., B.W., M.N., C.C., A.A., C.A., C.D. and S.S.; resources, C.Z., J.-A.S. and I.A.; data curation, V.M.S., C.Z., J.-A.S., I.A., B.W., M.N., C.C., A.A., C.A., C.D. and S.S.; writing—original draft preparation, V.M.S., writing—review and editing, V.M.S., C.Z. and I.A.; supervision, C.Z. and I.A.; funding acquisition, C.Z., J.-A.S. and I.A. All authors have read and agreed to the published version of the manuscript.
Funding
LABEX LIFESENSES [reference ANR-10-LABX-65] supported by French state funds managed by the Agence Nationale de la Recherche within the Investissements d’Avenir program [ANR-11-IDEX-0004-0]; IHU FOReSIGHT [ANR-18-IAHU-0001] supported by French state funds managed by the Agence Nationale de la Recherche within the Investissements d’Avenir program; funding from RHU-Light4deaf [ANR-15-RHU-0001]; Foundation Fighting Blindness center grant [C-CMM-0907-0428-INSERM04], BR-GE-0619-0761-INSERM and fellowship award (MN) [CD-CL-0619-0759-INSERM]. Retina France, U UNADEV (Union Nationale des Aveugles et Déficients Visuels) in partnership with ITMO NNP/AVIESAN (alliance nationale pour les sciences de la vie et de la santé) for research on visual disorders.
Institutional Review Board Statement
Research procedures adhered to the tenets of the Declaration of Helsinki and were approved by the local Ethics Committee (CPP, Ile de France V, Project number 06693, N◦EUDRACT 2006-A00347-44, 11 December 2006).
Informed Consent Statement
Prior to testing, written informed consent was obtained from the parents and participant who was under 18. No compensation or incentive was offered to the subject to participate in the study.
Data Availability Statement
All data are contained within the article or Supplementary Materials.
Acknowledgments
The authors thank the family in the study and the clinical staff of National Rare Disease Center REFERET and INSERM-DGOS CIC 1423 for collecting phenotypic data. We are thankful to Robert Duvoisin for his English revisions.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Grant, C.A.; Berson, E.L. Treatable Forms of Retinitis Pigmentosa Associated with Systemic Neurological Disorders. Int Ophthalmol. Clin. 2001, 41, 103–110. [Google Scholar] [CrossRef]
- Weleber, R.G.; Kurz, D.E.; Trzupek, K.M. Treatment of Retinal and Choroidal Degenerations and Dystrophies: Current Status and Prospects for Gene-Based Therapy. Ophthalmol. Clin. N. Am. 2003, 16, 583–593. [Google Scholar] [CrossRef]
- Chowers, I.; Banin, E.; Merin, S.; Cooper, M.; Granot, E. Long-Term Assessment of Combined Vitamin A and E Treatment for the Prevention of Retinal Degeneration in Abetalipoproteinaemia and Hypobetalipoproteinaemia Patients. Eye 2001, 15, 525–530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balfoort, B.M.; Buijs, M.J.N.; Ten Asbroek, A.L.M.A.; Bergen, A.A.B.; Boon, C.J.F.; Ferreira, E.A.; Houtkooper, R.H.; Wagenmakers, M.A.E.M.; Wanders, R.J.A.; Waterham, H.R.; et al. A Review of Treatment Modalities in Gyrate Atrophy of the Choroid and Retina (GACR). Mol. Genet. Metab. 2021, 134, 96–116. [Google Scholar] [CrossRef] [PubMed]
- Casalino, G.; Pierro, L.; Manitto, M.P.; Michaelides, M.; Bandello, F. Resolution of Cystoid Macular Edema Following Arginine-Restricted Diet and Vitamin B6 Supplementation in a Case of Gyrate Atrophy. J. AAPOS 2018, 22, 321–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rüether, K.; Baldwin, E.; Casteels, M.; Feher, M.D.; Horn, M.; Kuranoff, S.; Leroy, B.P.; Wanders, R.J.; Wierzbicki, A.S. Adult Refsum Disease: A Form of Tapetoretinal Dystrophy Accessible to Therapy. Surv. Ophthalmol. 2010, 55, 531–538. [Google Scholar] [CrossRef]
- Benson, M.D.; MacDonald, I.M.; Sheehan, M.; Jain, S. Improved Electroretinographic Responses Following Dietary Intervention in a Patient with Refsum Disease. JIMD Rep. 2020, 55, 32–37. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [Green Version]
- Seeliger, M.W.; Biesalski, H.K.; Wissinger, B.; Gollnick, H.; Gielen, S.; Frank, J.; Beck, S.; Zrenner, E. Phenotype in Retinol Deficiency Due to a Hereditary Defect in Retinol Binding Protein Synthesis. Investig. Ophthalmol. Vis. Sci. 1999, 40, 3–11. [Google Scholar]
- Cehajic-Kapetanovic, J.; Jasani, K.M.; Shanks, M.; Clouston, P.; MacLaren, R.E. A Novel Homozygous c.67C>T Variant in Retinol Binding Protein 4 (RBP4) Associated with Retinitis Pigmentosa and Childhood Acne Vulgaris. Ophthalmic. Genet. 2020, 41, 288–292. [Google Scholar] [CrossRef]
- Roman, A.J.; Cideciyan, A.V.; Aleman, T.S.; Jacobson, S.G. Full-Field Stimulus Testing (FST) to Quantify Visual Perception in Severely Blind Candidates for Treatment Trials. Physiol. Meas. 2007, 28, N51–N56. [Google Scholar] [CrossRef] [PubMed]
- Klein, M.; Birch, D.G. Psychophysical Assessment of Low Visual Function in Patients with Retinal Degenerative Diseases (RDDs) with the Diagnosys Full-Field Stimulus Threshold (D-FST). Doc. Ophthalmol. 2009, 119, 217–224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiser, P.D.; Palczewski, K. Retinoids and Retinal Diseases. Annu. Rev. Vis. Sci. 2016, 2, 197–234. [Google Scholar] [CrossRef] [Green Version]
- Monaco, H.L.; Rizzi, M.; Coda, A. Structure of a Complex of Two Plasma Proteins: Transthyretin and Retinol-Binding Protein. Science 1995, 268, 1039–1041. [Google Scholar] [CrossRef]
- Kawaguchi, R.; Yu, J.; Honda, J.; Hu, J.; Whitelegge, J.; Ping, P.; Wiita, P.; Bok, D.; Sun, H. A Membrane Receptor for Retinol Binding Protein Mediates Cellular Uptake of Vitamin A. Science 2007, 315, 820–825. [Google Scholar] [CrossRef] [PubMed]
- Quadro, L.; Hamberger, L.; Colantuoni, V.; Gottesman, M.E.; Blaner, W.S. Understanding the Physiological Role of Retinol-Binding Protein in Vitamin A Metabolism Using Transgenic and Knockout Mouse Models. Mol. Asp. Med. 2003, 24, 421–430. [Google Scholar] [CrossRef]
- Quadro, L. Impaired Retinal Function and Vitamin A Availability in Mice Lacking Retinol-Binding Protein. EMBO J. 1999, 18, 4633–4644. [Google Scholar] [CrossRef] [PubMed]
- Du, M.; Phelps, E.; Balangue, M.J.; Dockins, A.; Moiseyev, G.; Shin, Y.; Kane, S.; Otalora, L.; Ma, J.-X.; Farjo, R.; et al. Transgenic Mice Over-Expressing RBP4 Have RBP4-Dependent and Light-Independent Retinal Degeneration. Investig. Ophthalmol. Vis. Sci. 2017, 58, 4375–4383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cukras, C.; Gaasterland, T.; Lee, P.; Gudiseva, H.V.; Chavali, V.R.M.; Pullakhandam, R.; Maranhao, B.; Edsall, L.; Soares, S.; Reddy, G.B.; et al. Exome Analysis Identified a Novel Mutation in the RBP4 Gene in a Consanguineous Pedigree with Retinal Dystrophy and Developmental Abnormalities. PLoS ONE 2012, 7, e50205. [Google Scholar] [CrossRef]
- Khan, K.N.; Carss, K.; Raymond, F.L.; Islam, F.; Nihr BioResource-Rare Diseases Consortium, null; Moore, A.T.; Michaelides, M.; Arno, G. Vitamin A Deficiency Due to Bi-Allelic Mutation of RBP4: There’s More to It than Meets the Eye. Ophthalmic. Genet. 2017, 38, 465–466. [Google Scholar] [CrossRef]
- Riggs, L.A. Electroretinography in Cases of Night Blindness. Am. J. Ophthalmol. 1954, 38, 70–78. [Google Scholar] [CrossRef]
- Driessen, C.A.; Winkens, H.J.; Hoffmann, K.; Kuhlmann, L.D.; Janssen, B.P.; Van Vugt, A.H.; Van Hooser, J.P.; Wieringa, B.E.; Deutman, A.F.; Palczewski, K.; et al. Disruption of the 11-Cis-Retinol Dehydrogenase Gene Leads to Accumulation of Cis-Retinols and Cis-Retinyl Esters. Mol. Cell. Biol. 2000, 20, 4275–4287. [Google Scholar] [CrossRef] [Green Version]
- Jang, G.F.; Van Hooser, J.P.; Kuksa, V.; McBee, J.K.; He, Y.G.; Janssen, J.J.; Driessen, C.A.; Palczewski, K. Characterization of a Dehydrogenase Activity Responsible for Oxidation of 11-Cis-Retinol in the Retinal Pigment Epithelium of Mice with a Disrupted RDH5 Gene. A Model for the Human Hereditary Disease Fundus Albipunctatus. J. Biol. Chem. 2001, 276, 32456–32465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burstedt, M.; Jonsson, F.; Köhn, L.; Burstedt, M.; Kivitalo, M.; Golovleva, I. Genotype-Phenotype Correlations in Bothnia Dystrophy Caused by RLBP1 Gene Sequence Variations. Acta Ophthalmol. 2013, 91, 437–444. [Google Scholar] [CrossRef] [PubMed]
- Dessalces, E.; Bocquet, B.; Bourien, J.; Zanlonghi, X.; Verdet, R.; Meunier, I.; Hamel, C.P. Early-Onset Foveal Involvement in Retinitis Punctata Albescens With Mutations in RLBP1. JAMA Ophthalmol. 2013, 131, 1314. [Google Scholar] [CrossRef] [Green Version]
- Burstedt, M.S.I.; Sandgren, O.; Golovleva, I.; Wachtmeister, L. Effects of Prolonged Dark Adaptation in Patients with Retinitis Pigmentosa of Bothnia Type: An Electrophysiological Study. Doc. Ophthalmol. 2008, 116, 193–205. [Google Scholar] [CrossRef]
- Souied, E.; Soubrane, G.; Benlian, P.; Coscas, G.J.; Gerber, S.; Munnich, A.; Kaplan, J. Retinitis Punctata Albescens Associated With the Arg135Trp Mutation in the Rhodopsin Gene. Am. J. Ophthalmol. 1996, 121, 19–25. [Google Scholar] [CrossRef]
- Kajiwara, K.; Sandberg, M.A.; Berson, E.L.; Dryja, T.P. A Null Mutation in the Human Peripherin/RDS Gene in a Family with Autosomal Dominant Retinitis Punctata Albescens. Nat. Genet. 1993, 3, 208–212. [Google Scholar] [CrossRef]
- Littink, K.W.; van Genderen, M.M.; van Schooneveld, M.J.; Visser, L.; Riemslag, F.C.C.; Keunen, J.E.E.; Bakker, B.; Zonneveld, M.N.; den Hollander, A.I.; Cremers, F.P.M.; et al. A Homozygous Frameshift Mutation in LRAT Causes Retinitis Punctata Albescens. Ophthalmology 2012, 119, 1899–1906. [Google Scholar] [CrossRef]
- Talib, M.; van Schooneveld, M.J.; van Duuren, R.J.G.; Van Cauwenbergh, C.; Ten Brink, J.B.; De Baere, E.; Florijn, R.J.; Schalij-Delfos, N.E.; Leroy, B.P.; Bergen, A.A.; et al. Long-Term Follow-Up of Retinal Degenerations Associated With LRAT Mutations and Their Comparability to Phenotypes Associated With RPE65 Mutations. Transl. Vis. Sci. Technol. 2019, 8, 24. [Google Scholar] [CrossRef] [Green Version]
- Schatz, P.; Preising, M.; Lorenz, B.; Sander, B.; Larsen, M.; Rosenberg, T. Fundus Albipunctatus Associated with Compound Heterozygous Mutations in RPE65. Ophthalmology 2011, 118, 888–894. [Google Scholar] [CrossRef] [Green Version]
- Ramtohul, P.; Denis, D. RPE65-Mutation Associated Fundus Albipunctatus with Cone Dystrophy. Ophthalmol. Retina 2019, 3, 535. [Google Scholar] [CrossRef]
- Apushkin, M.A.; Fishman, G.A. Improvement in Visual Function and Fundus Findings for a Patient with Vitamin A-Deficient Retinopathy. Retina 2005, 25, 650–652. [Google Scholar] [CrossRef] [PubMed]
- Genead, M.A.; Fishman, G.A.; Lindeman, M. Fundus White Spots and Acquired Night Blindness Due to Vitamin A Deficiency. Doc. Ophthalmol. 2009, 119, 229–233. [Google Scholar] [CrossRef] [PubMed]
- Aleman, T.S.; Garrity, S.T.; Brucker, A.J. Retinal Structure in Vitamin A Deficiency as Explored with Multimodal Imaging. Doc. Ophthalmol. 2013, 127, 239–243. [Google Scholar] [CrossRef]
- Cvekl, A.; Wang, W.-L. Retinoic Acid Signaling in Mammalian Eye Development. Exp. Eye Res. 2009, 89, 280–291. [Google Scholar] [CrossRef] [Green Version]
- MacGilchrist, A.J.; Mills, P.R.; Noble, M.; Foulds, W.S.; Simpson, J.A.; Watkinson, G. Abetalipoproteinaemia in Adults: Role of Vitamin Therapy. J. Inherit. Metab. Dis. 1988, 11, 184–190. [Google Scholar] [CrossRef]
- Bishara, S.; Merin, S.; Cooper, M.; Azizi, E.; Delpre, G.; Deckelbaum, R.J. Combined Vitamin A and E Therapy Prevents Retinal Electrophysiological Deterioration in Abetalipoproteinaemia. Br. J. Ophthalmol. 1982, 66, 767–770. [Google Scholar] [CrossRef] [PubMed]
- Biesalski, H.K.; Frank, J.; Beck, S.C.; Heinrich, F.; Illek, B.; Reifen, R.; Gollnick, H.; Seeliger, M.W.; Wissinger, B.; Zrenner, E. Biochemical but Not Clinical Vitamin A Deficiency Results from Mutations in the Gene for Retinol Binding Protein. Am. J. Clin. Nutr. 1999, 69, 931–936. [Google Scholar] [CrossRef]
- Hathcock, J.N.; Hattan, D.G.; Jenkins, M.Y.; McDonald, J.T.; Sundaresan, P.R.; Wilkening, V.L. Evaluation of Vitamin A Toxicity. Am. J. Clin. Nutr. 1990, 52, 183–202. [Google Scholar] [CrossRef]
- During, A.; Doraiswamy, S.; Harrison, E.H. Xanthophylls Are Preferentially Taken up Compared with β-Carotene by Retinal Cells via a SRBI-Dependent Mechanism. J. Lipid Res. 2008, 49, 1715–1724. [Google Scholar] [CrossRef] [Green Version]
- Audo, I. An Unusual Retinal Phenotype Associated With a Novel Mutation in RHO. Arch. Ophthalmol. 2010, 128, 1036. [Google Scholar] [CrossRef] [Green Version]
- McCulloch, D.L.; Marmor, M.F.; Brigell, M.G.; Hamilton, R.; Holder, G.E.; Tzekov, R.; Bach, M. ISCEV Standard for Full-Field Clinical Electroretinography (2015 Update). Doc. Ophthalmol. 2015, 130, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Audo, I.; Lancelot, M.; Mohand-Saïd, S.; Antonio, A.; Germain, A.; Sahel, J.; Bhattacharya, S.S.; Zeitz, C. Novel C2orf71 Mutations Account for ∼1% of Cases in a Large French ArRP Cohort. Hum. Mutat. 2011, 32, E2091–E2103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Audo, I.; Bujakowska, K.M.; Léveillard, T.; Mohand-Saïd, S.; Lancelot, M.-E.; Germain, A.; Antonio, A.; Michiels, C.; Saraiva, J.-P.; Letexier, M.; et al. Development and Application of a Next-Generation-Sequencing (NGS) Approach to Detect Known and Novel Gene Defects Underlying Retinal Diseases. Orphanet. J. Rare Dis. 2012, 7, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graham, T.E.; Wason, C.J.; Blüher, M.; Kahn, B.B. Shortcomings in Methodology Complicate Measurements of Serum Retinol Binding Protein (RBP4) in Insulin-Resistant Human Subjects. Diabetologia 2007, 50, 814–823. [Google Scholar] [CrossRef] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).