
In Vitro Selection and Characterization of HIV-1 Variants with Increased Resistance to LP-40, Enfuvirtide-Based Lipopeptide Inhibitor


Abstract
:1. Introduction
2. Results
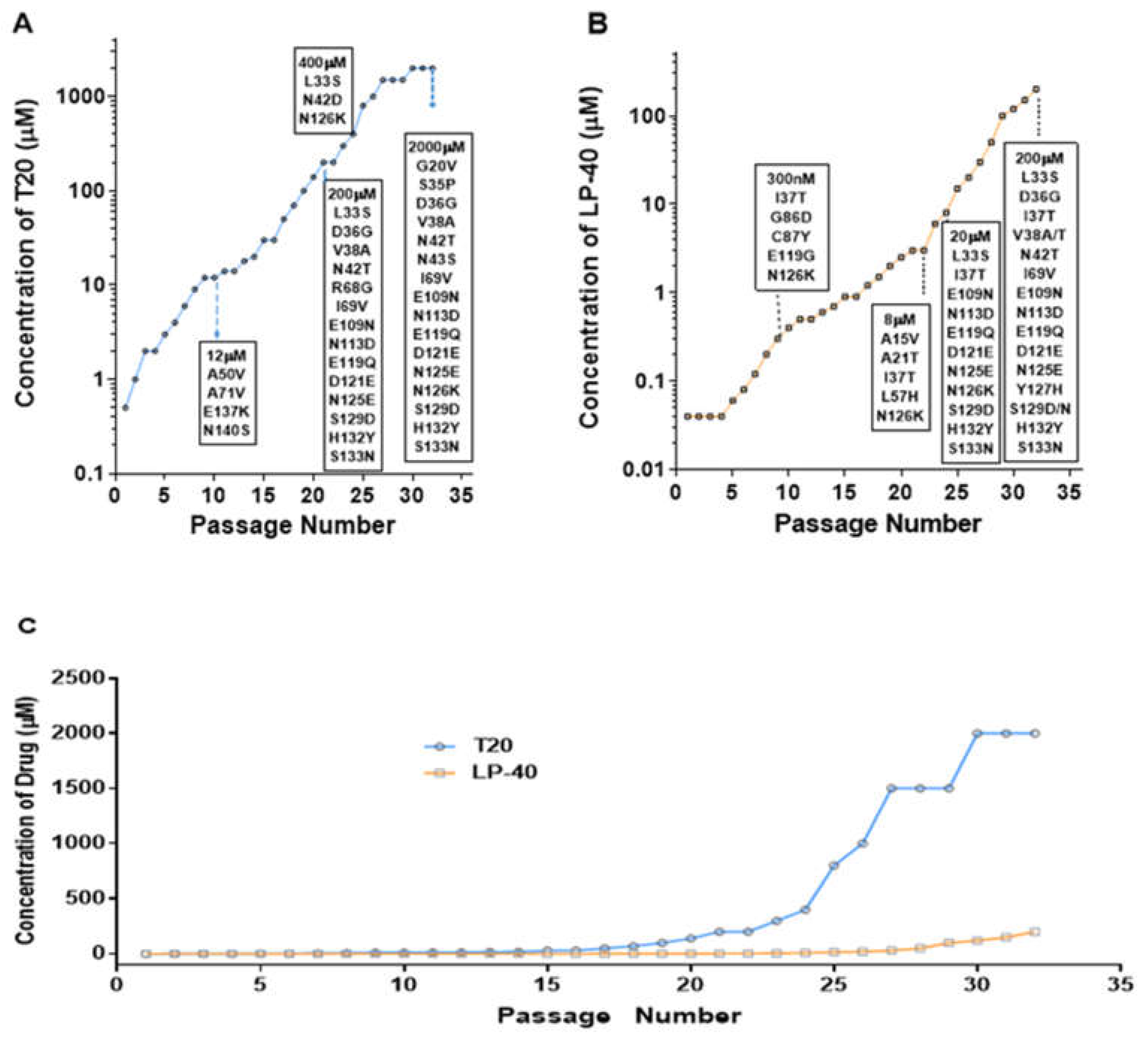
2.1. Selection of LP-40-Resistant HIV-1 In Vitro
2.2. Selection of T20-Resistant HIV-1 In Vitro
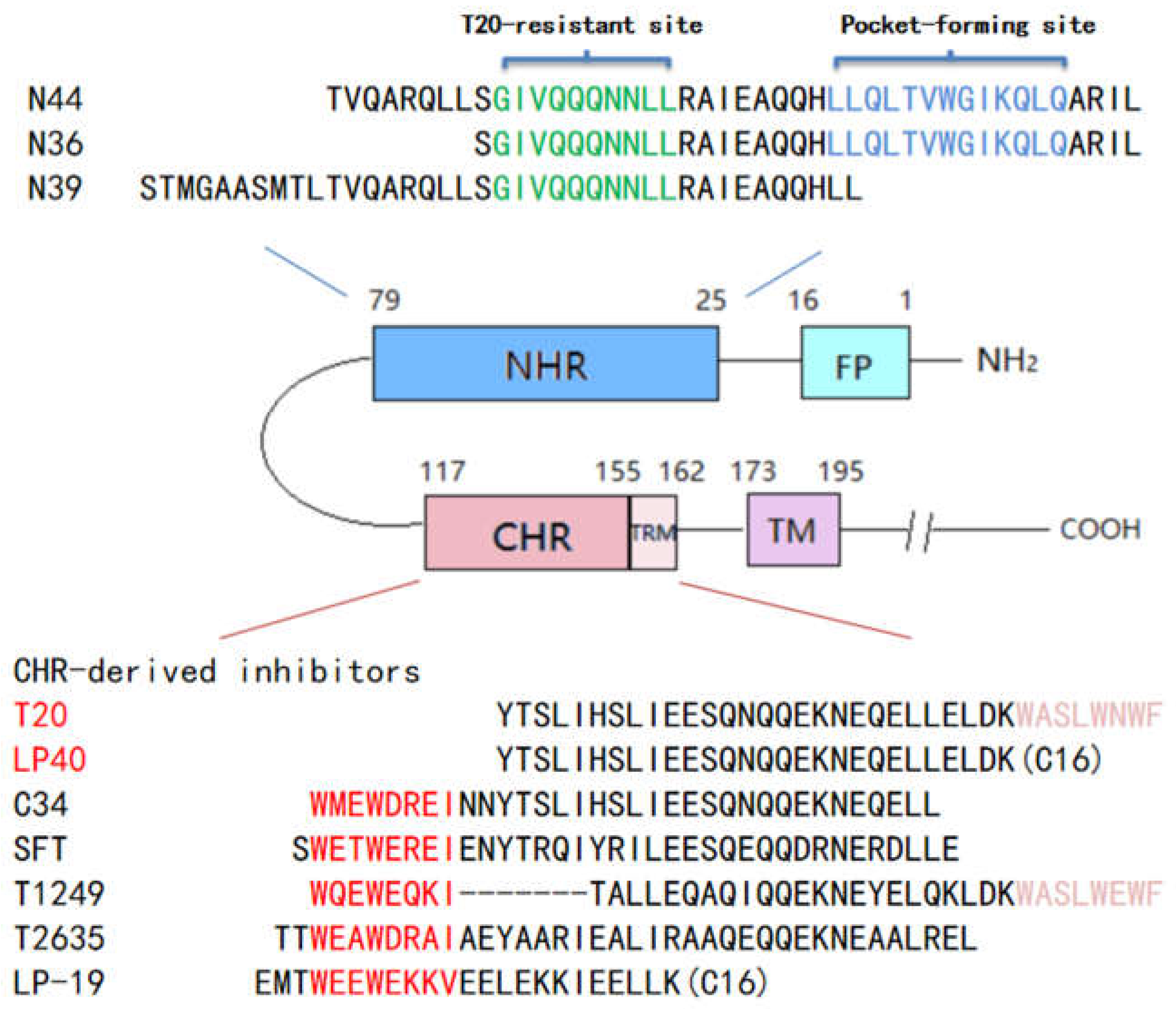
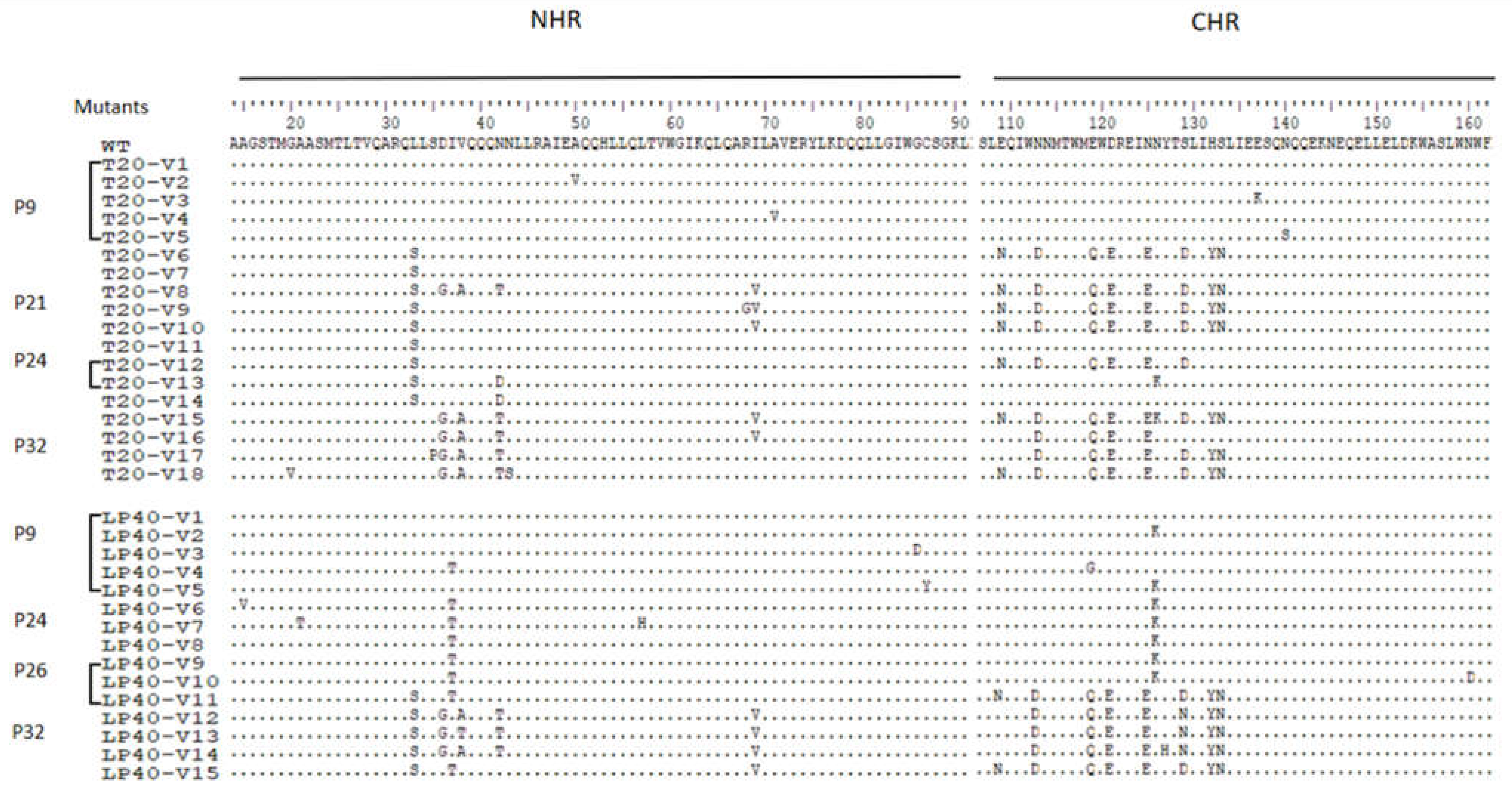
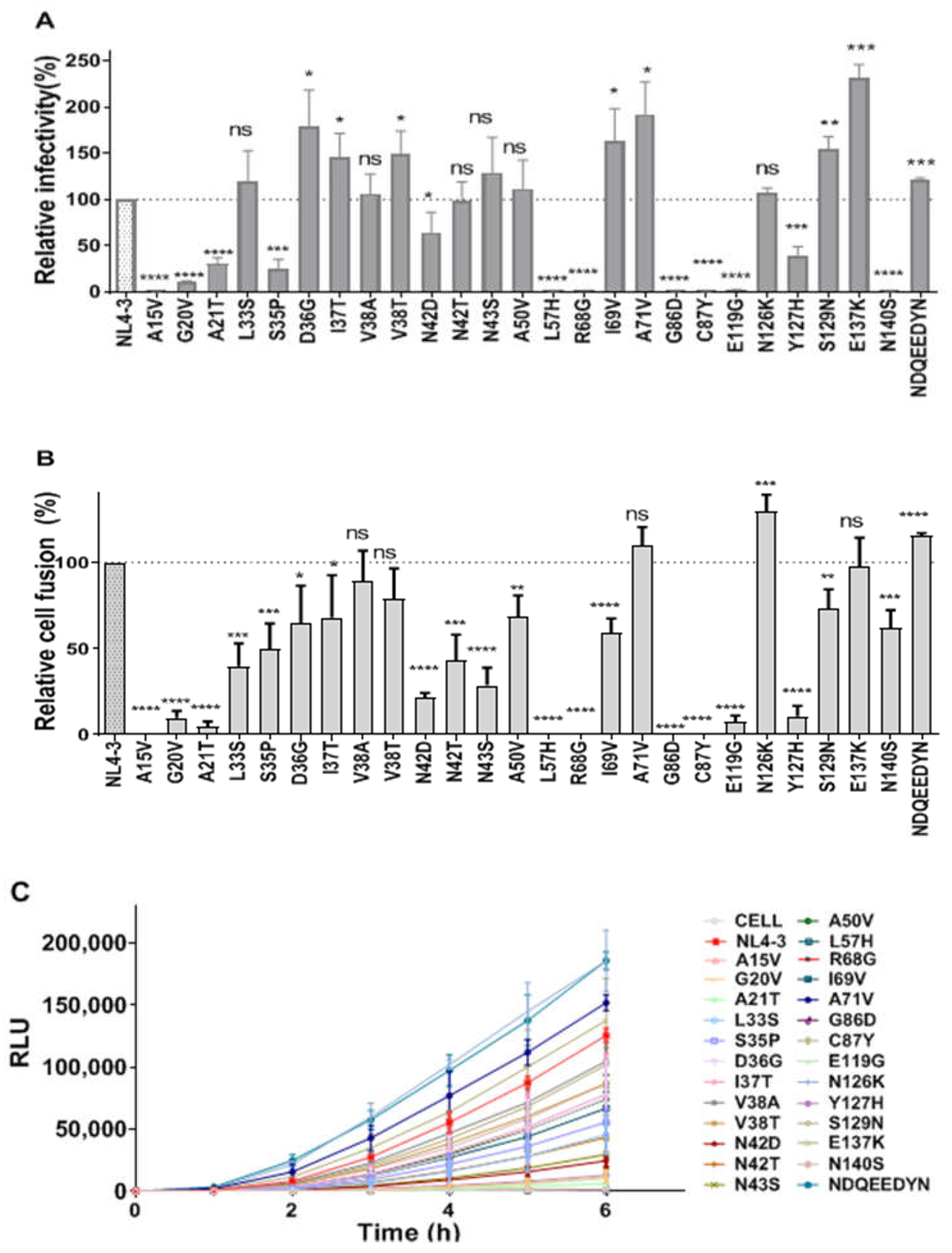
2.3. Construction of a Series of Mutant Pseudoviruses to Simulate Viruses under Drug Pressure, and Analysis of the Effect of a Series of Mutations on the Function of Viral Envelope Proteins
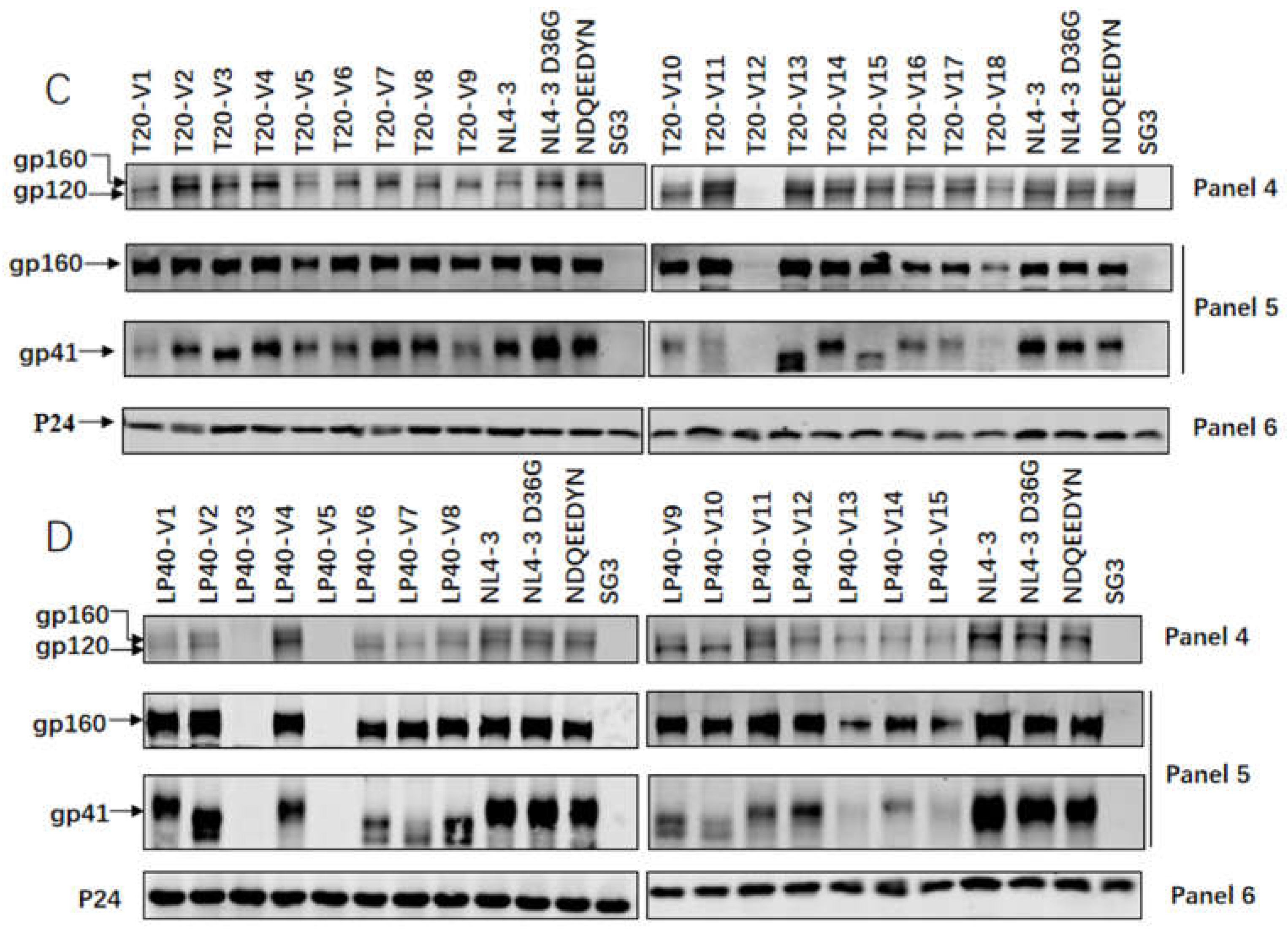
2.4. Expression and Processing of a Series of Mutations in the Env Glycoprotein
2.5. Characteristics of Resistance of Serial Mutations to T20 and LP-40
2.6. Effects of Single-Point Mutations on Viral Entry and Fusion
2.7. Cross-Resistance to T20 and LP40 of Single-Point Mutations on Pseudoviruses
2.8. Identification of the Sensitivity of LP-40-Induced Resistance Mutations to Serial Membrane Fusion Inhibitors
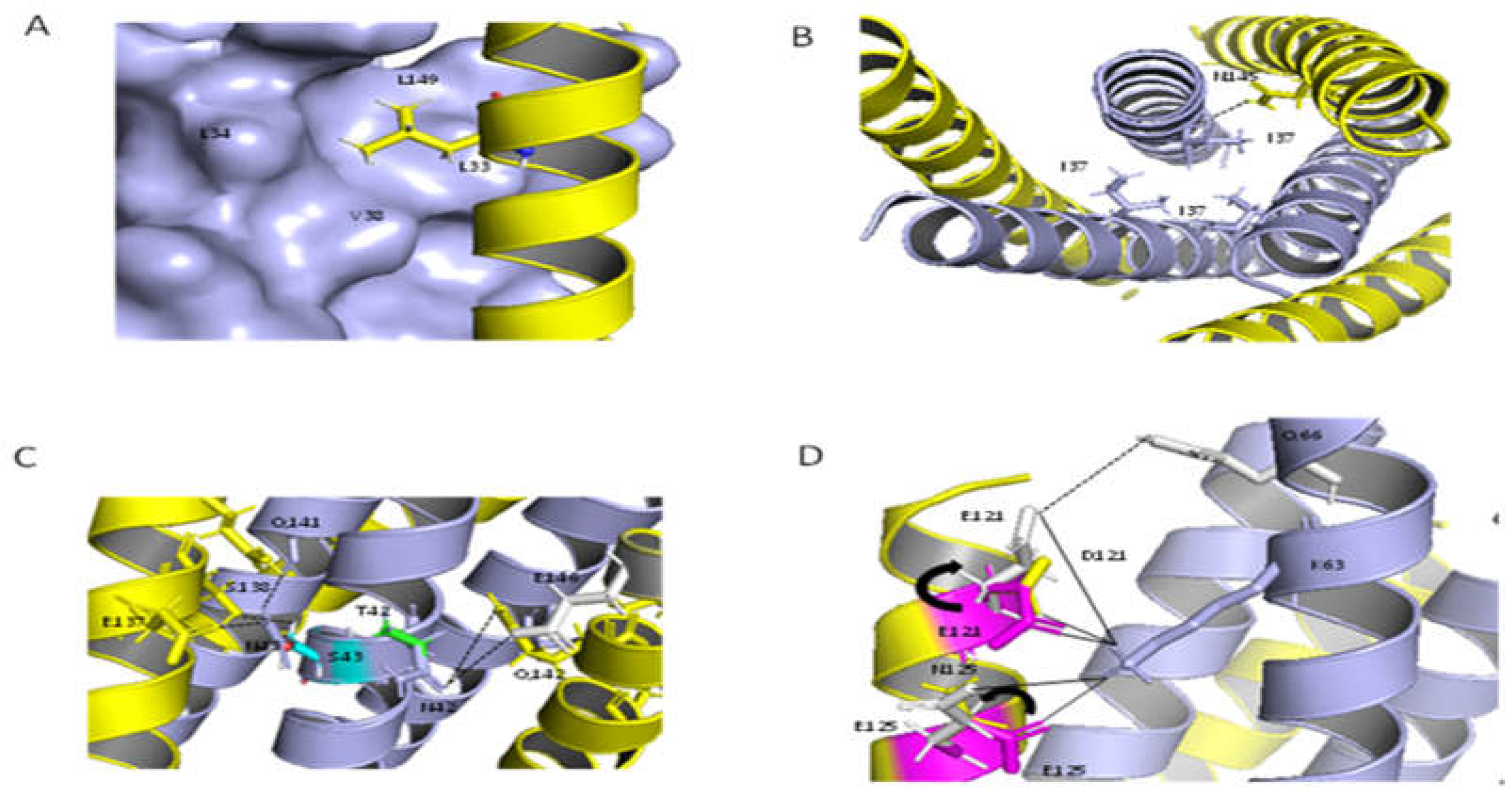
2.9. Structural Analysis of the Resistant Sites
3. Discussion
4. Materials and Methods
4.1. Peptide Synthesis
4.2. Selection of T20- or LP40-Resistant Viruses
4.3. Mutation Determination and Site-Directed Mutagenesis
4.4. Single-Cycle Infection Assay
4.5. DSP-Based Cell–Cell Fusion Assay
4.6. Western Blotting
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Root, M.J.; Kay, M.S.; Kim, P.S. Protein design of an HIV-1 entry inhibitor. Science 2001, 291, 884–888. [Google Scholar] [CrossRef] [Green Version]
- Weiss, R.A. How Does Hiv Cause Aids. Science 1993, 260, 1273–1279. [Google Scholar] [CrossRef]
- Tan, K.M.; Liu, J.-H.; Wang, J.-H.; Shen, S.; Lu, M. Atomic structure of a thermostable subdomain of HIV-1 gp41. Proc. Natl. Acad. Sci. USA 1997, 94, 12303–12308. [Google Scholar] [CrossRef] [Green Version]
- Lu, M.; Kim, P.S. A trimeric structural subdomain of the HIV-1 transmembrane glycoprotein. J. Biomol. Struct. Dyn. 1997, 15, 465–471. [Google Scholar] [CrossRef]
- Chan, D.C.; Fass, D.; Berger, J.M.; Kim, P.S. Core structure of gp41 from the HIV envelope glycoprotein. Cell 1997, 89, 263–273. [Google Scholar] [CrossRef] [Green Version]
- Munch, J.; Ständker, L.; Adermann, K.; Schulz, A.; Schindler, M.; Chinnadurai, R.; Kirchhoff, F. Discovery and optimization of a natural HIV-1 entry inhibitor targeting the gp41 fusion peptide. Cell 2007, 129, 263–275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eggink, D.; Bontjer, I.; De Taeye, S.W.; Langedijk, J.P.; Berkhout, B.; Sanders, R.W. HIV-1 anchor inhibitors and membrane fusion inhibitors target distinct but overlapping steps in virus entry. J. Biol. Chem. 2019, 294, 5736–5746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bewley, C.A.; Louis, J.M.; Ghirlando, R.; Clore, G.M. Design of a novel peptide inhibitor of HIV fusion that disrupts the internal trimeric coiled-coil of gp41. J. Biol. Chem. 2002, 277, 14238–14245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wild, C.T.; Shugars, D.C.; Greenwell, T.K.; McDanal, C.B.; Matthews, T.J. Peptides Corresponding to a Predictive Alpha-Helical Domain of Human-Immunodeficiency-Virus Type-1 Gp41 Are Potent Inhibitors of Virus-Infection. Proc. Natl. Acad. Sci. USA 1994, 91, 9770–9774. [Google Scholar] [CrossRef] [Green Version]
- Lalezari, J.P.; Eron, J.J.; Carlson, M.; Cohen, C.; DeJesus, E.; Arduino, R.C.; Kilby, J.M. A phase II clinical study of the long-term safety and antiviral activity of enfuvirtide-based antiretroviral therapy. AIDS 2003, 17, 691–698. [Google Scholar] [CrossRef]
- Rimsky, L.T.; Shugars, D.C.; Matthews, T.J. Determinants of human immunodeficiency virus type 1 resistance to gp41-derived inhibitory peptides. J. Virol. 1998, 72, 986–993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eron, J.J.; Gulick, R.M.; Bartlett, J.A.; Merigan, T.; Arduino, R.; Kilby, J.M.; Miralles, G.D. Short-term safety and antiretroviral activity of T-1249, a second-generation fusion inhibitor of HIV. J. Infect. Dis. 2004, 189, 1075–1083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chinnadurai, R.; Rajan, D.; Münch, J.; Kirchhoff, F. Human immunodeficiency virus type 1 variants resistant to first- and second-version fusion inhibitors and cytopathic in ex vivo human lymphoid tissue. J. Virol. 2007, 81, 6563–6572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gulick, R.M.; Mellors, J.W.; Havlir, D.; Eron, J.J.; Gonzalez, C.; McMahon, D.; Condra, J.H. Treatment with indinavir, zidovudine, and lamivudine in adults with human immunodeficiency virus infection and prior antiretroviral therapy. N. Engl. J. Med. 1997, 337, 734–739. [Google Scholar] [CrossRef] [Green Version]
- Ding, X.; Zhang, X.; Chong, H.; Zhu, Y.; Wei, H.; Wu, X.; He, J.; Wang, X.; He, Y. Enfuvirtide (T20)-Based Lipopeptide Is a Potent HIV-1 Cell Fusion Inhibitor: Implications for Viral Entry and Inhibition. J. Virol. 2017, 91, e00831-17. [Google Scholar] [CrossRef] [Green Version]
- Ingallinella, P.; Bianchi, E.; Ladwa, N.A.; Wang, Y.-J.; Hrin, R.; Veneziano, M.; Bonelli, F.; Ketas, T.J.; Moore, J.P.; Miller, M.D.; et al. Addition of a cholesterol group to an HIV-1 peptide fusion inhibitor dramatically increases its antiviral potency. Proc. Natl. Acad. Sci. USA 2009, 106, 5801–5806. [Google Scholar] [CrossRef] [Green Version]
- Wexler-Cohen, Y.; Ashkenazi, A.; Viard, M.; Blumenthal, R.; Shai, Y. Virus-cell and cell-cell fusion mediated by the HIV-1 envelope glycoprotein is inhibited by short gp41 N-terminal membrane-anchored peptides lacking the critical pocket domain. FASEB J. 2010, 24, 4196–4202. [Google Scholar] [CrossRef] [Green Version]
- Alam, S.M.; Paleos, C.A.; Liao, H.-X.; Scearce, R.; Robinson, J.; Haynes, B.F. An inducible HIV type 1 gp41 HR-2 peptide-binding site on HIV type 1 envelope gp120. AIDS Res. Hum. Retrovir. 2004, 20, 836–845. [Google Scholar] [CrossRef]
- Wu, X.Y.; Liu, Z.; Ding, X.; Yu, D.; Wei, H.; Qin, B.; Zhu, Y.; Chong, H.; Cui, S.; He, Y. Mechanism of HIV-1 Resistance to an Electronically Constrained alpha-Helical Peptide Membrane Fusion Inhibitor. J. Virol. 2018, 92, e02044-17. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Ding, X.; Zhu, Y.; Chong, H.; Cui, S.; He, J.; Wang, X.; He, Y. Structural and functional characterization of HIV-1 cell fusion inhibitor T20. AIDS 2019, 33, 1–11. [Google Scholar] [CrossRef]
- Yao, X.; Chong, H.; Zhang, C.; Waltersperger, S.; Wang, M.; Cui, S.; He, Y. Broad Antiviral Activity and Crystal Structure of HIV-1 Fusion Inhibitor Sifuvirtide. J. Biol. Chem. 2012, 287, 6788–6796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fung, H.B.; Guo, Y. Enfuvirtide: A fusion inhibitor for the treatment of HIV infection. Clin. Ther. 2004, 26, 352–378. [Google Scholar] [CrossRef]
- Greenberg, M.L.; Cammack, N. Resistance to enfuvirtide, the first HIV fusion inhibitor. J. Antimicrob. Chemother. 2004, 54, 333–340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perez Plasencia, A.; Martinez Sanchez, B. Entry inhibitors: Enfuvirtide, a reality for antiretroviral therapy. Farm. Hosp. 2004, 28, 106–115. [Google Scholar] [PubMed]
- Xu, L.; Pozniak, A.; Wildfire, A.; Stanfield-Oakley, S.A.; Mosier, S.M.; Ratcliffe, D.; Workman, J.; Joall, A.; Myers, R.; Smit, E.; et al. Emergence and evolution of enfuvirtide resistance following long-term therapy involves heptad repeat 2 mutations within gp41. Antimicrob. Agents Chemother. 2005, 49, 1113–1119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chong, H.; Xue, J.; Xiong, S.; Cong, Z.; Ding, X.; Zhu, Y.; He, Y. A Lipopeptide HIV-1/2 Fusion Inhibitor with Highly Potent In Vitro, Ex Vivo, and In Vivo Antiviral Activity. J. Virol. 2017, 91, e00288-17. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.H.; Shan, M.; Li, L.; Lu, L.; Meng, S.; Chen, C.; He, Y.; Jiang, S.; Zhang, L. In Vitro Selection and Characterization of HIV-1 Variants with Increased Resistance to Sifuvirtide, a Novel HIV-1 Fusion Inhibitor. J. Biol. Chem. 2011, 286, 3277–3287. [Google Scholar] [CrossRef] [Green Version]
- Chong, H.; Qiu, Z.; Sun, J.; Qiao, Y.; Li, X.; He, Y. Two M-T hook residues greatly improve the antiviral activity and resistance profile of the HIV-1 fusion inhibitor SC29EK. Retrovirology 2014, 11, 40. [Google Scholar] [CrossRef] [Green Version]
- Su, Y.; Chong, H.; Xiong, S.; Qiao, Y.; Qiu, Z.; He, Y. Genetic Pathway of HIV-1 Resistance to Novel Fusion Inhibitors Targeting the Gp41 Pocket. J. Virol. 2015, 89, 12467–12479. [Google Scholar] [CrossRef] [Green Version]
- Su, Y.; Chong, H.; Qiu, Z.; Xiong, S.; He, Y. Mechanism of HIV-1 Resistance to Short-Peptide Fusion Inhibitors Targeting the Gp41 Pocket. J. Virol. 2015, 89, 5801–5811. [Google Scholar] [CrossRef] [Green Version]
- Chong, H.; Yao, X.; Sun, J.; Qiu, Z.; Zhang, M.; Waltersperger, S.; Wang, M.; Cui, S.; He, Y. The M-T hook structure is critical for design of HIV-1 fusion inhibitors. J. Biol. Chem. 2012, 287, 34558–34568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, D.; Su, Y.; Ding, X.; Zhu, Y.; Qin, B.; Chong, H.; Cui, S.; He, Y. Structural and Functional Characterization of the Secondary Mutation N126K Selected by Various HIV-1 Fusion Inhibitors. Viruses 2020, 12, 326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Y.M.; Zhang, X.; Ding, X.; Chong, H.; Cui, S.; He, J.; Wang, X.; He, Y. Exceptional potency and structural basis of a T1249-derived lipopeptide fusion inhibitor against HIV-1, HIV-2, and simian immunodeficiency virus. J. Biol. Chem. 2018, 293, 5323–5334. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| T20 | LP-40 | |||
|---|---|---|---|---|
| Pseudovirus | IC50 (nM) | Fold | IC50 (nM) | Fold |
| NL4-3 | 55.01 ± 32.65 | 1.00 | 29.68 ± 4.52 | 1.00 |
| T20-V1 | 57.69 ± 7.23 | 1.05 | 40.07 ± 8.62 | 1.35 |
| T20-V2 | 146.70 ± 39.40 | 2.67 | 59.24 ± 15.90 | 2.00 |
| T20-V3 | 78.92 ± 12.10 | 1.43 | 70.51 ± 8.56 | 2.38 |
| T20-V4 | 53.13 ± 14.90 | 0.97 | 76.63 ± 19.66 | 2.58 |
| T20-V6 | 3853.00 ± 516.06 | 70.04 | 3513.22 ± 631.31 | 118.37 |
| T20-V7 | 2808.17 ± 457.53 | 51.05 | 2725.11 ± 211.71 | 91.82 |
| T20-V8 | 5275.34 ± 1100.34 | 95.90 | >6250 | >227.43 |
| T20-V10 | 3335.50 ± 432.90 | 60.63 | 6080.78 ± 1172.18 | 204.88 |
| T20-V13 | 4010.67 ± 570.83 | 72.91 | 3185.11 ± 206.85 | 107.32 |
| T20-V14 | 4429.42 ± 966.54 | 80.52 | 2895.78 ± 85.08 | 97.57 |
| T20-V15 | 2089.66 ± 247.94 | 37.99 | >6250 | >227.43 |
| T20-V16 | 1741.55 ± 229.44 | 31.66 | >6250 | >227.43 |
| T20-V17 | 2891.28 ± 881.88 | 52.56 | 5167.89 ± 1020.06 | 174.12 |
| T20-V18 | 6146.11 ± 475.36 | 111.73 | >6250 | >227.43 |
| LP40-V1 | 59.76 ± 16.33 | 1.09 | 44.85 ± 4.39 | 1.51 |
| LP40-V2 | 71.37 ± 4.32 | 1.30 | 373.61 ± 29.84 | 12.59 |
| LP40-V8 | 1151.16 ± 137.30 | 20.93 | 2883.89 ± 1070.02 | 97.17 |
| LP40-V9 | 1270.70 ± 314.74 | 23.10 | 2885.45 ± 430.88 | 97.22 |
| LP40-V10 | 1261.73 ± 264.08 | 22.94 | 2746.00 ± 634.40 | 92.52 |
| LP40-V11 | 4348.78 ± 521.53 | 79.05 | 3333.78 ± 977.77 | 112.32 |
| LP40-V12 | 5868.67 ± 978.46 | 106.68 | >6250 | >227.43 |
| LP40-V13 | 5167.09 ± 2461.48 | 93.93 | >6250 | >227.43 |
| LP40-V14 | 4911.25 ± 1996.92 | 89.28 | >6250 | >227.43 |
| T20 | LP-40 | |||
|---|---|---|---|---|
| Pseudovirus | IC50 (nM) | Fold | IC50 (nM) | Fold |
| NL4-3 | 67.34 ± 15.33 | 1.00 | 31.54 ± 10.42 | 1.00 |
| G20V | 49.12 ± 10.94 | 0.73 | 204.78 ± 47.22 | 6.49 |
| A21T | 58.85 ± 9.24 | 0.87 | 26.75 ± 8.36 | 0.85 |
| L33S | 3448.00 ± 564.06 | 51.20 | 2115.78 ± 533.73 | 67.08 |
| S35P | 137.56 ± 10.60 | 2.04 | 21.63 ± 5.15 | 0.69 |
| D36G | 6.00 ± 1.68 | 0.09 | 2.22 ± 0.57 | 0.07 |
| I37T | 710.62 ± 156.89 | 10.55 | 514.91 ± 74.31 | 16.33 |
| V38A | 1544.24 ± 21.96 | 22.93 | 3487.11 ± 423.38 | 110.56 |
| V38T | 991.11 ± 215.54 | 14.72 | 1451.90 ± 236.99 | 46.03 |
| N42D | 164.10 ± 26.82 | 2.44 | 109.08 ± 18.40 | 3.46 |
| N42T | 413.44 ± 81.98 | 6.14 | 1267.43 ± 167.03 | 40.18 |
| N43S | 540.06 ± 91.86 | 8.02 | 356.87 ± 117.38 | 11.31 |
| A50V | 162.15 ± 69.39 | 2.41 | 58.83 ± 13.68 | 1.87 |
| I69V | 71.39 ± 23.19 | 1.06 | 29.40 ± 4.59 | 0.93 |
| A71V | 46.50 ± 12.52 | 0.69 | 43.25 ± 8.19 | 1.37 |
| N126K | 116.81 ± 37.35 | 1.73 | 304.39 ± 50.53 | 9.65 |
| Y127H | 23.71 ± 2.63 | 0.35 | 5.74 ± 1.86 | 0.18 |
| S129N | 70.32 ± 19.83 | 1.04 | 34.03 ± 10.04 | 1.08 |
| E137K | 68.50 ± 20.14 | 1.02 | 42.64 ± 11.73 | 1.35 |
| T20 | LP40 | C34 | SFT | T1249 | T2635 | LP-19 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Pseudovirus | IC50 | Fold | IC50 | Fold | IC50 | Fold | IC50 | Fold | IC50 | Fold | IC50 | Fold | IC50 | Fold |
| NL4-3 | 71.88 | 1.00 | 27.32 | 1.00 | 0.80 | 1.00 | 1.56 | 1.00 | 1.14 | 1.00 | 0.73 | 1.00 | 0.0800 | 1.00 |
| I37T | 710.62 | 9.89 | 514.91 | 18.85 | 14.33 | 17.84 | 11.25 | 7.21 | 1.38 | 1.21 | 0.97 | 1.33 | 0.1298 | 1.62 |
| L33S | 3448.00 | 47.97 | 2115.78 | 77.43 | 5.55 | 6.90 | 4.67 | 3.00 | 13.01 | 11.41 | 1.43 | 1.97 | 0.1289 | 1.61 |
| I69V | 71.39 | 0.99 | 29.40 | 1.08 | 1.38 | 1.72 | 1.67 | 1.07 | 0.93 | 0.82 | 0.75 | 1.04 | 0.0800 | 1.00 |
| L33S/I37T | 5847.22 | 81.35 | 2979.00 | 109.04 | 110.54 | 137.60 | 22.97 | 14.72 | 81.48 | 73.47 | 1.51 | 2.07 | 0.1000 | 1.25 |
| V38A/N42T | 3348.55 | 46.59 | 4074.78 | 149.15 | 109.91 | 137.39 | 116.19 | 74.48 | 27.10 | 23.77 | 0.56 | 0.77 | 0.0530 | 0.66 |
| L33S/V38A/N42T | 4915.34 | 68.38 | 4990.44 | 182.67 | 912.42 | 1140.53 | 260.02 | 166.68 | 258.83 | 227.04 | 1.30 | 1.78 | 0.0444 | 0.56 |
| NDQEEDYN | 62.45 | 0.87 | 100.20 | 3.67 | 6.68 | 8.32 | 8.03 | 5.15 | 1.07 | 0.94 | 1.94 | 2.67 | 0.1345 | 1.68 |
| NDQEEDYN/L33S/I37T | 3794.89 | 52.80 | 3278.89 | 120.00 | 679.76 | 846.17 | 110.30 | 70.71 | 165.92 | 145.54 | 5.44 | 7.49 | 0.2433 | 3.04 |
| NDQEEDYN/L33S/V38A/N42T | 4781.45 | 66.52 | >6250 | >228.77 | 2497.78 | 3122.23 | 738.73 | 473.54 | 343.28 | 301.12 | 5.73 | 7.85 | 0.1180 | 1.48 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hu, Y.; Yu, W.; Geng, X.; Zhu, Y.; Chong, H.; He, Y. In Vitro Selection and Characterization of HIV-1 Variants with Increased Resistance to LP-40, Enfuvirtide-Based Lipopeptide Inhibitor. Int. J. Mol. Sci. 2022, 23, 6638. https://doi.org/10.3390/ijms23126638
Hu Y, Yu W, Geng X, Zhu Y, Chong H, He Y. In Vitro Selection and Characterization of HIV-1 Variants with Increased Resistance to LP-40, Enfuvirtide-Based Lipopeptide Inhibitor. International Journal of Molecular Sciences. 2022; 23(12):6638. https://doi.org/10.3390/ijms23126638
Chicago/Turabian StyleHu, Yue, Wenjiang Yu, Xiuzhu Geng, Yuanmei Zhu, Huihui Chong, and Yuxian He. 2022. "In Vitro Selection and Characterization of HIV-1 Variants with Increased Resistance to LP-40, Enfuvirtide-Based Lipopeptide Inhibitor" International Journal of Molecular Sciences 23, no. 12: 6638. https://doi.org/10.3390/ijms23126638