
Effects of miRNA-149-5p and Platelet-Activating Factor-Receptor Signaling on the Growth and Targeted Therapy Response on Lung Cancer Cells


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Functional PAFR Activation Augments the Proliferation of NSCLC Cell Lines
2.2. miR-149-5p Inhibits the Proliferation of A549 Cells in a Dose-Dependent Manner
2.3. miR-149-5p Overexpression Attenuates CPAF-Mediated Increased Proliferation of A549 Cells
2.4. Effects of PAFR Antagonist on miR-149-5p-Mediated Reduced Cell Proliferation
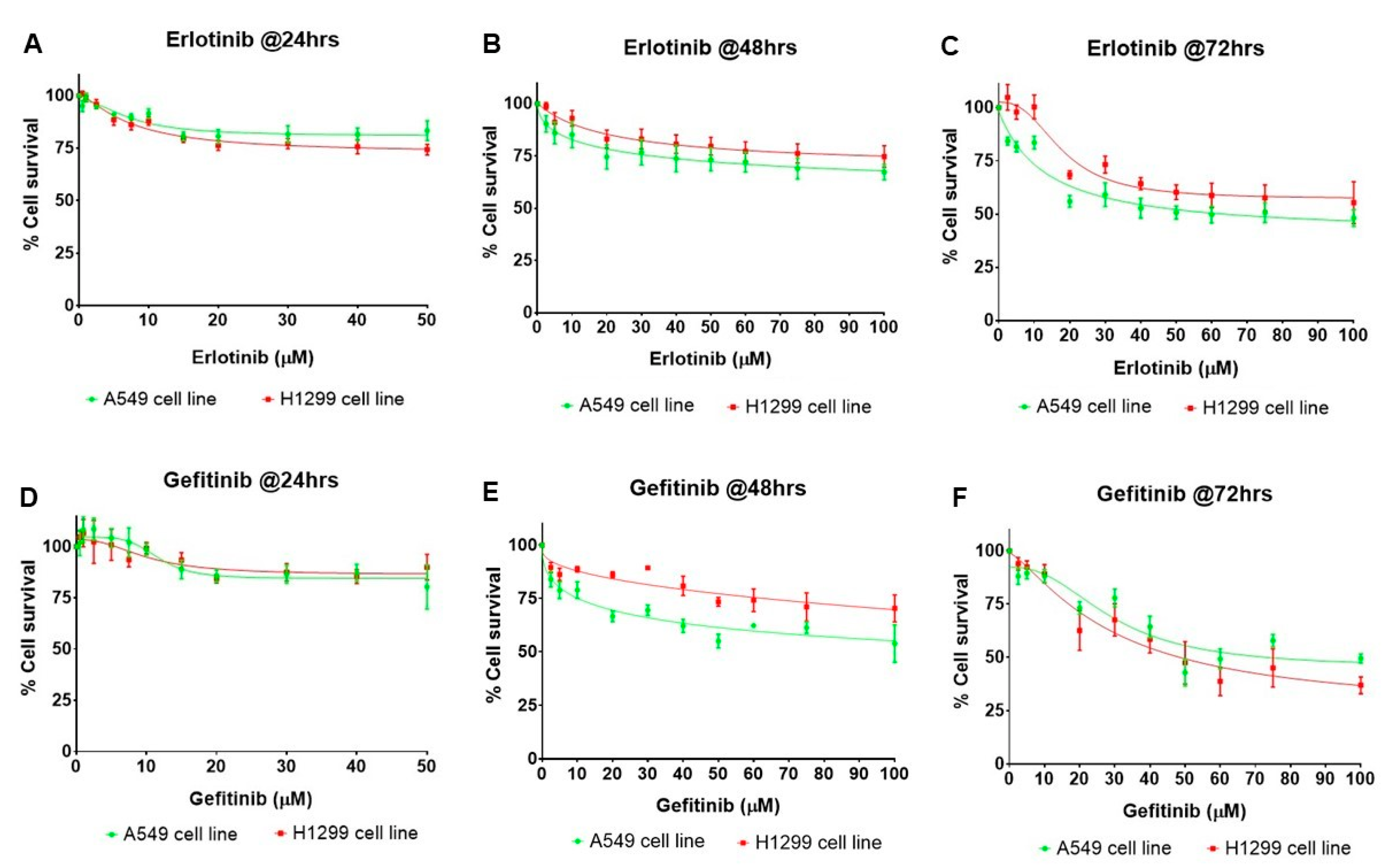
2.5. Erlotinib and Gefitinib Inhibit the Growth of NSCLC Cell Lines in a Dose- and Time-Dependent Manner
2.6. Effects of PAFR Activation, PAFR Antagonist, and miR-149-5p Overexpression on the Cytotoxic Responses of Erlotinib and Gefitinib
3. Discussion
4. Materials and Methods
4.1. Reagents and Cells
4.2. Quantitative Real-Time PCR (qPCR) Analysis
4.3. MTT Assay
4.4. Crystal Violet Assay
4.5. Cell Survival Assay
4.6. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| PAFR | platelet-activating factor receptor |
| miR | microRNA |
| Scr ctrl | scrambled-miRNA control |
| NSCLC | non-small-cell lung cancer |
| CPAF | carbamyl-PAF |
| TKIs | tyrosine kinase inhibitors |
| Gef | gefitinib |
| Erlo | erlotinib |
| CV | crystal violet |
| DMSO | dimethyl sulfoxide |
| ETOH | ethanol |
| SRB | sulforhodamine B |
| FOXM1 | forkhead box protein M1 |
| STAT3 | signal transducer and activator of transcription 3 |
References
- Catalanotto, C.; Cogoni, C.; Zardo, G. MicroRNA in Control of Gene Expression: An Overview of Nuclear Functions. Int. J. Mol. Sci. 2016, 17, 1712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adams, B.D.; Kasinski, A.L.; Slack, F.J. Aberrant regulation and function of microRNAs in cancer. Curr. Biol. 2014, 24, R762–R776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, M.; Li, J.; Ding, X.; He, M.; Cheng, S.Y. microRNA and cancer. AAPS J. 2010, 12, 309–317. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.L.; Tsai, Y.M.; Lien, C.T.; Kuo, P.L.; Hung, A.J. The Roles of MicroRNA in Lung Cancer. Int. J. Mol. Sci. 2019, 20, 1611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nie, H.; Xie, X.; Zhang, D.; Zhou, Y.; Li, B.; Li, F.; Li, F.; Cheng, Y.; Mei, H.; Meng, H.; et al. Use of lung-specific exosomes for miRNA-126 delivery in non-small cell lung cancer. Nanoscale 2020, 12, 877–887. [Google Scholar] [CrossRef]
- Chen, J.; Liu, A.; Wang, Z.; Wang, B.; Chai, X.; Lu, W.; Cao, T.; Li, R.; Wu, M.; Lu, Z.; et al. LINC00173.v1 promotes angiogenesis and progression of lung squamous cell carcinoma by sponging miR-511-5p to regulate VEGFA expression. Mol. Cancer 2020, 19, 98. [Google Scholar] [CrossRef]
- Padda, J.; Khalid, K.; Khedr, A.; Patel, V.; Al-Ewaidat, O.A.; Tasnim, F.; Padda, S.; Cooper, A.C.; Jean-Charles, G. Exosome-Derived microRNA: Efficacy in Cancer. Cureus 2021, 13, e17441. [Google Scholar] [CrossRef]
- Ali Syeda, Z.; Langden, S.; Munkhzul, C.; Lee, M.; Song, S.J. Regulatory Mechanism of MicroRNA Expression in Cancer. Int. J. Mol. Sci. 2020, 21, 1723. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Cai, N.; Zhao, L. MicroRNA-1 regulates the growth and chemosensitivity of breast cancer cells by targeting MEK/ERK pathway. J. BUON 2020, 25, 2215–2220. [Google Scholar]
- Zhao, Z.; Sun, W.; Guo, Z.; Zhang, J.; Yu, H.; Liu, B. Mechanisms of lncRNA/microRNA interactions in angiogenesis. Life Sci. 2020, 254, 116900. [Google Scholar] [CrossRef]
- Zhao, L.; Liu, L.; Dong, Z.; Xiong, J. miR-149 suppresses human non-small cell lung cancer growth and metastasis by inhibiting the FOXM1/cyclin D1/MMP2 axis. Oncol. Rep. 2017, 38, 3522–3530. [Google Scholar] [PubMed]
- Ke, Y.; Zhao, W.; Xiong, J.; Cao, R. miR-149 Inhibits Non-Small-Cell Lung Cancer Cells EMT by Targeting FOXM1. Biochem. Res. Int. 2013, 2013, 506731. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Yu, D.; Zhu, L.; Zhong, S.; Zhao, J.; Tang, J. miR-149 in Human Cancer: A Systemic Review. J. Cancer 2018, 9, 375–388. [Google Scholar] [CrossRef] [PubMed]
- Nasim, F.; Sabath, B.F.; Eapen, G.A. Lung cancer. Med. Clin. N. Am. 2019, 103, 463–473. [Google Scholar] [CrossRef]
- Latimer, K.M.; Mott, T.F. Lung cancer: Diagnosis, treatment principles, and screening. Am. Fam. Physician 2015, 91, 250–256. [Google Scholar]
- Rosell, R.; Dafni, U.; Felip, E.; Curioni-Fontecedro, A.; Gautschi, O.; Peters, S.; Massutí, B.; Palmero, R.; Aix, S.P.; Carcereny, E.; et al. Erlotinib and bevacizumab in patients with advanced non-small-cell lung cancer and activating EGFR mutations (BELIEF): An international, multicentre, single-arm, phase 2 trial. Lancet Respir. Med. 2017, 5, 435–444. [Google Scholar] [CrossRef]
- Chung, C. Tyrosine kinase inhibitors for epidermal growth factor receptor gene mutation-positive non-small cell lung cancers: An update for recent advances in therapeutics. J. Oncol. Pharm. Pract. 2016, 22, 461–476. [Google Scholar] [CrossRef]
- Hackler, P.C.; Reuss, S.; Konger, R.L.; Travers, J.B.; Sahu, R.P. Systemic Platelet-activating Factor Receptor Activation Augments Experimental Lung Tumor Growth and Metastasis. Cancer Growth Metastasis 2014, 7, 27–32. [Google Scholar] [CrossRef] [Green Version]
- da Silva, A., Jr.; Chammas, R.; Lepique, A.P.; Jancar, S. Platelet-activating factor (PAF) receptor as a promising target for cancer cell repopulation after radiotherapy. Oncogenesis 2017, 6, e296. [Google Scholar] [CrossRef] [Green Version]
- Sahu, R.P.; Ocana, J.A.; Harrison, K.A.; Ferracini, M.; Touloukian, C.E.; Al-Hassani, M.; Sun, L.; Loesch, M.; Murphy, R.C.; Althouse, S.K.; et al. Chemotherapeutic agents subvert tumor immunity by generating agonists of platelet-activating factor. Cancer Res. 2014, 74, 7069–7078. [Google Scholar] [CrossRef] [Green Version]
- Sahu, R.P.; Harrison, K.A.; Weyerbacher, J.; Murphy, R.C.; Konger, R.L.; Garrett, J.E.; Chin-Sinex, H.J.; Johnston, M.E., 2nd; Dynlacht, J.R.; Mendonca, M.; et al. Radiation therapy generates platelet-activating factor agonists. Oncotarget 2016, 7, 20788–20800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zappa, C.; Mousa, S.A. Non-small cell lung cancer: Current treatment and future advances. Transl. Lung Cancer Res. 2016, 5, 288–300. [Google Scholar] [CrossRef] [Green Version]
- Pallis, A.G.; Gridelli, C.; Wedding, U.; Faivre-Finn, C.; Veronesi, G.; Jaklitsch, M.; Luciani, A.; O’Brien, M. Management of elderly patients with NSCLC; updated expert’s opinion paper: EORTC Elderly Task Force, Lung Cancer Group and International Society for Geriatric Oncology. Ann. Oncol. 2014, 25, 1270–1283. [Google Scholar] [CrossRef] [PubMed]
- Howe, G.A.; Xiao, B.; Zhao, H.; Al-Zahrani, K.N.; Hasim, M.S.; Villeneuve, J.; Sekhon, H.S.; Goss, G.D.; Sabourin, L.A.; Dimitroulakos, J.; et al. Focal Adhesion Kinase Inhibitors in Combination with Erlotinib Demonstrate Enhanced Anti-Tumor Activity in Non-Small Cell Lung Cancer. PLoS ONE 2016, 11, e0150567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, J.; Kang, M.; Nam, S.H.; Lee, G.-H.; Kim, H.-J.; Ryu, J.; Cheong, J.G.; Jung, J.W.; Kim, T.Y.; Lee, H.-Y.; et al. Bidirectional signaling between TM4SF5 and IGF1R promotes resistance to EGFR kinase inhibitors. Lung Cancer 2015, 90, 22–31. [Google Scholar] [CrossRef]
- Lin, C.; Qin, Y.; Zhang, H.; Gao, M.Y.; Wang, Y.F. EGF upregulates RFPL3 and hTERT via the MEK signaling pathway in non small cell lung cancer cells. Oncol. Rep. 2018, 40, 29–38. [Google Scholar] [CrossRef]
- Chen, J.; Lan, T.; Zhang, W.; Dong, L.; Kang, N.; Zhang, S.; Fu, M.; Liu, B.; Liu, K.; Zhang, C.; et al. Platelet-activating factor receptor-mediated PI3K/AKT activation contributes to the malignant development of esophageal squamous cell carcinoma. Oncogene 2015, 34, 5114–5127. [Google Scholar] [CrossRef]
- Lordan, R.; Tsoupras, A.; Zabetakis, J. The Potential Role of Dietary Platelet-Activating Factor Inhibitors in Cancer Prevention and Treatment. Adv. Nutr. 2019, 10, 148–164. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Lan, T.; Zhang, W.; Dong, L.; Kang, N.; Zhang, S.; Fu, M.; Liu, B.; Liu, K.; Zhan, Q. Feed-Forward Reciprocal Activation of PAFR and STAT3 Regulates Epithelial-Mesenchymal Transition in Non-Small Cell Lung Cancer. Cancer Res. 2015, 75, 4198–4210. [Google Scholar] [CrossRef] [Green Version]
- Cruz, C.S.D.; Tanoue, L.T.; Matthay, R.A. Lung cancer: Epidemiology, etiology, and prevention. Clin. Chest Med. 2011, 32, 605–644. [Google Scholar] [CrossRef] [Green Version]
- Chauhan, S.J.; Thyagarajan, A.; Chen, Y.; Travers, J.B.; Sahu, R.P. Platelet-Activating Factor-Receptor Signaling Mediates Targeted Therapies-Induced Microvesicle Particles Release in Lung Cancer Cells. Int. J. Mol. Sci. 2020, 21, 8517. [Google Scholar] [CrossRef] [PubMed]
- Raychaudhuri, P.; Park, H.J. FoxM1: A master regulator of tumor metastasis. Cancer Res. 2011, 71, 4329–4333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katoh, M.; Igarashi, M.; Fukuda, H.; Nakagama, H.; Katoh, M. Cancer genetics and genomics of human FOX family genes. Cancer Lett. 2013, 328, 198–206. [Google Scholar] [CrossRef] [PubMed]
- Kim, I.M.; Ackerson, T.; Ramakrishna, S.; Tretiakova, M.; Wang, I.C.; Kalin, T.V.; Major, M.L.; Gusarova, G.A.; Yoder, H.M.; Costa, R.H.; et al. The Forkhead Box m1 transcription factor stimulates the proliferation of tumor cells during development of lung cancer. Cancer Res. 2006, 66, 2153–2161. [Google Scholar] [CrossRef] [Green Version]
- Kong, X.; Li, L.; Li, Z.; Le, X.; Huang, C.; Jia, Z.; Cui, J.; Huang, S.; Wang, L.; Xie, K. Dysregulated expression of FOXM1 isoforms drives progression of pancreatic cancer. Cancer Res. 2013, 73, 3987–3996. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.; Xie, D.; Cui, J.; Li, Q.; Gao, Y.; Xie, K. FOXM1c promotes pancreatic cancer epithelial-to-mesenchymal transition and metastasis via upregulation of expression of the urokinase plasminogen activator system. Clin. Cancer Res. 2014, 20, 1477–1488. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.; Zhang, X.; Jiang, L.; Zhang, L.; Xiang, M.; Ren, H. FoxM1 Induced Paclitaxel Resistance via Activation of the FoxM1/PHB1/RAF-MEK-ERK Pathway and Enhancement of the ABCA2 Transporter. Mol. Ther. Oncolytics 2019, 14, 196–212. [Google Scholar] [CrossRef] [Green Version]
- Pei, Y.; Barber, L.A.; Murphy, R.C.; Johnson, C.A.; Kelley, S.W.; Dy, L.C.; Fertel, R.H.; Nguyen, T.M.; Williams, D.A.; Travers, J.B. Activation of the epidermal platelet-activating factor receptor results in cytokine and cyclooxygenase-2 biosynthesis. J. Immunol. 1998, 161, 1954–1961. [Google Scholar]
- Sahu, R.P. Expression of the platelet-activating factor receptor enhances benzyl isothiocyanate-induced apoptosis in murine and human melanoma cells. Mol. Med. Rep. 2015, 12, 394–400. [Google Scholar] [CrossRef] [Green Version]
- Crystal Violet Staining. Available online: https://openwetware.org/wiki/Crystal_Violet_Staining (accessed on 13 November 2019).








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chauhan, S.J.; Thyagarajan, A.; Sahu, R.P. Effects of miRNA-149-5p and Platelet-Activating Factor-Receptor Signaling on the Growth and Targeted Therapy Response on Lung Cancer Cells. Int. J. Mol. Sci. 2022, 23, 6772. https://doi.org/10.3390/ijms23126772
Chauhan SJ, Thyagarajan A, Sahu RP. Effects of miRNA-149-5p and Platelet-Activating Factor-Receptor Signaling on the Growth and Targeted Therapy Response on Lung Cancer Cells. International Journal of Molecular Sciences. 2022; 23(12):6772. https://doi.org/10.3390/ijms23126772
Chicago/Turabian StyleChauhan, Shreepa J., Anita Thyagarajan, and Ravi P. Sahu. 2022. "Effects of miRNA-149-5p and Platelet-Activating Factor-Receptor Signaling on the Growth and Targeted Therapy Response on Lung Cancer Cells" International Journal of Molecular Sciences 23, no. 12: 6772. https://doi.org/10.3390/ijms23126772
APA StyleChauhan, S. J., Thyagarajan, A., & Sahu, R. P. (2022). Effects of miRNA-149-5p and Platelet-Activating Factor-Receptor Signaling on the Growth and Targeted Therapy Response on Lung Cancer Cells. International Journal of Molecular Sciences, 23(12), 6772. https://doi.org/10.3390/ijms23126772