Our results show that, in rats treated with an MCD diet, the progression from steatosis to steatohepatitis significantly affects serum and tissue levels of Zn and Fe; we also documented the upregulation of MMPs and a decrease in TIMPs and RECK, as well as the existence of significant correlations between Zn and Fe versus both molecules involved in the control of the ECM and pro-inflammation markers such as IL-1beta and TNF-alpha. In addition, we found significant correlations between Zn and Fe versus specific fatty acids.
NAFLD is the most common cause of chronic liver disease, and the activation of inflammatory pathways could contribute to disease pathogenesis. In the estimation of the World Health Organization (2016), around 1.9 billion adults (people over 18 years of age) were overweight, and more than 600 million were obese [
11]. It is expected that, by 2030, more than 2.16 billion people will be overweight and 1.12 billion obese [
12], of which one-fourth of the adult population will be affected by NAFLD [
1]. Thus, there is an urgent need for effective interventions to prevent the NAFLD progression from steatosis to steatohepatitis [
2].
The development of an appropriate animal model is crucial in developing a treatment for this rapidly growing disease. The mouse/rat model diets can be identified in the following two types: the MCD diet and the Western-style diet (WD). In the MCD diet, the absence of methionine leads to hepatic injury, inflammation and fibrosis, and the deficiency of choline leads to macrovesicular steatosis [
13]. In the MCD-fed animals, the absence of systemic insulin resistance and the presence of hepatic insulin resistance were found [
14]; thus, this model may help to delineate NASH pathogenesis in the absence of confounding parameters and to identify the involvement of non-insulin resistant mechanisms [
15]. Furthermore, the MCD-fed animal model provides the histological hallmark of NASH because of its vulnerability to transition from simple steatosis to steatohepatitis [
16], becoming a widely used model for the study of the progression of NASH [
17].
In detail, choline, an essential nutrient, is necessary for the de novo synthesis of phosphatidylcholine, needed for the export of triglycerides out of hepatocytes via very-low-density lipoprotein packaging [
18]. Humans can become depleted of choline and previous data showed that choline deficiency in patients that need parenteral nutrition results in the development of hepatic steatosis, similar to various animal models [
19]. Furthermore, methionine is an essential amino acid that plays a key role in many cellular functions because it is used for protein synthesis and as an intermediate in S-adenosylmethionine (SAM) and GSH synthesis, two important metabolites in cellular homeostasis and hepatocyte function [
20].
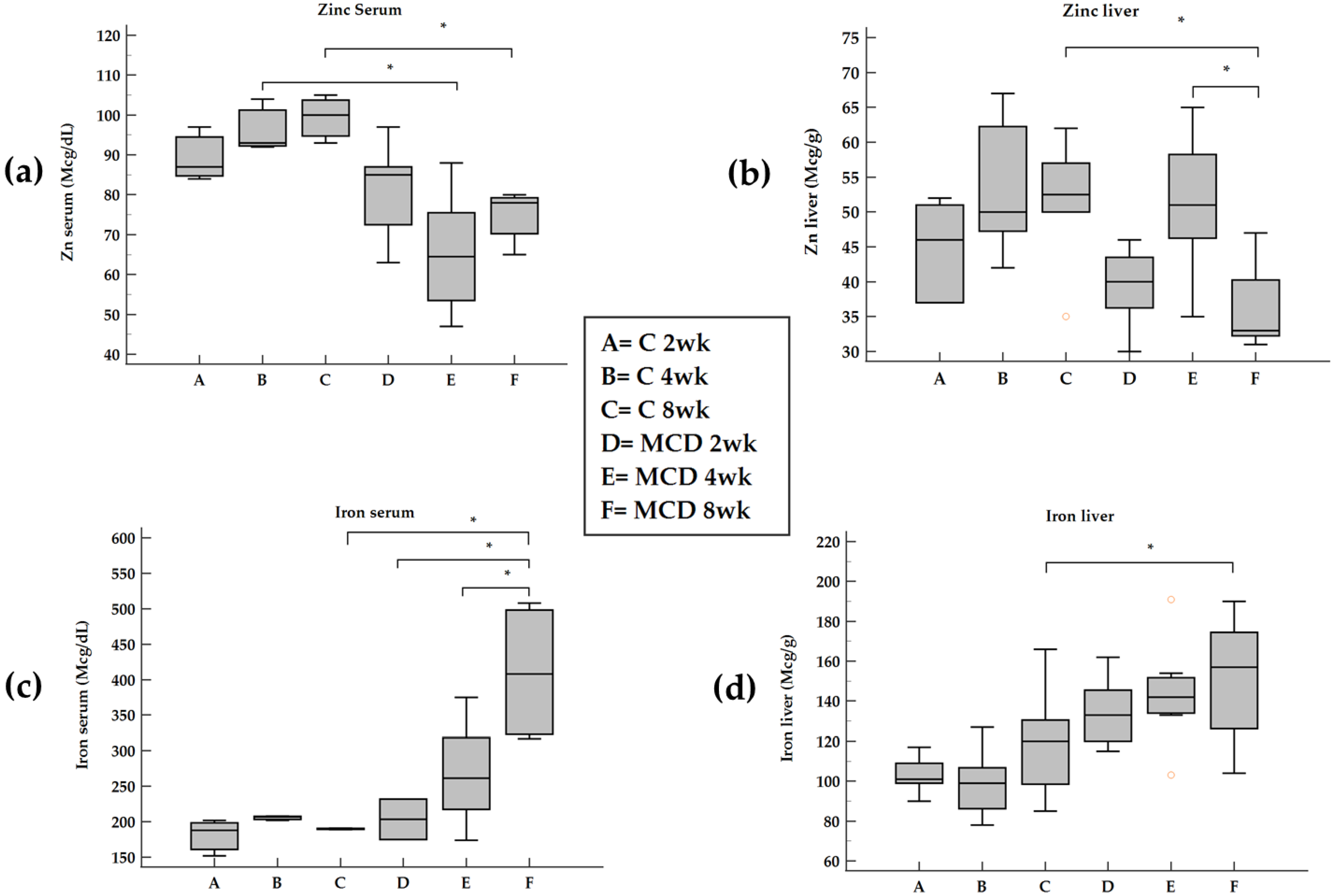
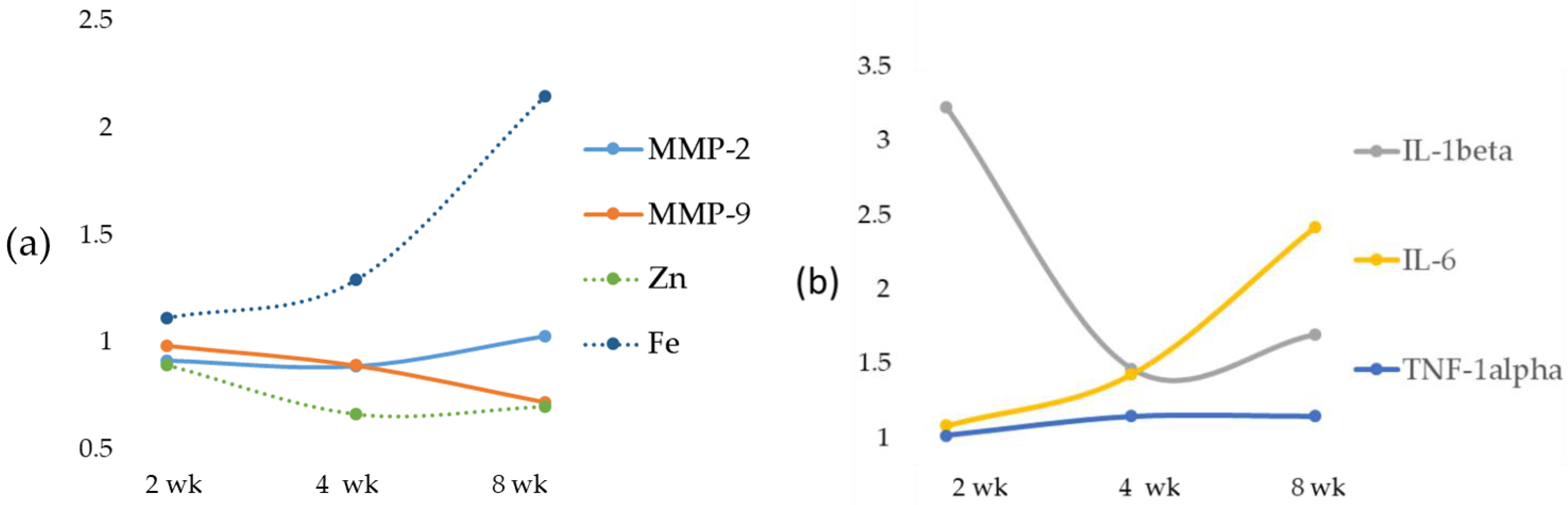
Here we report that in rats treated with an MCD diet, a time-dependent decrease in Zn and an increase in Fe occurred both in serum and in the liver. Furthermore, the co-existence of Fe and Zn changes can contribute to the liver’s development of fibrosis as supported by the relationships with these two oligo-elements versus molecules involved in ECM modulation.
3.1. Zn and Fe and MMPs
Serum and hepatic Zn concentrations are decreased in chronic liver diseases, and Zn depletion has been suggested as a cause of hepatic fibrosis [
21]. The probable underlying mechanism is that the MCD diet induces an inflammatory state in which the expression of gelatinases increases [
22].
Inflammation has been considered the main cause of chronic diseases, and Zn can influence the production and signaling of numerous inflammatory cytokines in a variety of cell types [
23]. Studies evaluating simultaneously circulating cytokines and Zn status showed that the reduced circulating Zn correlates with increased IL-6, IL-8 and TNF-α levels [
24]. In support of these findings, in our data, an inverse correlation between serum Zn and IL-1beta and TNF-alpha was found. On the contrary, no correlation was found between Zn and IL-6. During the progression from NAFLD to NASH, we observed an opposite trend of Zn in serum and tissues with a switch at 4 weeks attributable to an inverse relationship between hepatic Zn content and serum Zn levels [
25], with a mechanism probably modulated by interleukins. In fact, in infectious diseases, interleukins released from activated phagocytic cells have been shown to reduce plasma Zn concentrations by redistributing it from plasma to the liver [
23]. Zn deficiency caused by chronic liver disease also evokes metabolic alterations such as insulin resistance, Fe overload and hepatic steatosis [
26].
Zn also competes with Fe and a weak inverse correlation between serum Zn and ferritin was found in HCV-related chronic liver disease patients associated with NAFLD [
27]. On the other hand, it has been established that Zn and Fe may compete for access to transporters as follows: divalent metal transporter 1 (DMT1) is a possible candidate Zn-Fe transporter, as well as Zip14, is another possible candidate for transport of both Zn and Fe in the liver [
28,
29]. Zip14 has been documented to mediate Fe not bound to transferrin in the liver, which could lead to Fe overload [
30]. Both in vivo and in vitro experiments in the liver demonstrated that Zn transporter Zip14 is regulated by IL-6, likely contributing to the hypozincemia observed during inflammation [
31]. Zip14, one of the major cytokine-responsive transporters localized at the plasma membrane of hepatocytes, could be thus involved in the MCD diet model as an increase in IL-6 occurs. Although in tissues different from the liver, IL-1beta has also been demonstrated to downregulate a number of Zn-transporters, most strikingly ZnT8 [
32].
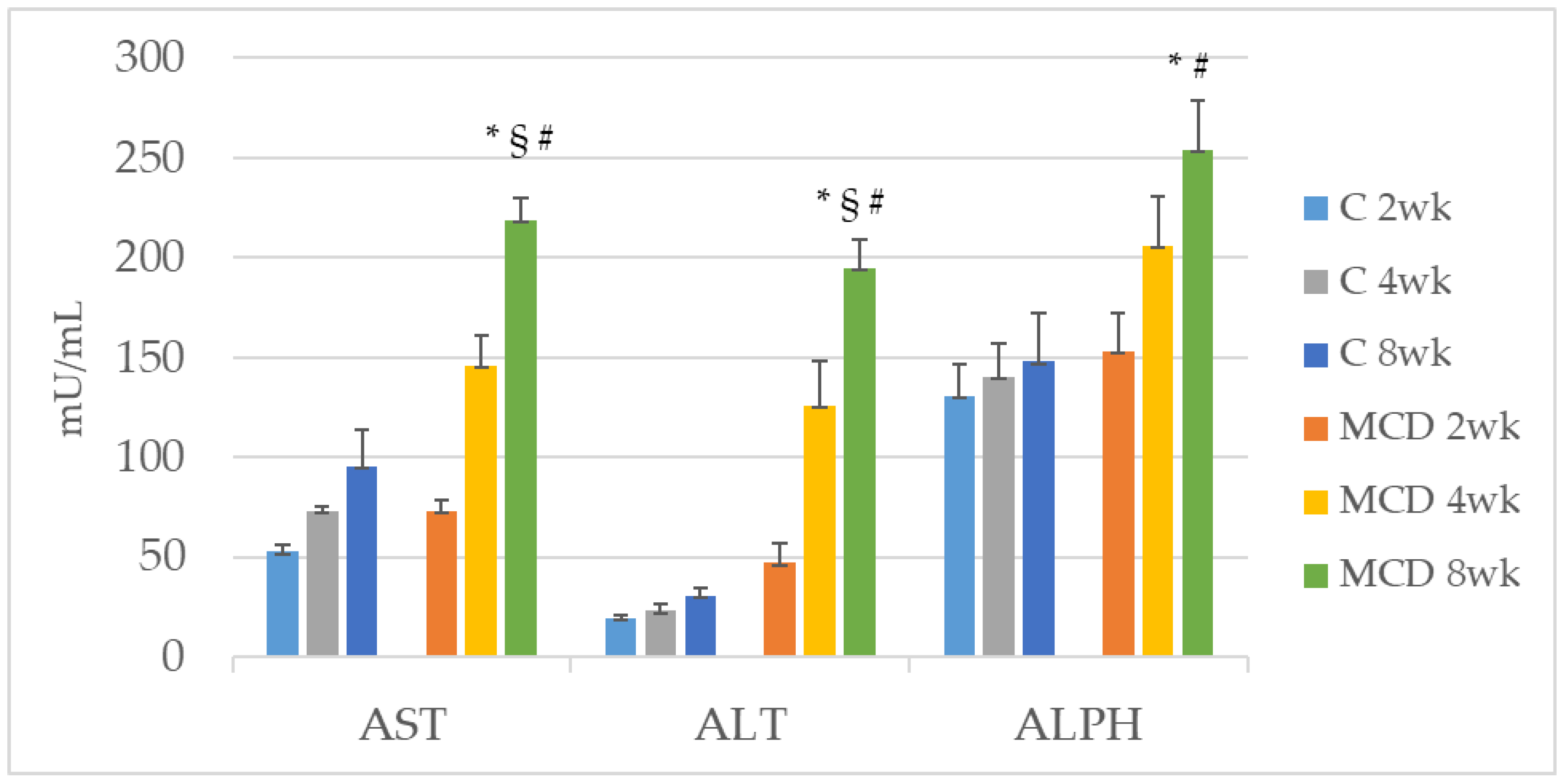
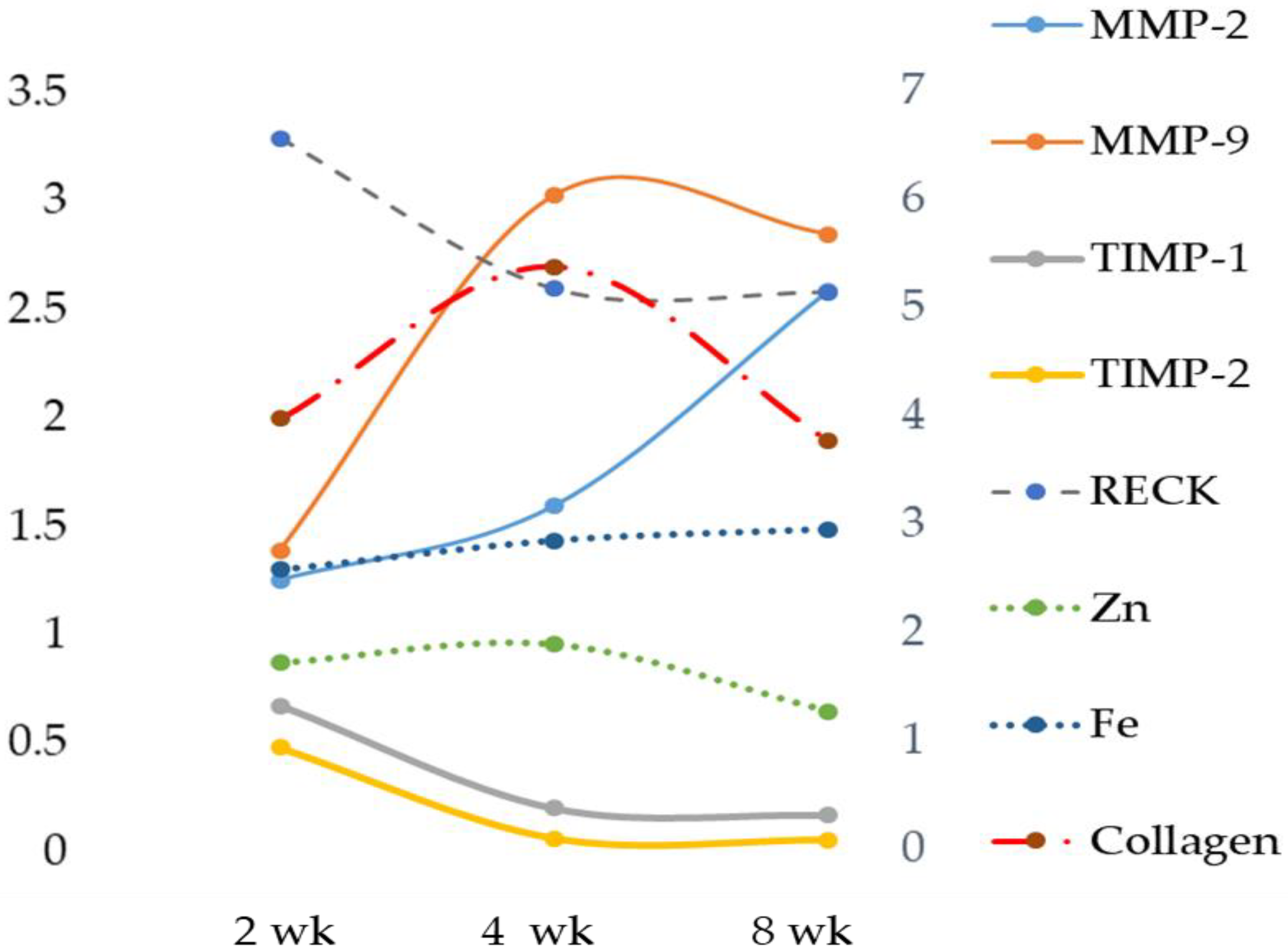
The role of Zn as an anti-inflammatory and antioxidant agent would justify the reduction of the expression of MMP-2 and MMP-9 [
22]. Vice versa, during NAFLD progression from steatosis to steatohepatitis, a Zn decrease is correlated with an increase in gelatinase activity [
33]. The reported data in our MCD model showed a significant increasing trend in both liver MMP-2 and MMP-9 when compared with their respective control and MCD 2 wk, while the result of serum MMP analysis showed no significant change in gelatinase activity. In spite of the MMP-2 and MMP-9 increase, no significant correlation was found between liver or serum Zn and gelatinases. Concurrently, a significant reduction of TIMP-1 and a tendency toward reduction of TIMP-2 were found. In addition, the novel matrix metalloproteinase regulator, RECK [
34], was significantly reduced after 8 weeks of the MCD diet. A great deal of interest has been shown in tissue inhibitor metalloproteinase (TIMP) levels as non-invasive biomarkers for the diagnosis of NAFLD [
35].
In the observational one-point study by Abdelaziz, the authors evaluated the status of TIMP-1 and TIMP-2 in patients with NAFLD and compared these levels with those of obese and control patients [
35]. They showed that serum levels of both TIMPs were significantly elevated in NASH patients as compared to the control group. These results are not consistent with ours. Both TIMP-1 and TIMP-2 had significant diagnostic ability in detecting advanced liver disease but the status of TIMP-1 and TIMP-2, in the algorithm of NAFLD diagnosis, is yet to be established. Furthermore, we evaluated TIMPs in the tissue, not in the plasma, and it is known that TIMPs prevent cellular MMP’s fibrinolytic action in a stoichiometric manner [
36].
Oxidative stress plays an important role in the pathophysiology of NAFLD/NASH. For the purpose of evaluating lipid peroxidation, various markers of oxidative stress and related antioxidants have been evaluated over the years [
37]. Among the oxidative stress biomarkers examined, TBARS, MDA, CYP2E1 and 4-HNE were found to be incisive; their steady increase during the transition from NAFLD to NASH has been reported in both clinical and experimental settings [
38]. In the present study, the increased TBARS and ROS values of the MCD rats correlated negatively with tissue concentrations of Zn as expected. In addition, TBARS and ROS were also correlated with gelatinases.
We documented, using rats submitted to the MCD diet, a time-dependent increase in serum Fe that occurs by a systemic insulin-independent mechanism. This finding is in agreement with a previous study on the MCD model where hepatic Fe overload was associated with a trend toward increased perivenular fibrosis [
39].
The role of hepatic Fe in the progression of steatosis to steatohepatitis remains controversial, but the hypothesis that Fe may play a role in the pathogenesis of NASH is increasingly emerging [
40].
The mechanism for this progression is not yet known, but a “two hit theory” is generally accepted. The first hit is considered to be insulin resistance, resulting in an excessive accumulation of fat in the liver. The second hit is the generation of oxidative stress by a number of factors, including hepatic Fe overload, which leads to cytotoxicity and inflammation [
41]. However, further studies are needed to define the contribution of hepatic Fe to severe disease progression [
42].
Fe overload has been reported in 30–70% of patients with non-alcoholic fatty liver disease (NAFLD) and non-alcoholic steatohepatitis (NASH) [
43]. Fe could potentially play a supporting role in lipid peroxidation and fibrogenesis during the development and progression of NASH by inducing fibrosis-promoting signals in the parenchymal (hepatocytes) and non-parenchymal cells (hepatic stellate cells-HSCs, liver sinusoidal endothelial cells, Kupffer cells) [
4]. Liver injury stimulates the non-parenchymal cells to secrete several profibrogenic cytokines. Pro-inflammatory mediators cause quiescent HSCs to differentiate into myofibroblast-like cells to produce extracellular matrix (ECM) components, which are subsequently degraded by matrix metalloproteinases (MMPs). In turn, MMP activity is inhibited and modulated by specific tissue inhibitors (TIMPs), produced by the activated HSCs. Subsequently, the activated HSCs either undergo apoptosis and/or revert to their original quiescent phenotype, thereby terminating a well-regulated and reversible healing process [
44]. Prolonged liver injury via chronic inflammation, infection and/or oxidative stress leads to continuous stimulation of the wound healing mechanism whereby the HSCs remain persistently activated, leading to excessive deposition of ECM [
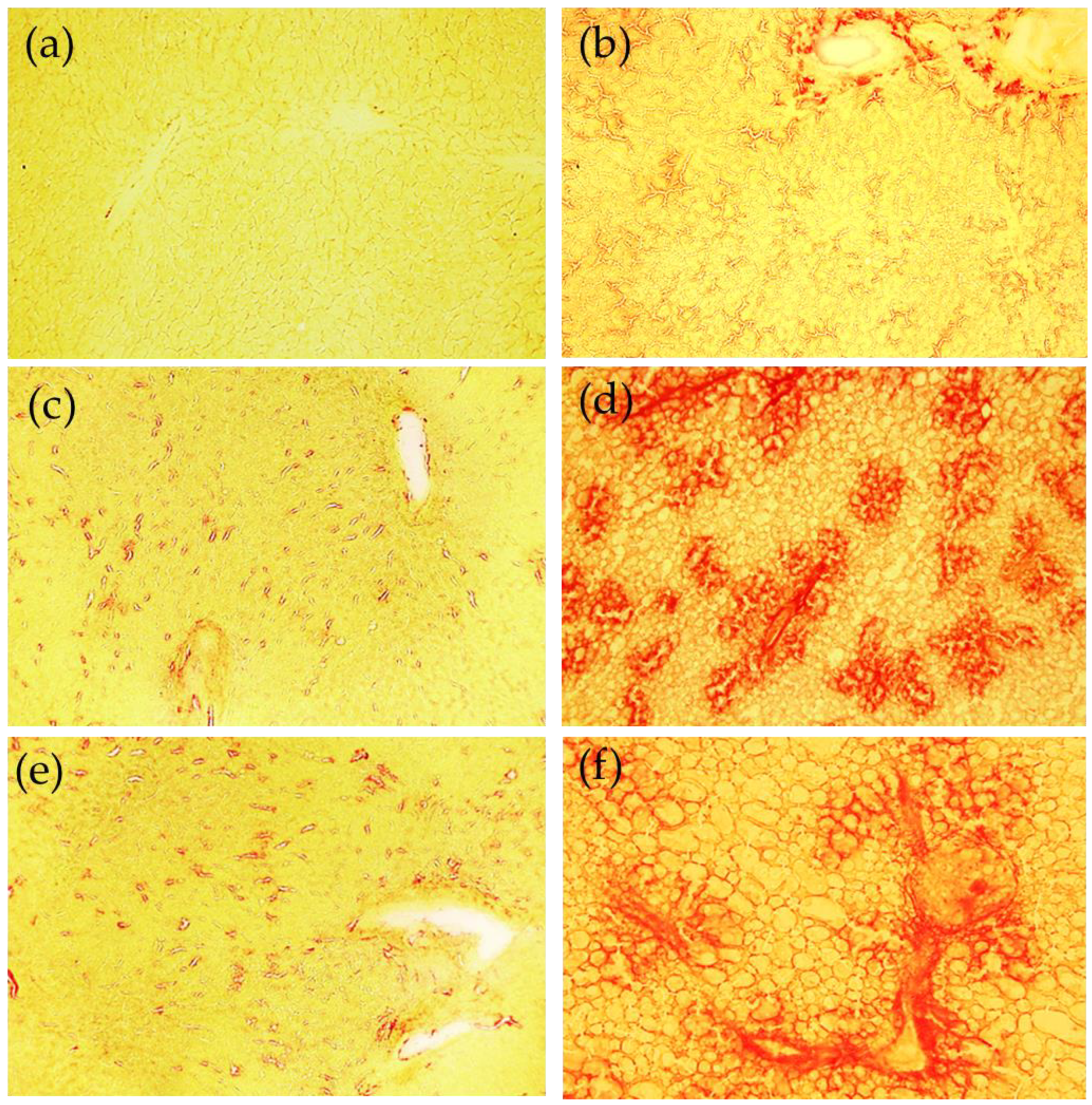
45]. The fibrotic responses are collectively mediated by multiple mechanisms.
Excess Fe accelerates the Fenton reaction to generate ROS, which can oxidize lipids, proteins and nucleic acids, thereby promoting fibrosis initiation and/or fibrosis progression. ROS, in turn, induces lipid peroxidation [
46]. The by-products of lipid peroxidation detected in the liver of Fe-loaded rats can act as profibrogenic stimuli [
47]. In support of this, we detected a significant and positive correlation between liver Fe and TBARS. Sensitized hepatocytes produce a pool of pro-fibrogenic and pro-inflammatory mediators such as IL-6, Il-1beta and TNF-alpha. Cytokines, particularly pro-inflammatory cytokines, have been shown to play an important role in the pathological progression of NASH [
48]. This was evident in our MCD-induced NASH rat model due to the significant increase in the levels of the pro-inflammatory cytokine IL-6.
Another mechanism involved is Fe-mediated ECM remodeling. Studies have shown that Fe promotes collagen crosslinking. This form of collagen is more resistant to proteolytic degradation by MMPs [
49]. However, a previous in vitro study excluded Fe as a major participant in collagen crosslinking since the Fe chelator deferoxamine did not alter collagen modifications [
50]. As a result, to date, the exact effect of Fe on collagen maturation is still unclear.
Our data suggest that Fe plays a role in matrix degradation by stimulating some metalloprotease activities. In our MCD model, a positive correlation was found comparing hepatic Fe versus gelatinases MMP-9 and MMP-2. In particular, during hepatic fibrosis, the metalloproteinase mainly expressed is MMP-2, and our results are in accordance with data obtained by Gardi et al. [
51]. We also evaluated the inhibition of MMPs activity by measuring both the expression of TIMPs and RECK, a novel matrix metalloproteinase regulator [
34]. A significant decrease was detected for TIMP-1 and TIMP-2, and a time-dependent decrease was found for RECK. This newly discovered MMP inhibitor does not show similarities to TIMPs in amino acid sequence and can inhibit at least three members of the MMP family as follows: MMP-2, MMP-9 and MT1-MMP, playing an important role in the process of degradation of ECM. The statistical analysis showed a strong inverse correlation between Fe and specific MMP inhibitors TIMP-1 and TIMP-2 and a significant positive correlation with RECK, despite the latter’s role being comparable to that of TIMPs. Anyway, to date, direct evidence of the role of Fe on RECK is lacking.
3.2. Zn and Fe and Fatty Acids
Linoleic acid (LA), an omega-6 fatty acid and α-linolenic acid (ALA), an omega-3 fatty acid, are considered essential fatty acids (EFA) because they cannot be synthesized by humans, and a relationship between Zn and EFA deficiency has been proposed [
52].
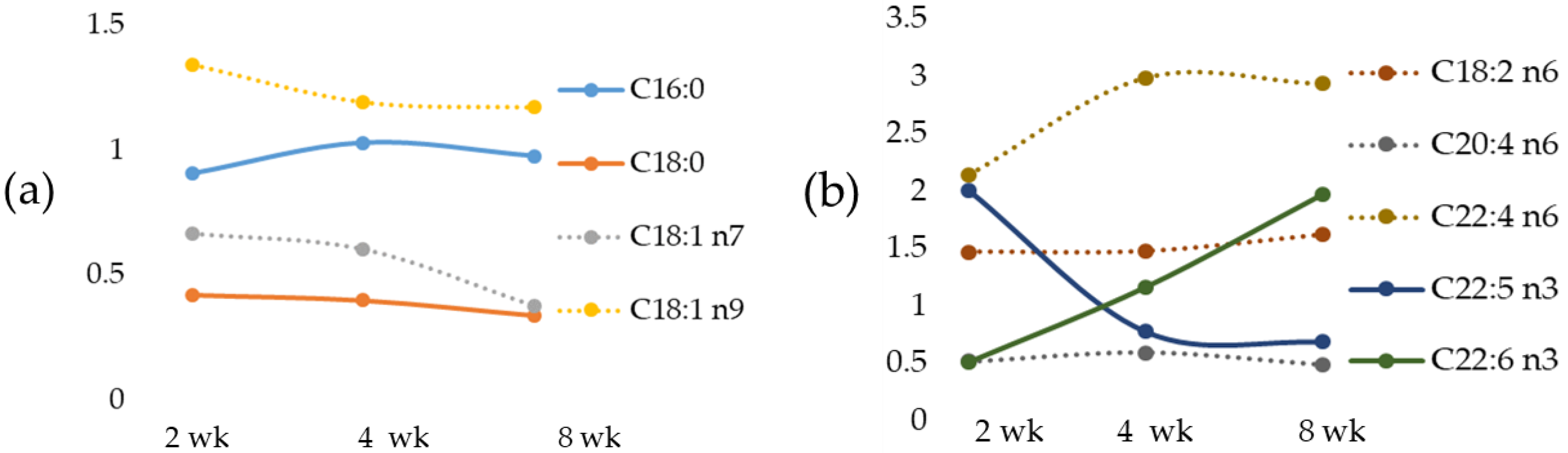
We documented, in the MCD diet rat model, a time-dependent change of the most important fatty acids that occurred early in fatty acid accumulation, in agreement with data previously reported in mice fed with the MCD diet [
53]. We have documented a tissue increase in DHA after an 8-wk MCD diet, also found in the liver of high-fat diet mice [
54]. Of note, the significant correlation between Zn and fatty acid composition reported in the present study suggests that in the presence of a consistent Zn decrease a change in lipid composition occurs; on the contrary, a moderate Zn deficiency, as that observed in weanling rats fed diets rich in Cocoa Butter or Safflower Oil, was not able to alter significantly liver lipid concentrations and fatty acid composition [
55]. We also documented in the MCD rats a correlation between Fe versus the most relevant fatty acids. Linoleic acid, an EFA, was inversely correlated with Zn and positively with Fe. Particularly, an increase in LA occurred with a peak at 2-wk MCD diet.
Arachidonic acid (AA), as a pro-inflammatory precursor, is associated with metabolic disorders [
56]. We documented low AA in the livers of MCD 2 wk rats and a significant increase in the MCD 8 wk group; our results are in agreement with those observed in mice treated with the MCD diet [
53]. Similar results have been also found in rats during high-fat diet (HFD)-induced NAFLD development [
56]. Based on these results, we may propose that changes in AA content may be an early indicator of inflammation in NAFLD progression.
Docosapentaenoic acid, or DPA, has been reported to have a major role in reducing inflammation [
57]. In the MCD diet model, a strong reduction in DPA has been found at 8 weeks, and this event is associated with an increase in IL-6.
A recent study, based on combined classical histology, pathological evaluation and gene expression analysis, reported that the MCD diet model recapitulates the liver manifestation of human NASH [
14]. Although the MCD diet does not provoke systemic insulin resistance, which is important in the pathogenesis of NAFLD in humans, this model may be potentially used for the investigation of the lipotoxic effects of fatty acids. Moreover, the MCD diet model produces reproducible and rapid changes similar to those observed in human NASH, and changes in fatty acids already occurred after 2 weeks.
The findings of this study reinforce the roles of the high prevalence of Zn deficiency and Fe overload among subjects undergoing NAFLD, also found in the MCD experimental model. The results obtained indicate that the assessment of Zn and Fe status should provide a standard parameter of nutritional status screening in NAFLD patients. Although further research is required, Zn supplementation and a control of Fe overload in NAFD patients might be beneficial for the prevention and attenuation of adverse health outcomes.
Diet influences the progression of NAFLD; following a western diet or simply a high-fat diet may contribute to the worsening of NAFLD and further progression to NASH and cirrhosis in later stages. A systematic review found that saturated fatty acids, trans-fats, animal proteins and simple sugars have a harmful effect on the liver [
58]. Dietary changes can slow these events and prevent lasting liver damage. For example, Zn supplementation has beneficial metabolic effects in patients with NAFLD risk factors. Plasma Zn levels should be evaluated in patients with NAFLD and NAFLD risk factors, specifically in those with advanced hepatic fibrosis/cirrhosis [
59]. In addition, the Mediterranean diet is the gold standard for both treatment and prevention [
60]. This dietary pattern is characterized by a high intake of olive oil, which is rich in monounsaturated fat. It has also been shown that diets enriched with omega-3 polyunsaturated fatty acids (PUFA) ameliorate steatohepatitis, together with a reduction in intrahepatic triglyceride content [
61]; this is strongly recommended since a lower consumption of omega-3 PUFA was found in NASH patients [
62].


 ,
, 











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}