
Optineurin Deficiency and Insufficiency Lead to Higher Microglial TDP-43 Protein Levels

 , , and
, , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
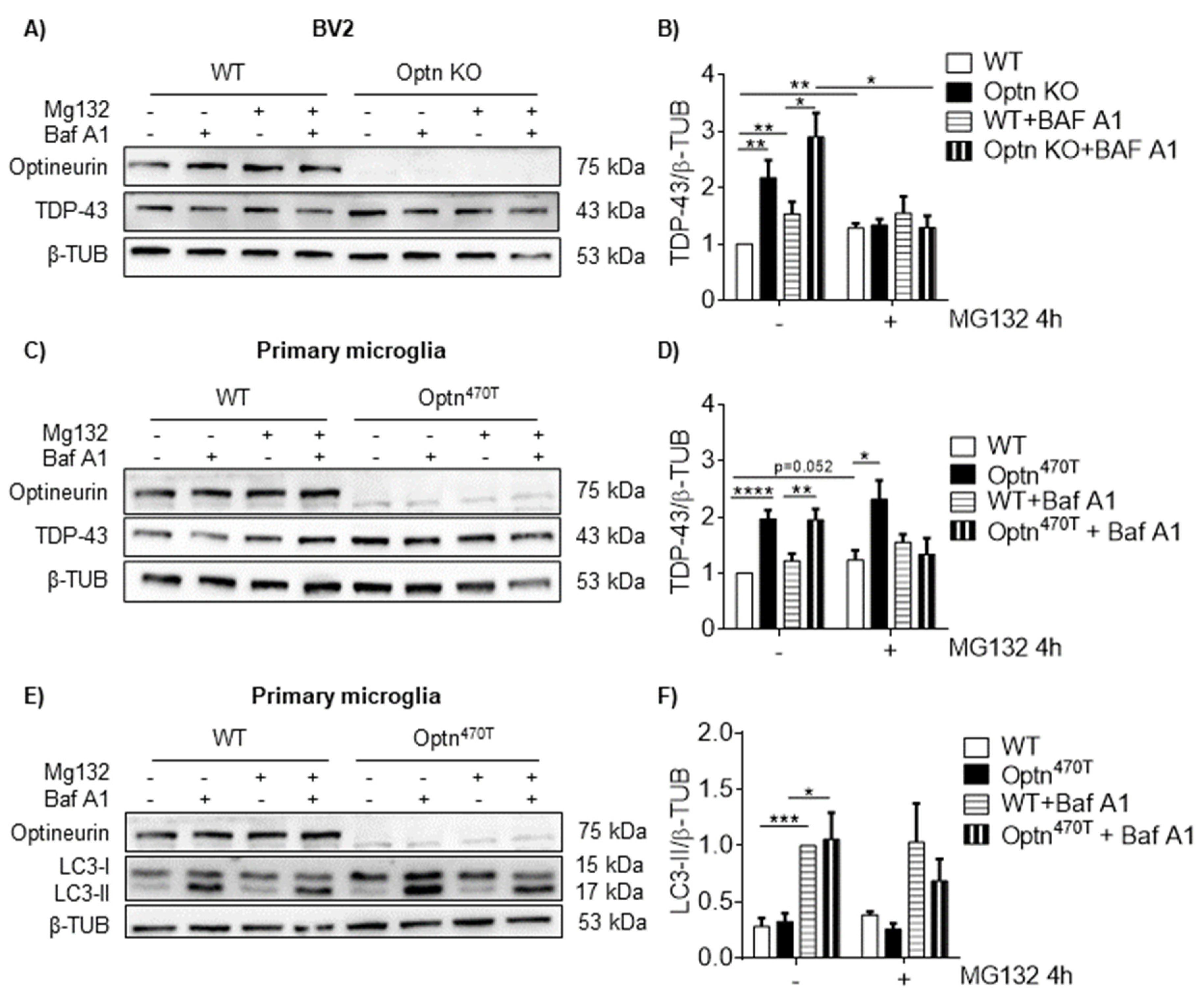
2.1. Optineurin Deficiency Led to an Increase of TDP-43 in BV2 Microglial Cell Line
2.2. Optineurin Insufficiency Led to an Increase of TDP-43 and G3BP1 in Primary Microglia and Macrophages
2.3. Block in Autophagy Is Not the Reason for Increased TDP-43 in BV2 Optn KO and Optn470T Primary Microglia
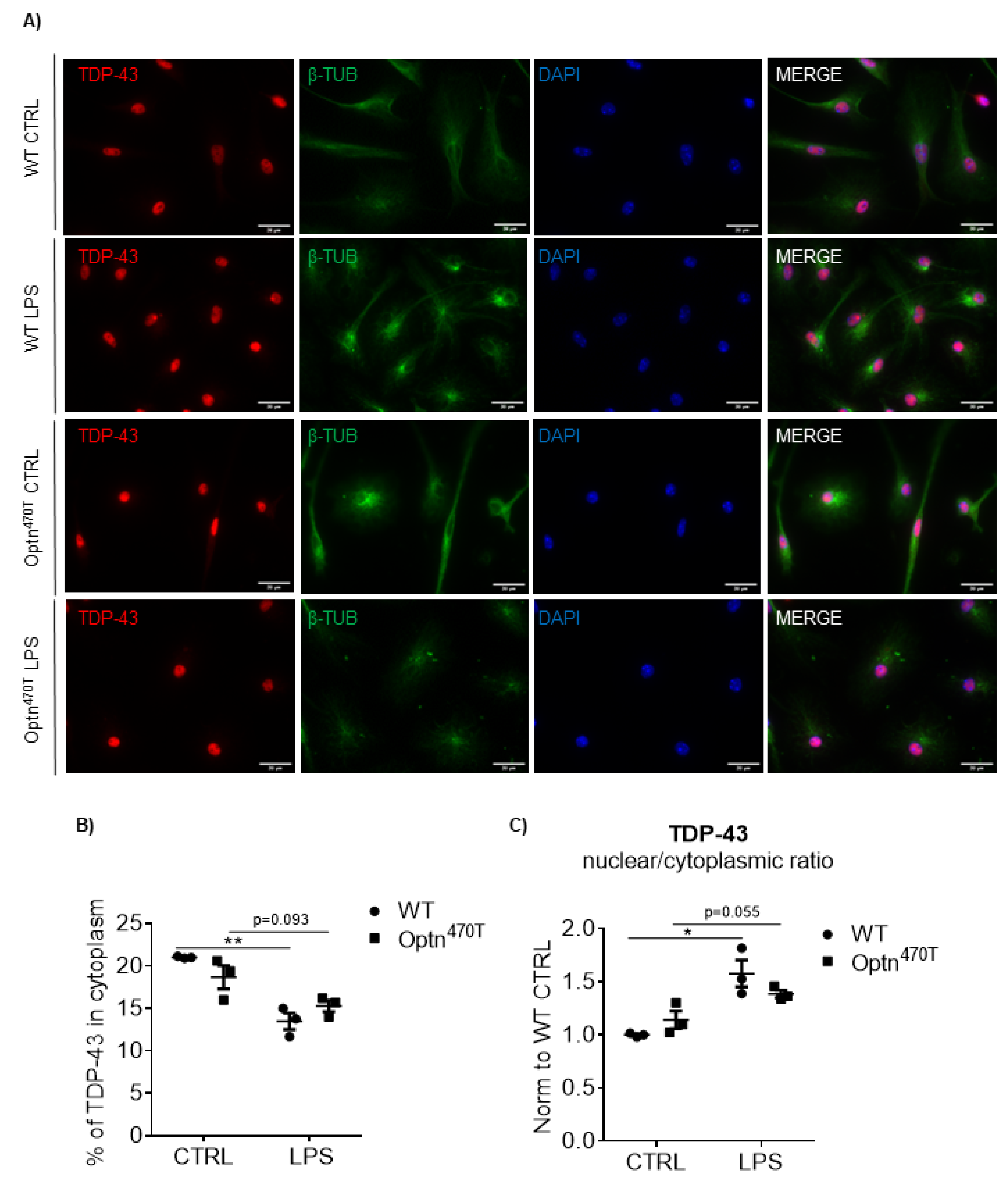
2.4. LPS Increased TDP-43 in WT Cells, but Failed to Increase the Already Elevated TDP-43 Levels in Optineurin KO and Insufficient Cells
2.5. Increased TDP-43 Does Not Form Aggregates in Microglia
3. Discussion
4. Materials and Methods
4.1. Mice
4.2. Reagents
4.3. Establishing Optn KO Microglial and Neuronal Cell Lines
4.4. Primary Microglia and Bone Marrow-Derived Macrophages (BMDMs) Isolation and Cultivation
4.5. Cell Culture and Treatment
4.6. Protein Isolation and Western Blot Analysis
4.7. Quantitative Real-Time PCR (RT-PCR)
4.8. Immunofluorescence Analysis
4.9. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Hardiman, O.; Al-Chalabi, A.; Chio, A.; Corr, E.M.; Logroscino, G.; Robberecht, W.; Shaw, P.J.; Simmons, Z.; van den Berg, L.H. Amyotrophic Lateral Sclerosis. Nat. Rev. Dis. Primers 2017, 3, 17071. [Google Scholar] [CrossRef] [PubMed]
- Béland, L.-C.; Markovinovic, A.; Jakovac, H.; De Marchi, F.; Bilic, E.; Mazzini, L.; Kriz, J.; Munitic, I. Immunity in Amyotrophic Lateral Sclerosis: Blurred Lines between Excessive Inflammation and Inefficient Immune Responses. Brain Commun. 2020, 2, fcaa124. [Google Scholar] [CrossRef]
- Geloso, M.C.; Corvino, V.; Marchese, E.; Serrano, A.; Michetti, F.; D’Ambrosi, N. The Dual Role of Microglia in ALS: Mechanisms and Therapeutic Approaches. Front. Aging Neurosci. 2017, 9, 242. [Google Scholar] [CrossRef] [Green Version]
- Noristani, H.N.; Sabourin, J.C.; Gerber, Y.N.; Teigell, M.; Sommacal, A.; dM Vivanco, M.; Weber, M.; Perrin, F.E. Brca1 Is Expressed in Human Microglia and Is Dysregulated in Human and Animal Model of ALS. Mol. Neurodegener. 2015, 10, 34. [Google Scholar] [CrossRef] [Green Version]
- Arai, T.; Hasegawa, M.; Akiyama, H.; Ikeda, K.; Nonaka, T.; Mori, H.; Mann, D.; Tsuchiya, K.; Yoshida, M.; Hashizume, Y.; et al. TDP-43 Is a Component of Ubiquitin-Positive Tau-Negative Inclusions in Frontotemporal Lobar Degeneration and Amyotrophic Lateral Sclerosis. Biochem. Biophys. Res. Commun. 2006, 351, 602–611. [Google Scholar] [CrossRef] [PubMed]
- Neumann, M.; Sampathu, D.M.; Kwong, L.K.; Truax, A.C.; Micsenyi, M.C.; Chou, T.T.; Bruce, J.; Schuck, T.; Grossman, M.; Clark, C.M.; et al. Ubiquitinated TDP-43 in Frontotemporal Lobar Degeneration and Amyotrophic Lateral Sclerosis. Science 2006, 314, 130–133. [Google Scholar] [CrossRef] [Green Version]
- Mackenzie, I.R.; Rademakers, R. The Role of Transactive Response DNA-Binding Protein-43 in Amyotrophic Lateral Sclerosis and Frontotemporal Dementia. Curr. Opin. Neurol. 2008, 21, 693–700. [Google Scholar] [CrossRef] [Green Version]
- Ratti, A.; Buratti, E. Physiological Functions and Pathobiology of TDP-43 and FUS/TLS Proteins. J. Neurochem. 2016, 138, 95–111. [Google Scholar] [CrossRef]
- Ciryam, P.; Lambert-Smith, I.A.; Bean, D.M.; Freer, R.; Cid, F.; Tartaglia, G.G.; Saunders, D.N.; Wilson, M.R.; Oliver, S.G.; Morimoto, R.I.; et al. Spinal Motor Neuron Protein Supersaturation Patterns Are Associated with Inclusion Body Formation in ALS. Proc. Natl. Acad. Sci. USA. 2017, 114, 3935–3943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, B.S.; Snead, D.; Lee, J.J.; McCaffery, J.M.; Shorter, J.; Gitler, A.D. TDP-43 Is Intrinsically Aggregation-Prone, and Amyotrophic Lateral Sclerosis-Linked Mutations Accelerate Aggregation and Increase Toxicity. J. Biol. Chem. 2009, 284, 20329–20339. [Google Scholar] [CrossRef] [Green Version]
- Prasad, A.; Bharathi, V.; Sivalingam, V.; Girdhar, A.; Patel, B.K. Molecular Mechanisms of TDP-43 Misfolding and Pathology in Amyotrophic Lateral Sclerosis. Front. Mol. Neurosci. 2019, 12, 25. [Google Scholar] [CrossRef] [PubMed]
- Araki, W.; Minegishi, S.; Motoki, K.; Kume, H.; Hohjoh, H.; Araki, Y.M.; Tamaoka, A. Disease-Associated Mutations of TDP-43 Promote Turnover of the Protein Through the Proteasomal Pathway. Mol. Neurobiol. 2014, 50, 1049–1058. [Google Scholar] [CrossRef]
- Scotter, E.L.; Vance, C.; Nishimura, A.L.; Lee, Y.-B.; Chen, H.-J.; Urwin, H.; Sardone, V.; Mitchell, J.C.; Rogelj, B.; Rubinsztein, D.C.; et al. Differential Roles of the Ubiquitin Proteasome System and Autophagy in the Clearance of Soluble and Aggregated TDP-43 Species. J. Cell Sci. 2014, 127, 1263–1278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aman, Y.; Schmauck-Medina, T.; Hansen, M.; Morimoto, R.I.; Simon, A.K.; Bjedov, I.; Palikaras, K.; Simonsen, A.; Johansen, T.; Tavernarakis, N.; et al. Autophagy in Healthy Aging and Disease. Nat. Aging. 2021, 1, 634–650. [Google Scholar] [CrossRef]
- Swarup, V.; Phaneuf, D.; Dupré, N.; Petri, S.; Strong, M.; Kriz, J.; Julien, J.-P. Deregulation of TDP-43 in Amyotrophic Lateral Sclerosis Triggers Nuclear Factor ΚB–Mediated Pathogenic Pathways. J. Exp. Med. 2011, 208, 2429–2447. [Google Scholar] [CrossRef] [PubMed]
- Correia, A.S.; Patel, P.; Dutta, K.; Julien, J.-P. Inflammation Induces TDP-43 Mislocalization and Aggregation. PLoS ONE 2015, 10, e0140248. [Google Scholar] [CrossRef] [Green Version]
- Maruyama, H.; Morino, H.; Ito, H.; Izumi, Y.; Kato, H.; Watanabe, Y.; Kinoshita, Y.; Kamada, M.; Nodera, H.; Suzuki, H.; et al. Mutations of Optineurin in Amyotrophic Lateral Sclerosis. Nature 2010, 465, 223–226. [Google Scholar] [CrossRef] [PubMed]
- Markovinovic, A.; Cimbro, R.; Ljutic, T.; Kriz, J.; Rogelj, B.; Munitic, I. Optineurin in Amyotrophic Lateral Sclerosis: Multifunctional Adaptor Protein at the Crossroads of Different Neuroprotective Mechanisms. Prog. Neurobiol. 2017, 154, 1–20. [Google Scholar] [CrossRef]
- Akutsu, M.; Dikic, I.; Bremm, A. Ubiquitin Chain Diversity at a Glance. J. Cell Sci. 2016, 129, 875–880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laplantine, E.; Fontan, E.; Chiaravalli, J.; Lopez, T.; Lakisic, G.; Véron, M.; Agou, F.; Israël, A. NEMO Specifically Recognizes K63-Linked Poly-Ubiquitin Chains through a New Bipartite Ubiquitin-Binding Domain. EMBO J. 2009, 28, 2885–2895. [Google Scholar] [CrossRef]
- Markovinovic, A.; Ljutic, T.; Béland, L.-C.; Munitic, I. Optineurin Insufficiency Disbalances Proinflammatory and Anti-Inflammatory Factors by Reducing Microglial IFN-β Responses. Neuroscience 2018, 388, 139–151. [Google Scholar] [CrossRef]
- Munitic, I.; Giardino Torchia, M.L.; Meena, N.P.; Zhu, G.; Li, C.C.; Ashwell, J.D. Optineurin Insufficiency Impairs IRF3 but Not NF-ΚB Activation in Immune Cells. J. Immunol. 2013, 191, 6231–6240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slowicka, K.; Vereecke, L.; Mc Guire, C.; Sze, M.; Maelfait, J.; Kolpe, A.; Saelens, X.; Beyaert, R.; van Loo, G. Optineurin Deficiency in Mice Is Associated with Increased Sensitivity to Salmonella but Does Not Affect Proinflammatory NF-ΚB Signaling. Eur. J. Immunol. 2016, 46, 971–980. [Google Scholar] [CrossRef] [Green Version]
- Gleason, C.E.; Ordureau, A.; Gourlay, R.; Arthur, J.S.C.; Cohen, P. Polyubiquitin Binding to Optineurin Is Required for Optimal Activation of TANK-Binding Kinase 1 and Production of Interferon β. J. Biol. Chem. 2011, 286, 35663–35674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meena, N.P.; Zhu, G.; Mittelstadt, P.R.; Giardino Torchia, M.L.; Pourcelot, M.; Arnoult, D.; Ashwell, J.D.; Munitic, I. The TBK1-Binding Domain of Optineurin Promotes Type I Interferon Responses. FEBS Lett. 2016, 590, 1498–1508. [Google Scholar] [CrossRef] [Green Version]
- Pourcelot, M.; Zemirli, N.; Silva Da Costa, L.; Loyant, R.; Garcin, D.; Vitour, D.; Munitic, I.; Vazquez, A.; Arnoult, D. The Golgi Apparatus Acts as a Platform for TBK1 Activation after Viral RNA Sensing. BMC Biol. 2016, 14, 69. [Google Scholar] [CrossRef] [PubMed]
- Korac, J.; Schaeffer, V.; Kovacevic, I.; Clement, A.M.; Jungblut, B.; Behl, C.; Terzic, J.; Dikic, I. Ubiquitin-Independent Function of Optineurin in Autophagic Clearance of Protein Aggregates. J. Cell Sci. 2012, 126, 580–592. [Google Scholar] [CrossRef] [Green Version]
- Lazarou, M.; Sliter, D.A.; Kane, L.A.; Sarraf, S.A.; Wang, C.; Burman, J.L.; Sideris, D.P.; Fogel, A.I.; Youle, R.J. The Ubiquitin Kinase PINK1 Recruits Autophagy Receptors to Induce Mitophagy. Nature 2015, 524, 309–314. [Google Scholar] [CrossRef] [Green Version]
- Wild, P.; Farhan, H.; McEwan, D.G.; Wagner, S.; Rogov, V.V.; Brady, N.R.; Richter, B.; Korac, J.; Waidmann, O.; Choudhary, C.; et al. Phosphorylation of the Autophagy Receptor Optineurin Restricts Salmonella Growth. Science 2011, 333, 228–233. [Google Scholar] [CrossRef] [Green Version]
- Tumbarello, D.A.; Waxse, B.J.; Arden, S.D.; Bright, N.A.; Kendrick-Jones, J.; Buss, F. Autophagy Receptors Link Myosin VI to Autophagosomes to Mediate Tom1-Dependent Autophagosome Maturation and Fusion with the Lysosome. Nat. Cell Biol. 2012, 14, 1024–1035. [Google Scholar] [CrossRef] [Green Version]
- Ito, H.; Nakamura, M.; Komure, O.; Ayaki, T.; Wate, R.; Maruyama, H.; Nakamura, Y.; Fujita, K.; Kaneko, S.; Okamoto, Y.; et al. Clinicopathologic Study on an ALS Family with a Heterozygous E478G Optineurin Mutation. Acta Neuropathol. 2011, 122, 223–229. [Google Scholar] [CrossRef] [PubMed]
- Kamada, M.; Izumi, Y.; Ayaki, T.; Nakamura, M.; Kagawa, S.; Kudo, E.; Sako, W.; Maruyama, H.; Nishida, Y.; Kawakami, H.; et al. Clinicopathologic Features of Autosomal Recessive Amyotrophic Lateral Sclerosis Associated with Optineurin Mutation: Autosomal Recessive OPTN-ALS. Neuropathology 2014, 34, 64–70. [Google Scholar] [CrossRef] [PubMed]
- Kurashige, T.; Kuramochi, M.; Ohsawa, R.; Yamashita, Y.; Shioi, G.; Morino, H.; Kamada, M.; Ayaki, T.; Ito, H.; Sotomaru, Y.; et al. Optineurin Defects Cause TDP43-Pathology with Autophagic Vacuolar Formation. Neurobiol. Dis. 2021, 148, 105215. [Google Scholar] [CrossRef] [PubMed]
- Wang, P. Acetylation-Induced TDP-43 Pathology Is Suppressed by an HSF1-Dependent Chaperone Program. Nat. Commun. 2017, 8, 82. [Google Scholar] [CrossRef] [PubMed]
- Wong, Y.C.; Holzbaur, E.L.F. Optineurin Is an Autophagy Receptor for Damaged Mitochondria in Parkin-Mediated Mitophagy That Is Disrupted by an ALS-Linked Mutation. Cell Biol. 2014, 111, 4439–4448. [Google Scholar] [CrossRef] [Green Version]
- Evans, C.S.; Holzbaur, E.L. Degradation of Engulfed Mitochondria Is Rate-Limiting in Optineurin-Mediated Mitophagy in Neurons. eLife 2020, 9, e50260. [Google Scholar] [CrossRef]
- Sidibé, H.; Khalfallah, Y.; Xiao, S.; Gómez, N.B.; Fakim, H.; Tank, E.M.H.; Di Tomasso, G.; Bareke, E.; Aulas, A.; McKeever, P.M.; et al. TDP-43 Stabilizes G3BP1 MRNA: Relevance to Amyotrophic Lateral Sclerosis/Frontotemporal Dementia. Brain 2021, 144, 3461–3476. [Google Scholar] [CrossRef]
- Brown, R.H.; Phil, D. Amyotrophic Lateral Sclerosis. N. Engl. J. Med. 2017, 377, 162–172. [Google Scholar] [CrossRef] [Green Version]
- De Marchi, F.; Munitic, I.; Amedei, A.; Berry, J.D.; Feldman, E.L.; Aronica, E.; Nardo, G.; Van Weehaeghe, D.; Niccolai, E.; Prtenjaca, N.; et al. Interplay between Immunity and Amyotrophic Lateral Sclerosis: Clinical Impact. Neurosci. Biobehav. Rev. 2021, 127, 958–978. [Google Scholar] [CrossRef]
- Ito, Y.; Ofengeim, D.; Najafov, A.; Das, S.; Saberi, S.; Li, Y.; Hitomi, J.; Zhu, H.; Chen, H.; Mayo, L.; et al. RIPK1 Mediates Axonal Degeneration by Promoting Inflammation and Necroptosis in ALS. Science 2016, 353, 603–608. [Google Scholar] [CrossRef] [Green Version]
- Root, J.; Merino, P.; Nuckols, A.; Johnson, M.; Kukar, T. Lysosome Dysfunction as a Cause of Neurodegenerative Diseases: Lessons from Frontotemporal Dementia and Amyotrophic Lateral Sclerosis. Neurobiol. Dis. 2021, 154, 105360. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, Y.; Tsuji, K.; Ayaki, T.; Mori, M.; Tokunaga, F.; Ito, H. Linear Polyubiquitin Chain Modification of TDP-43-Positive Neuronal Cytoplasmic Inclusions in Amyotrophic Lateral Sclerosis. J. Neuropathol. Exp. Neurol. 2020, 79, 256–265. [Google Scholar] [CrossRef] [PubMed]
- Shen, W.-C.; Li, H.-Y.; Chen, G.-C.; Chern, Y.; Tu, P. Mutations in the Ubiquitin-Binding Domain of OPTN/Optineurin Interfere with Autophagy-Mediated Degradation of Misfolded Proteins by a Dominant-Negative Mechanism. Autophagy 2015, 11, 685–700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.; Shao, Z.; Liu, X.; Hou, M.; Cheng, F.; Lei, D.; Yuan, H. The E50K Optineurin Mutation Impacts Autophagy-Mediated Degradation of TDP-43 and Leads to RGC Apoptosis in Vivo and in Vitro. Cell Death Discov. 2021, 7, 49. [Google Scholar] [CrossRef] [PubMed]
- Ling, S.-C.; Albuquerque, C.P.; Han, J.S.; Lagier-Tourenne, C.; Tokunaga, S.; Zhou, H.; Cleveland, D.W. ALS-Associated Mutations in TDP-43 Increase Its Stability and Promote TDP-43 Complexes with FUS/TLS. Proc. Natl. Acad. Sci. USA. 2010, 107, 13318–13323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buratti, E. Functional Significance of TDP-43 Mutations in Disease. In Advances in Genetics; Elsevier: Amsterdam, The Netherlands, 2015; Volume 91, pp. 1–53. [Google Scholar]
- Ayala, Y.M.; Zago, P.; D’Ambrogio, A.; Xu, Y.-F.; Petrucelli, L.; Buratti, E.; Baralle, F.E. Structural Determinants of the Cellular Localization and Shuttling of TDP-43. J. Cell Sci. 2008, 121, 3778–3785. [Google Scholar] [CrossRef] [Green Version]
- Polymenidou, M.; Lagier-Tourenne, C.; Hutt, K.R.; Huelga, S.C.; Moran, J.; Liang, T.Y.; Ling, S.-C.; Sun, E.; Wancewicz, E.; Mazur, C.; et al. Long Pre-MRNA Depletion and RNA Missplicing Contribute to Neuronal Vulnerability from Loss of TDP-43. Nat. Neurosci. 2011, 14, 459–468. [Google Scholar] [CrossRef] [Green Version]
- Sephton, C.F.; Good, S.K.; Atkin, S.; Dewey, C.M.; Mayer, P.; Herz, J.; Yu, G. TDP-43 Is a Developmentally Regulated Protein Essential for Early Embryonic Development. J. Biol. Chem. 2010, 285, 6826–6834. [Google Scholar] [CrossRef] [Green Version]
- Kakihana, T.; Takahashi, M.; Katsuragi, Y.; Yamashita, S.-I.; Sango, J.; Kanki, T.; Onodera, O.; Fujii, M. The Optineurin/TIA1 Pathway Inhibits Aberrant Stress Granule Formation and Reduces Ubiquitinated TDP-43. iScience 2021, 24, 102733. [Google Scholar] [CrossRef]
- De Marco, G.; Lomartire, A.; Calvo, A.; Risso, A.; De Luca, E.; Mostert, M.; Mandrioli, J.; Caponnetto, C.; Borghero, G.; Manera, U.; et al. Monocytes of Patients with Amyotrophic Lateral Sclerosis Linked to Gene Mutations Display Altered TDP-43 Subcellular Distribution. Neuropathol. Appl. Neurobiol. 2017, 43, 133–153. [Google Scholar] [CrossRef]
- Fernandes, N.; Nero, L.; Lyons, S.; Ivanov, P.; Mittelmeier, T.; Bolger, T.; Buchan, J. Stress Granule Assembly Can Facilitate but Is Not Required for TDP-43 Cytoplasmic Aggregation. Biomolecules 2020, 10, 1367. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Prtenjaca, N.; Rob, M.; Alam, M.S.; Markovinovic, A.; Stuani, C.; Buratti, E.; Munitic, I. Optineurin Deficiency and Insufficiency Lead to Higher Microglial TDP-43 Protein Levels. Int. J. Mol. Sci. 2022, 23, 6829. https://doi.org/10.3390/ijms23126829
Prtenjaca N, Rob M, Alam MS, Markovinovic A, Stuani C, Buratti E, Munitic I. Optineurin Deficiency and Insufficiency Lead to Higher Microglial TDP-43 Protein Levels. International Journal of Molecular Sciences. 2022; 23(12):6829. https://doi.org/10.3390/ijms23126829
Chicago/Turabian StylePrtenjaca, Nikolina, Matea Rob, Muhammad S. Alam, Andrea Markovinovic, Cristiana Stuani, Emanuele Buratti, and Ivana Munitic. 2022. "Optineurin Deficiency and Insufficiency Lead to Higher Microglial TDP-43 Protein Levels" International Journal of Molecular Sciences 23, no. 12: 6829. https://doi.org/10.3390/ijms23126829
APA StylePrtenjaca, N., Rob, M., Alam, M. S., Markovinovic, A., Stuani, C., Buratti, E., & Munitic, I. (2022). Optineurin Deficiency and Insufficiency Lead to Higher Microglial TDP-43 Protein Levels. International Journal of Molecular Sciences, 23(12), 6829. https://doi.org/10.3390/ijms23126829