
A Fully-Human Antibody Specifically Targeting a Membrane-Bound Fragment of CADM1 Potentiates the T Cell-Mediated Death of Human Small-Cell Lung Cancer Cells

 , and
, and 
Abstract
:1. Introduction
2. Results
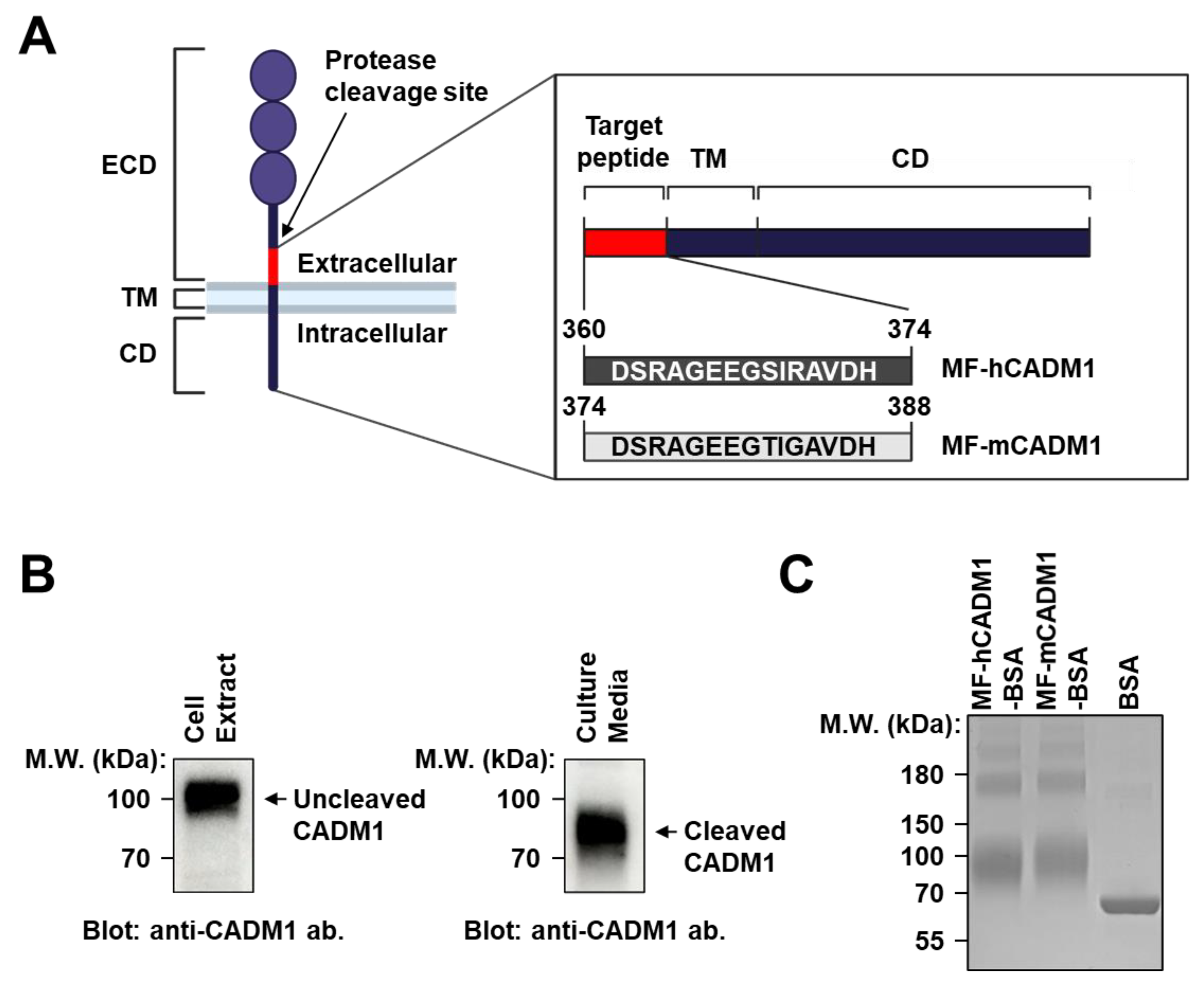
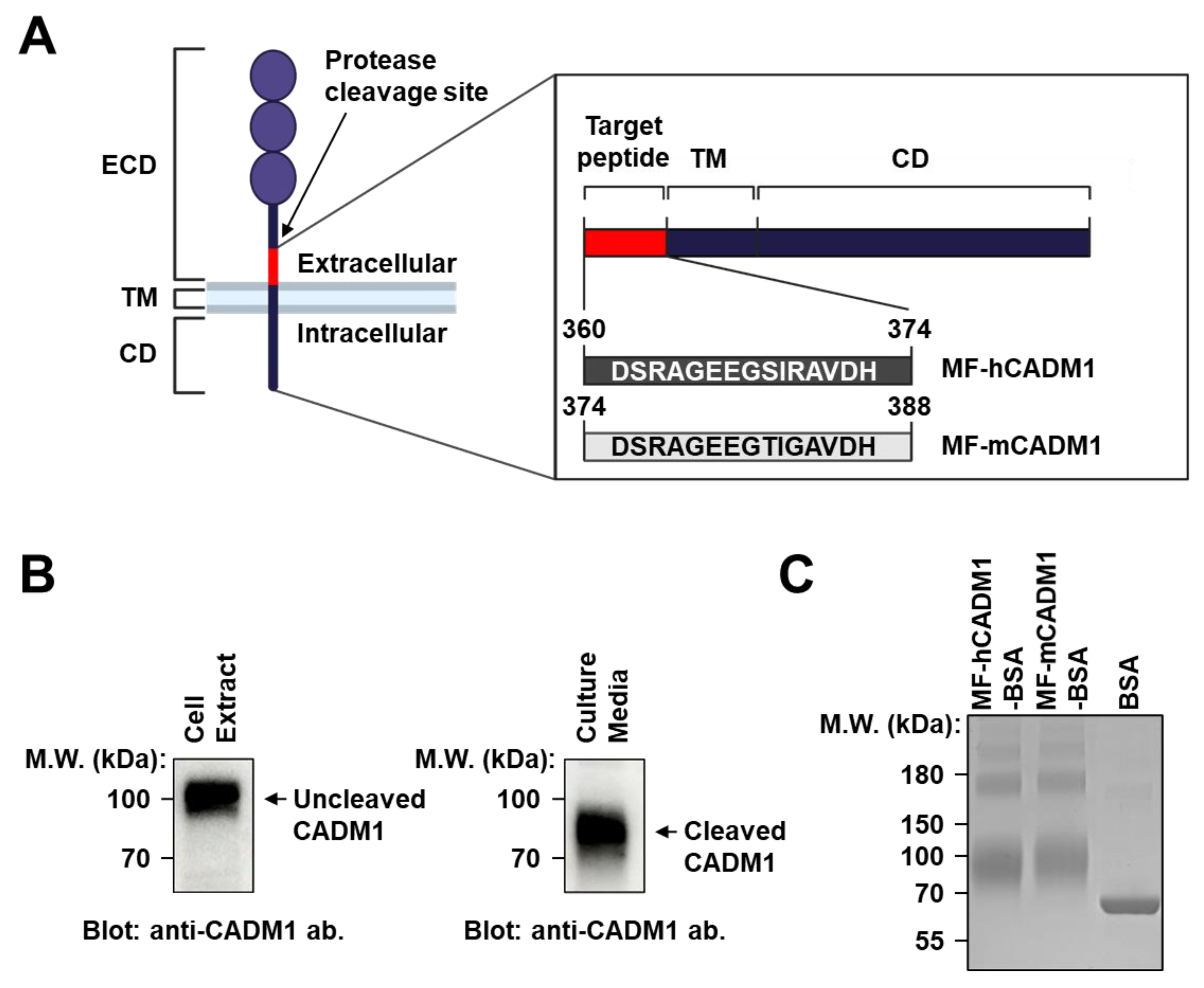
2.1. Preparation and Analysis of MF-CADM1 Peptide Conjugates for Antibody Selection
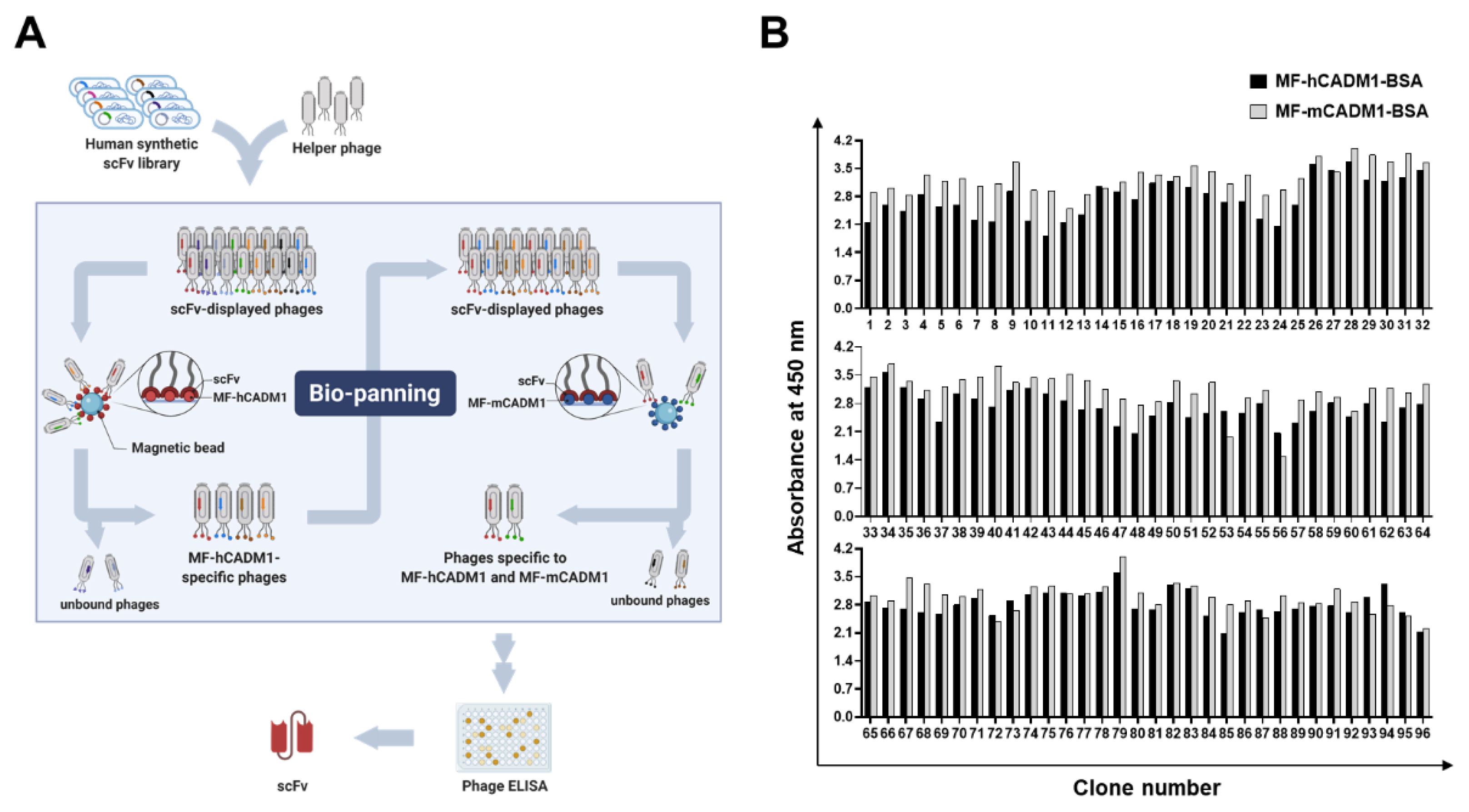
2.2. Selection of Fully Human scFvs Specific to Human and Mouse MF-CADM1
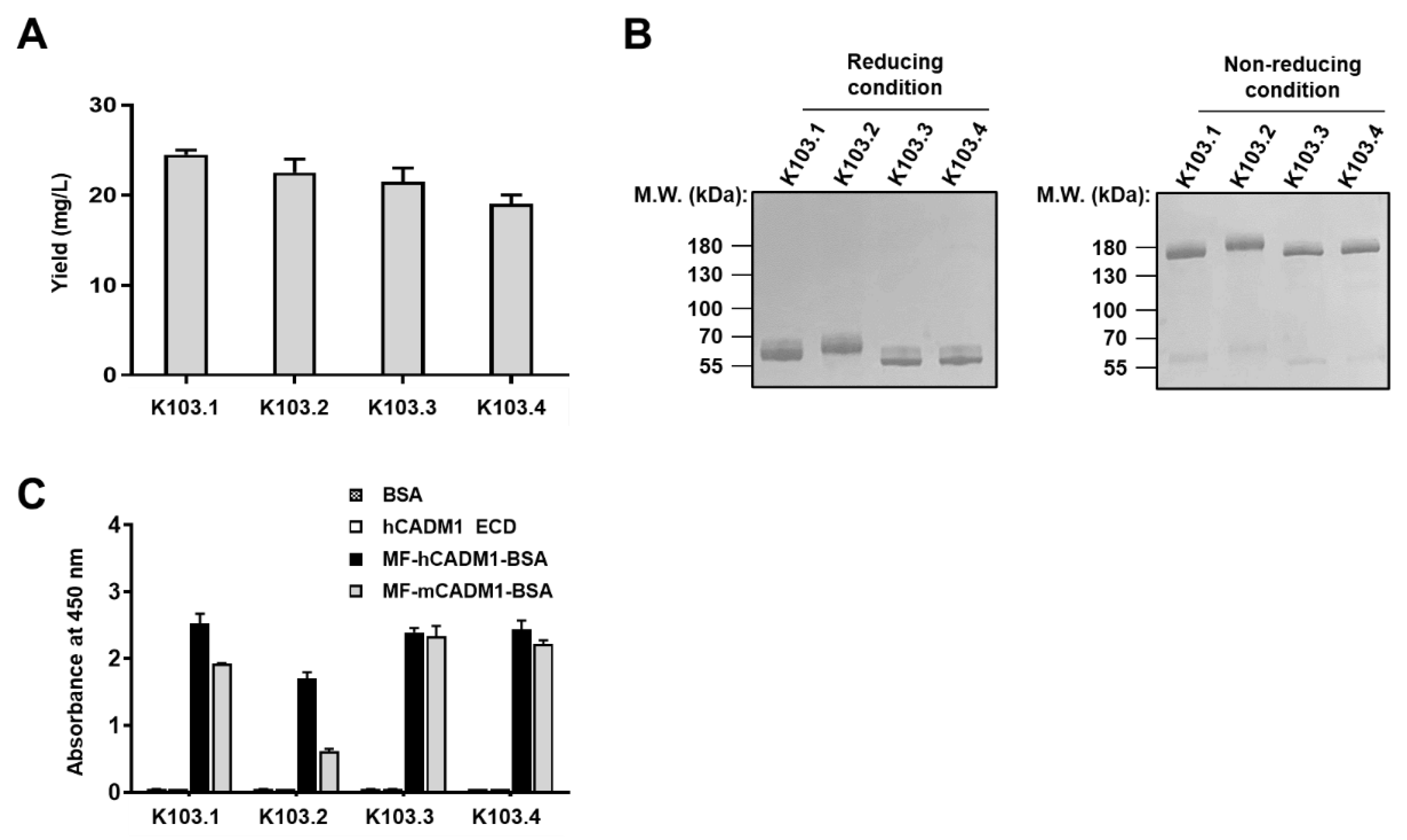
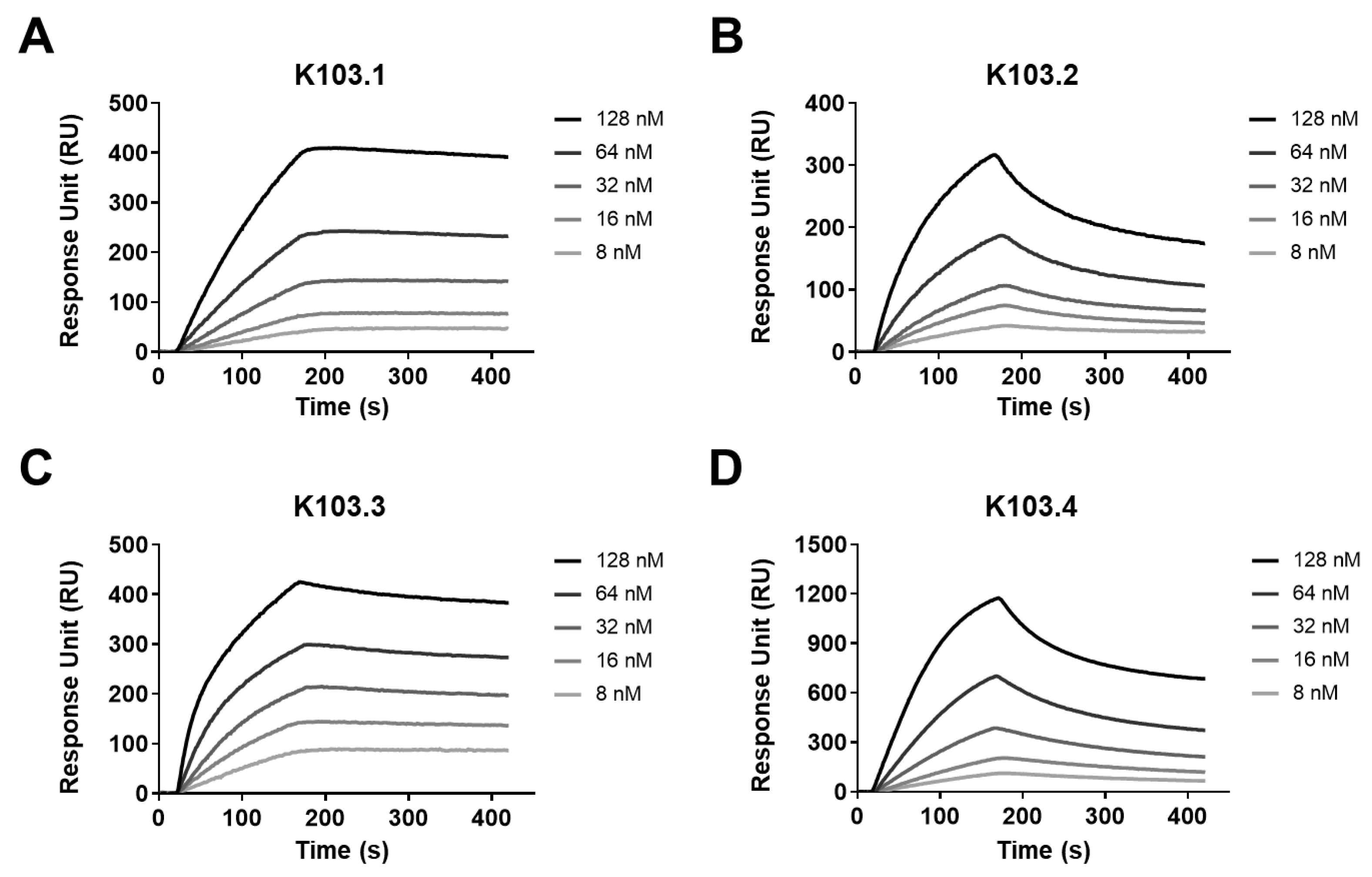
2.3. Biophysiochemical Characterization of the Selected scFv-Fc Antibodies
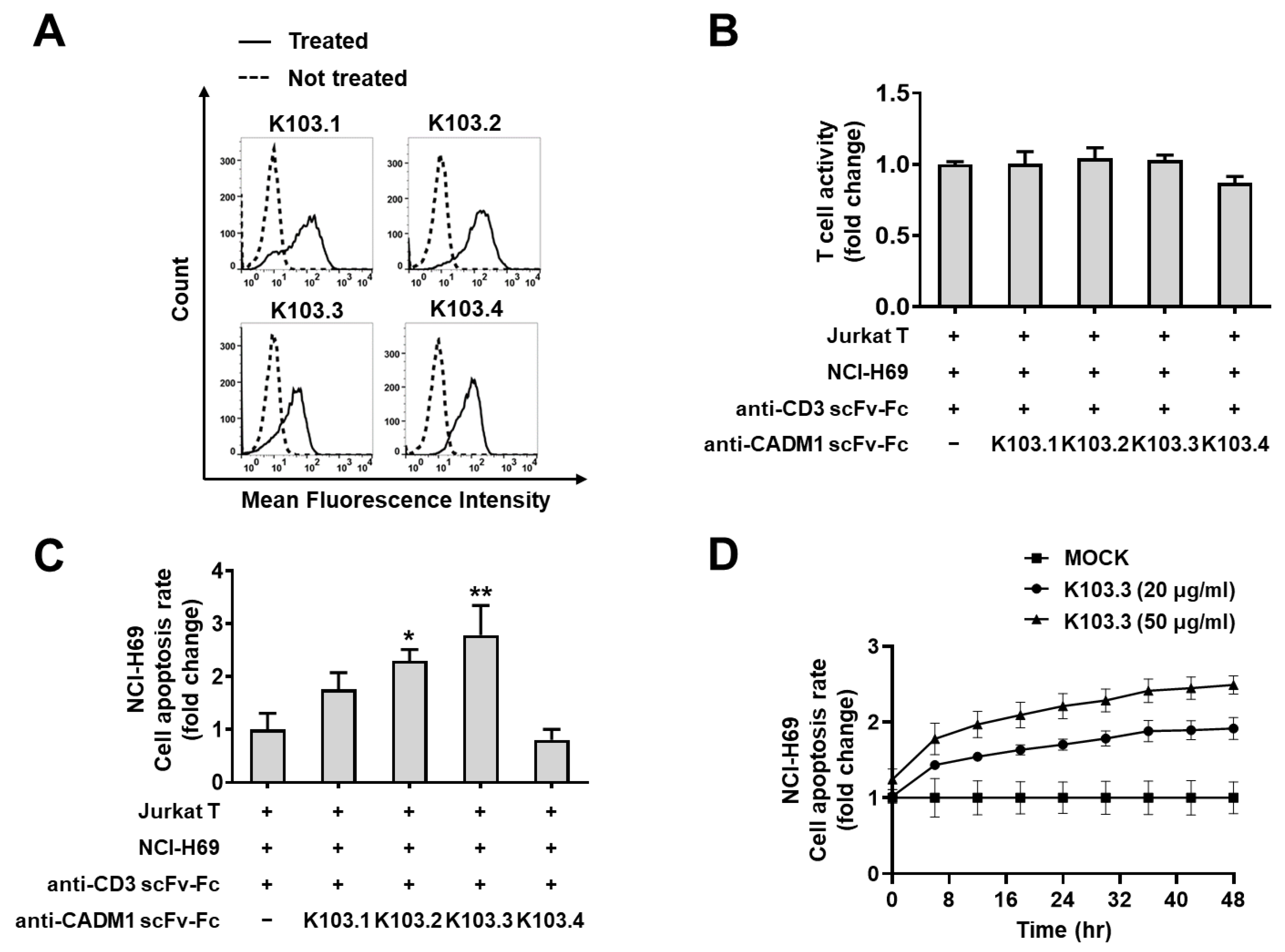
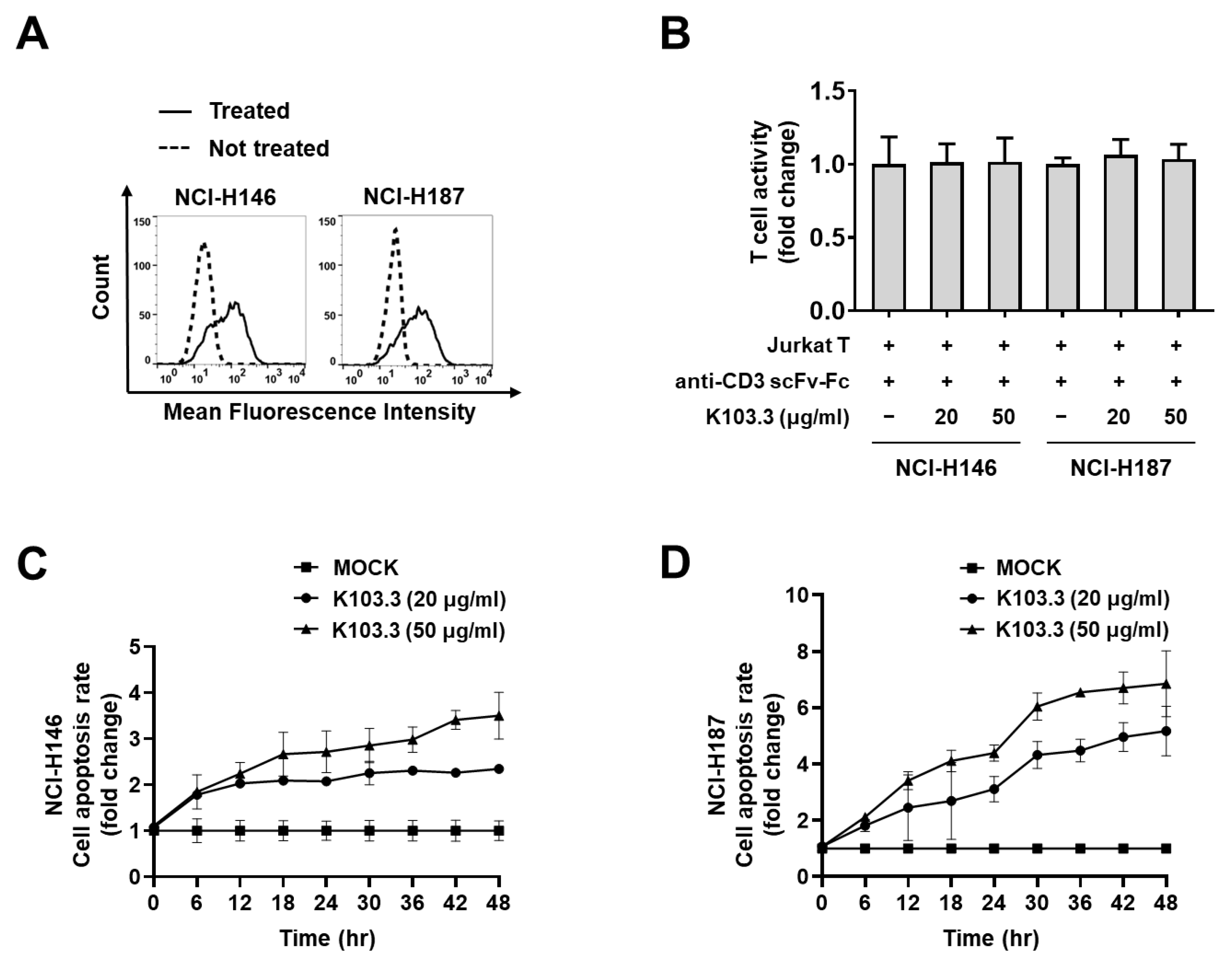
2.4. Effect of the Selected scFv-Fc Antibodies on Jurkat T Cell-Mediated SCLC Cell Death
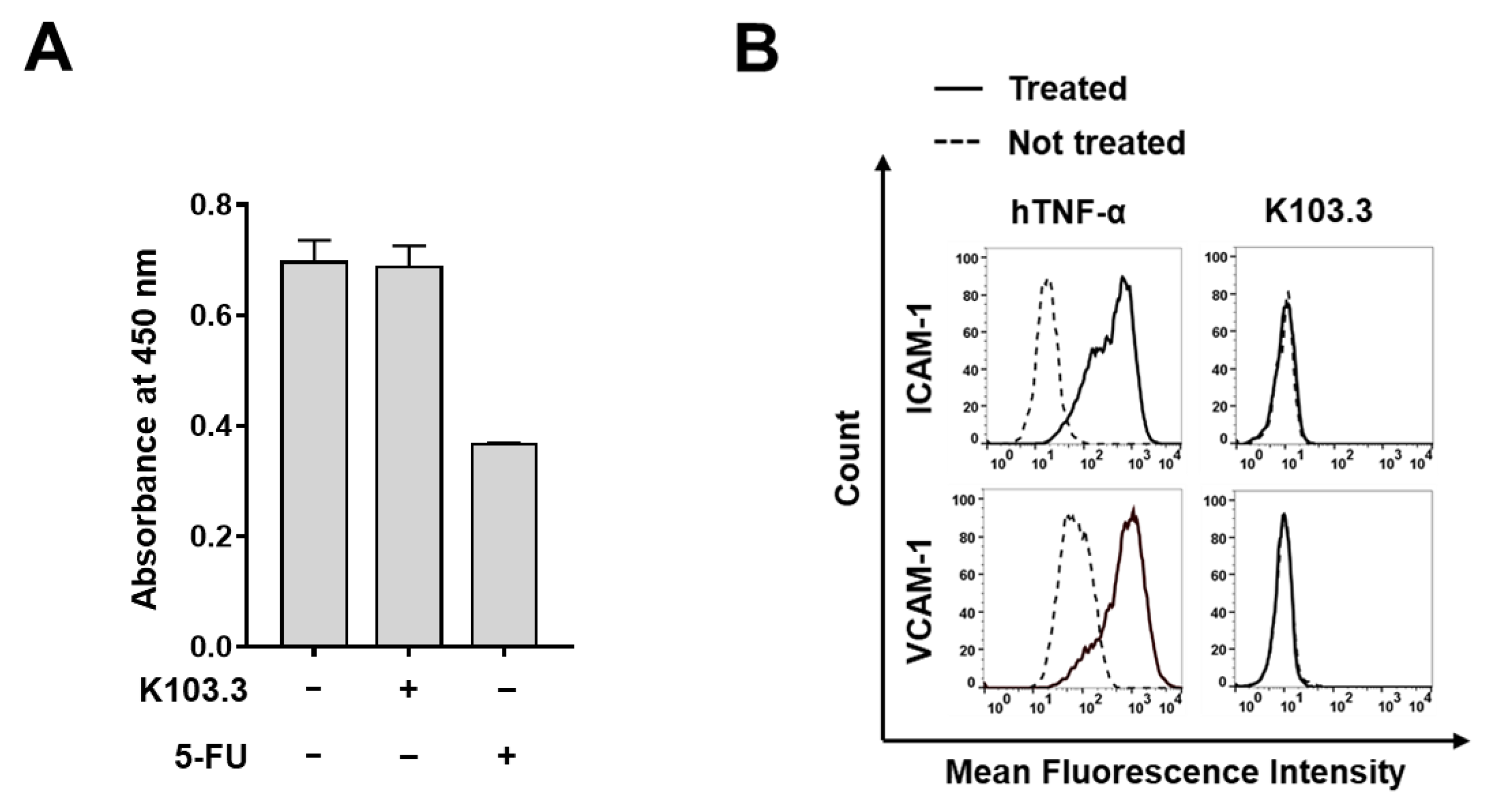
2.5. Effect of K103.3 on Endothelial Cell Toxicity
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Immunoblot Analysis
4.3. Analysis of MF-CADM1 Peptide-Conjugates and Purified Antibodies
4.4. Isolation of Fully Human scFv Antibodies Specific to MF-hCADM1 and MF-mCADM1
4.5. Phage ELISA
4.6. Generation of scFv-Fc Antibodies
4.7. ELISA
4.8. SPR
4.9. Flow Cytometry
4.10. T Cell-Mediated Tumor Cell Killing Assay
4.11. Cell Viability Assay
4.12. Statistical Methods
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Barta, J.A.; Powell, C.A.; Wisnivesky, J.P. Global Epidemiology of Lung Cancer. Ann. Glob. Health 2019, 85, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Rudin, C.M.; Brambilla, E.; Faivre-Finn, C.; Sage, J. Small-cell lung cancer. Nat. Rev. Dis. Prim. 2021, 7, 3. [Google Scholar] [CrossRef] [PubMed]
- Raso, M.G.; Bota-Rabassedas, N.; Wistuba, I.I. Pathology and Classification of SCLC. Cancers 2021, 13, 820. [Google Scholar] [CrossRef] [PubMed]
- Oronsky, B.; Reid, T.R.; Oronsky, A.; Carter, C.A. What’s New in SCLC? A Review. Neoplasia 2017, 19, 842–847. [Google Scholar] [CrossRef]
- National Organization for Rare Disorders (NORD). Small Cell Lung Cancer. Available online: https://rarediseases.org/rare-diseases/small-cell-lung-cancer/ (accessed on 2 March 2022).
- Kalemkerian, G.P. Staging and imaging of small cell lung cancer. Cancer Imaging 2012, 11, 253–258. [Google Scholar] [CrossRef]
- The American Cancer Society. What Is Lung Cancer? Available online: https://www.cancer.org/cancer/lung-cancer/about/what-is.html (accessed on 1 March 2022).
- Wang, Y.; Zou, S.; Zhao, Z.; Liu, P.; Ke, C.; Xu, S. New insights into small-cell lung cancer development and therapy. Cell Biol. Int. 2020, 44, 1564–1576. [Google Scholar] [CrossRef] [Green Version]
- Treatment Choices for Small Cell Lung Cancer, by Stage. Available online: https://www.cancer.org/cancer/lung-cancer/treating-small-cell/by-stage.html (accessed on 2 March 2022).
- Dayen, C.; Debieuvre, D.; Molinier, O.; Raffy, O.; Paganin, F.; Virally, J.; Larive, S.; Desurmont-Salasc, B.; Perrichon, M.; Martin, F.; et al. New insights into stage and prognosis in small cell lung cancer: An analysis of 968 cases. J. Thorac. Dis. 2017, 9, 5101–5111. [Google Scholar] [CrossRef] [Green Version]
- Quinteros, D.A.; Bermúdez, J.M.; Ravetti, S.; Cid, A.; Allemandi, D.A.; Palma, S.D. Therapeutic use of monoclonal antibodies: General aspects and challenges for drug delivery. In Nanostructures for Drug Delivery; Elsevier Inc.: Amsterdam, The Netherlands, 2017; pp. 807–833. [Google Scholar] [CrossRef]
- Lu, R.-M.; Hwang, Y.-C.; Liu, I.-J.; Lee, C.-C.; Tsai, H.-Z.; Li, H.-J.; Wu, H.-C. Development of therapeutic antibodies for the treatment of diseases. J. Biomed. Sci. 2020, 27, 1. [Google Scholar] [CrossRef] [PubMed]
- Zahavi, D.; Weiner, L. Monoclonal Antibodies in Cancer Therapy. Antibodies 2020, 9, 34. [Google Scholar] [CrossRef]
- Kuhn, C.; Weiner, H.L. Therapeutic anti-CD3 monoclonal antibodies: From bench to bedside. Immunotherapy 2016, 8, 889–906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaplon, H.; Chenoweth, A.; Crescioli, S.; Reichert, J.M. Antibodies to watch in 2022. MAbs 2022, 14, 2014296. [Google Scholar] [CrossRef] [PubMed]
- Mathieu, L.; Shah, S.; Pai-Scherf, L.; Larkins, E.; Vallejo, J.; Li, X.; Rodriguez, L.; Mishra-Kalyani, P.; Goldberg, K.B.; Kluetz, P.G.; et al. FDA Approval Summary: Atezolizumab and Durvalumab in Combination with Platinum-Based Chemotherapy in Extensive Stage Small Cell Lung Cancer. Oncol. 2021, 26, 433–438. [Google Scholar] [CrossRef] [PubMed]
- Regzedmaa, O.; Zhang, H.; Liu, H.; Chen, J. Immune checkpoint inhibitors for small cell lung cancer: Opportunities and challenges. OncoTargets Ther. 2019, 12, 4605–4620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horn, L.; Mansfield, A.S.; Szczęsna, A.; Havel, L.; Krzakowski, M.; Hochmair, M.J.; Huemer, F.; Losonczy, G.; Johnson, M.L.; Nishio, M.; et al. First-Line Atezolizumab plus Chemotherapy in Extensive-Stage Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 379, 2220–2229. [Google Scholar] [CrossRef]
- Paz-Ares, L.; Dvorkin, M.; Chen, Y.; Reinmuth, N.; Hotta, K.; Trukhin, D.; Statsenko, G.; Hochmair, M.J.; Özgüroğlu, M.; Ji, J.H.; et al. Durvalumab plus platinum–etoposide versus platinum–etoposide in first-line treatment of extensive-stage small-cell lung cancer (CASPIAN): A randomised, controlled, open-label, phase 3 trial. Lancet 2019, 394, 1929–1939. [Google Scholar] [CrossRef]
- van der Weyden, L.; Arends, M.J.; Rust, A.G.; Poulogiannis, G.; McIntyre, R.E.; Adams, D.J. Increased tumorigenesis associated with loss of the tumor suppressor gene Cadm1. Mol. Cancer 2012, 11, 29. [Google Scholar] [CrossRef] [Green Version]
- Hagiyama, M.; Kimura, R.; Yoneshige, A.; Inoue, T.; Otani, T.; Ito, A. Cell Adhesion Molecule 1 Contributes to Cell Survival in Crowded Epithelial Monolayers. Int. J. Mol. Sci. 2020, 21, 4123. [Google Scholar] [CrossRef]
- Hagiyama, M.; Ichiyanagi, N.; Kimura, K.B.; Murakami, Y.; Ito, A. Expression of a Soluble Isoform of Cell Adhesion Molecule 1 in the Brain and Its Involvement in Directional Neurite Outgrowth. Am. J. Pathol. 2009, 174, 2278–2289. [Google Scholar] [CrossRef] [Green Version]
- Koma, Y.; Furuno, T.; Hagiyama, M.; Hamaguchi, K.; Nakanishi, M.; Masuda, M.; Hirota, S.; Yokozaki, H.; Ito, A. Cell Adhesion Molecule 1 Is a Novel Pancreatic–Islet Cell Adhesion Molecule That Mediates Nerve–Islet Cell Interactions. Gastroenterology 2008, 134, 1544–1554. [Google Scholar] [CrossRef]
- Wakayama, T.; Iseki, S. Role of the spermatogenic–Sertoli cell interaction through cell adhesion molecule-1 (CADM1) in spermatogenesis. Anat. Sci. Int. 2009, 84, 112–121. [Google Scholar] [CrossRef] [PubMed]
- Moiseeva, E.P.; Straatman, K.R.; Leyland, M.L.; Bradding, P. CADM1 Controls Actin Cytoskeleton Assembly and Regulates Extracellular Matrix Adhesion in Human Mast Cells. PLoS ONE 2014, 9, e85980. [Google Scholar] [CrossRef] [Green Version]
- Prisco, A.R.; Hoffmann, B.R.; Kaczorowski, C.C.; McDermott-Roe, C.; Stodola, T.J.; Exner, E.C.; Greene, A.S. Tumor Necrosis Factor α Regulates Endothelial Progenitor Cell Migration via CADM1 and NF-kB. Stem Cells 2016, 34, 1922–1933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Si, X.; Xu, F.; Xu, F.; Wei, M.; Ge, Y.; Chenge, S. CADM1 inhibits ovarian cancer cell proliferation and migration by potentially regulating the PI3K/Akt/mTOR pathway. Biomed. Pharmacother. 2020, 123, 109717. [Google Scholar] [CrossRef]
- Saito, M.; Goto, A.; Abe, N.; Saito, K.; Maeda, D.; Ohtake, T.; Murakami, Y.; Takenoshita, S. Decreased expression of CADM1 and CADM4 are associated with advanced stage breast cancer. Oncol. Lett. 2018, 15, 2401–2406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.-L.; Zhou, R.; Liu, J.; Chang, Y.; Liu, S.; Wang, X.-B.; Huang, M.-F.; Zhao, Q. MicroRNA-196b inhibits late apoptosis of pancreatic cancer cells by targeting CADM1. Sci. Rep. 2017, 7, 11467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Funaki, T.; Ito, T.; Tanei, Z.-I.; Goto, A.; Niki, T.; Matsubara, D.; Murakami, Y. CADM1 promotes malignant features of small-cell lung cancer by recruiting 4.1R to the plasma membrane. Biochem. Biophys. Res. Commun. 2021, 534, 172–178. [Google Scholar] [CrossRef]
- Kikuchi, S.; Iwai, M.; Sakurai-Yageta, M.; Tsuboi, Y.; Ito, T.; Maruyama, T.; Tsuda, H.; Kanai, Y.; Onizuka, M.; Sato, Y.; et al. Expression of a splicing variant of the CADM1 specific to small cell lung cancer. Cancer Sci. 2012, 103, 1051–1057. [Google Scholar] [CrossRef]
- Nagara, Y.; Hagiyama, M.; Hatano, N.; Futai, E.; Suo, S.; Takaoka, Y.; Murakami, Y.; Ito, A.; Ishiura, S. Tumor suppressor cell adhesion molecule 1 (CADM1) is cleaved by a disintegrin and metalloprotease 10 (ADAM10) and subsequently cleaved by γ-secretase complex. Biochem. Biophys. Res. Commun. 2012, 417, 462–467. [Google Scholar] [CrossRef]
- Byers, L.A.; Rudin, C.M. Small cell lung cancer: Where do we go from here? Cancer 2015, 121, 664–672. [Google Scholar] [CrossRef]
- Guo, H.; Li, L.; Cui, J. Advances and challenges in immunotherapy of small cell lung cancer. Chin. J. Cancer Res. 2020, 32, 115–128. [Google Scholar] [CrossRef]
- Redman, J.M.; Hill, E.M.; AlDeghaither, D.; Weiner, L.M. Mechanisms of action of therapeutic antibodies for cancer. Mol. Immunol. 2015, 67, 28–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanneman, M.; Dranoff, G. Combining immunotherapy and targeted therapies in cancer treatment. Nat. Cancer 2012, 12, 237–251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castelli, M.S.; Mcgonigle, P.; Hornby, P.J. The pharmacology and therapeutic applications of monoclonal antibodies. Pharmacol. Res. Perspect. 2019, 7, e00535. [Google Scholar] [CrossRef] [PubMed]
- Waldman, A.D.; Fritz, J.M.; Lenardo, M.J. A guide to cancer immunotherapy: From T cell basic science to clinical practice. Nat. Rev. Immunol. 2020, 20, 651–668. [Google Scholar] [CrossRef] [PubMed]
- Hombach, A.; Köhler, H.; Rappl, G.; Abken, H. Human CD4+ T Cells Lyse Target Cells via Granzyme/Perforin upon Circumvention of MHC Class II Restriction by an Antibody-Like Immunoreceptor. J. Immunol. 2006, 177, 5668–5675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ki, M.K.; Jeoung, M.H.; Choi, J.R.; Rho, S.-S.; Kwon, Y.-G.; Shim, H.; Chung, J.; Hong, H.J.; Song, B.D.; Lee, S. Human antibodies targeting the C-type lectin-like domain of the tumor endothelial cell marker clec14a regulate angiogenic properties in vitro. Oncogene 2013, 32, 5449–5457. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibody (scFv-Fc) | KD (M) | Kon (1/M∙s) | Koff (1/s) |
|---|---|---|---|
| K103.1 | 4.49 × 10−9 | 4.63 × 104 | 2.07 × 10−4 |
| K103.2 | 3.91 × 10−8 | 5.04 × 104 | 1.97 × 10−3 |
| K103.3 | 2.25 × 10−9 | 1.38 × 105 | 3.11 × 10−4 |
| K103.4 | 4.12 × 10−8 | 4.59 × 104 | 1.89 × 10−3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, J.H.; Kim, J.W.; Yang, H.R.; Song, S.-W.; Lee, S.-J.; Jeon, Y.; Ju, A.; Lee, N.; Kim, M.-G.; Kim, M.; et al. A Fully-Human Antibody Specifically Targeting a Membrane-Bound Fragment of CADM1 Potentiates the T Cell-Mediated Death of Human Small-Cell Lung Cancer Cells. Int. J. Mol. Sci. 2022, 23, 6895. https://doi.org/10.3390/ijms23136895
Lee JH, Kim JW, Yang HR, Song S-W, Lee S-J, Jeon Y, Ju A, Lee N, Kim M-G, Kim M, et al. A Fully-Human Antibody Specifically Targeting a Membrane-Bound Fragment of CADM1 Potentiates the T Cell-Mediated Death of Human Small-Cell Lung Cancer Cells. International Journal of Molecular Sciences. 2022; 23(13):6895. https://doi.org/10.3390/ijms23136895
Chicago/Turabian StyleLee, Ji Hyun, Ji Woong Kim, Ha Rim Yang, Seong-Won Song, Song-Jae Lee, Yeongha Jeon, Anna Ju, Narim Lee, Min-Gu Kim, Minjoo Kim, and et al. 2022. "A Fully-Human Antibody Specifically Targeting a Membrane-Bound Fragment of CADM1 Potentiates the T Cell-Mediated Death of Human Small-Cell Lung Cancer Cells" International Journal of Molecular Sciences 23, no. 13: 6895. https://doi.org/10.3390/ijms23136895
APA StyleLee, J. H., Kim, J. W., Yang, H. R., Song, S.-W., Lee, S.-J., Jeon, Y., Ju, A., Lee, N., Kim, M.-G., Kim, M., Hwang, K., Yoon, J. H., Shim, H., & Lee, S. (2022). A Fully-Human Antibody Specifically Targeting a Membrane-Bound Fragment of CADM1 Potentiates the T Cell-Mediated Death of Human Small-Cell Lung Cancer Cells. International Journal of Molecular Sciences, 23(13), 6895. https://doi.org/10.3390/ijms23136895