
Bioenergetic Status of the Intestinal and Hepatic Cells after Short Term Exposure to Fumonisin B1 and Aflatoxin B1




Abstract
:1. Introduction
2. Results
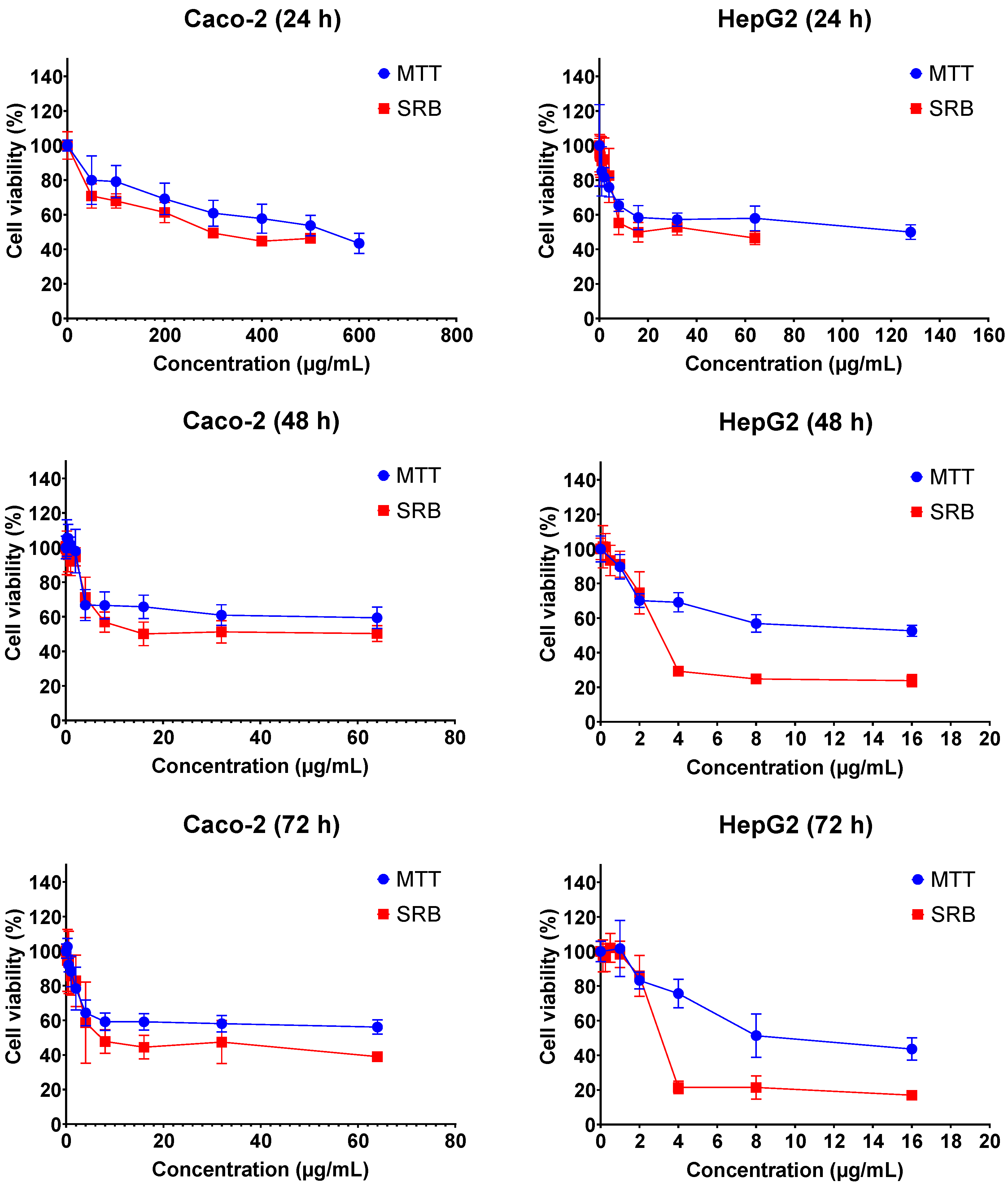
2.1. Cytotoxicity of FB1 and AFB1 on Caco-2 and HepG2 upon Different Exposure Times
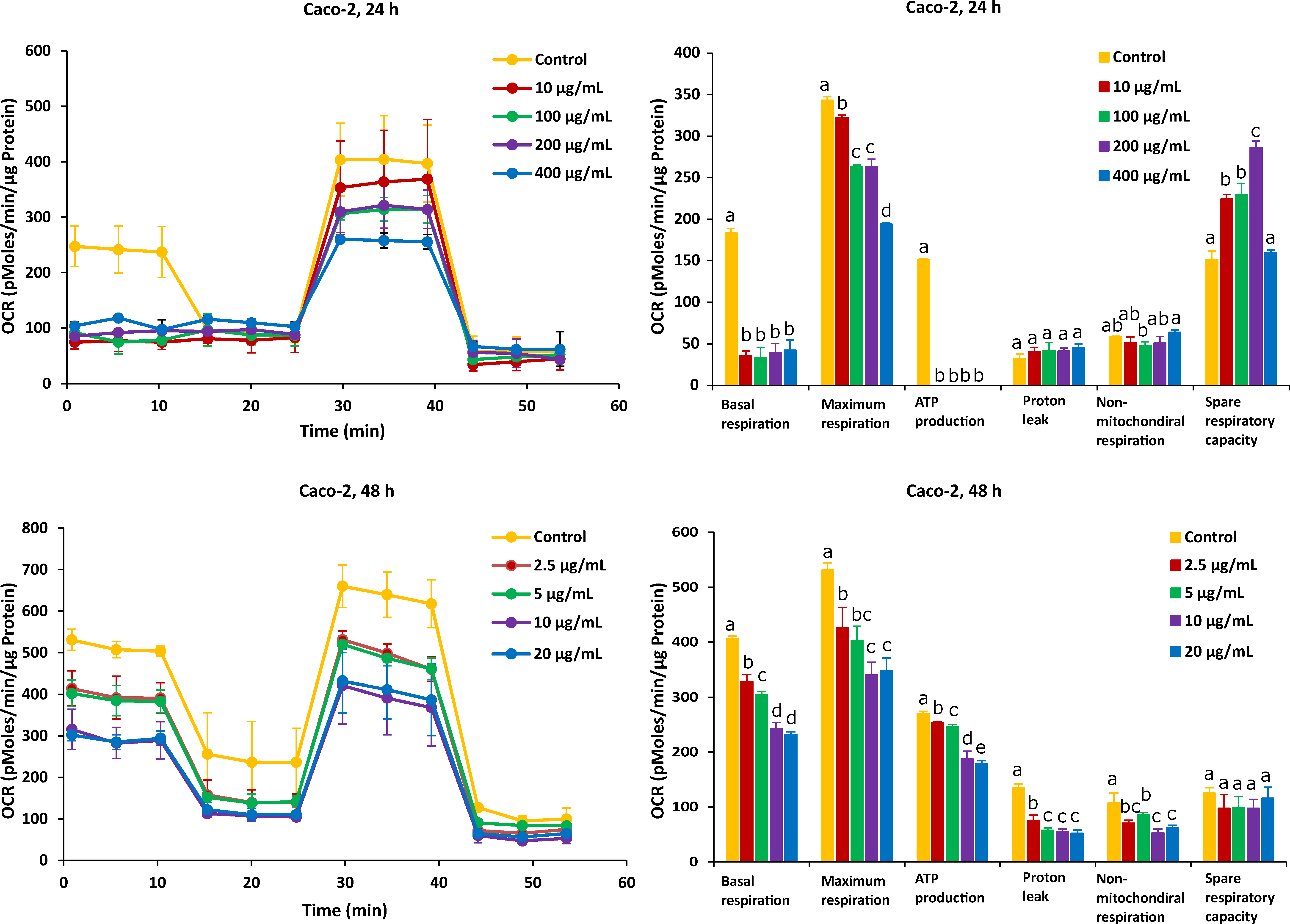
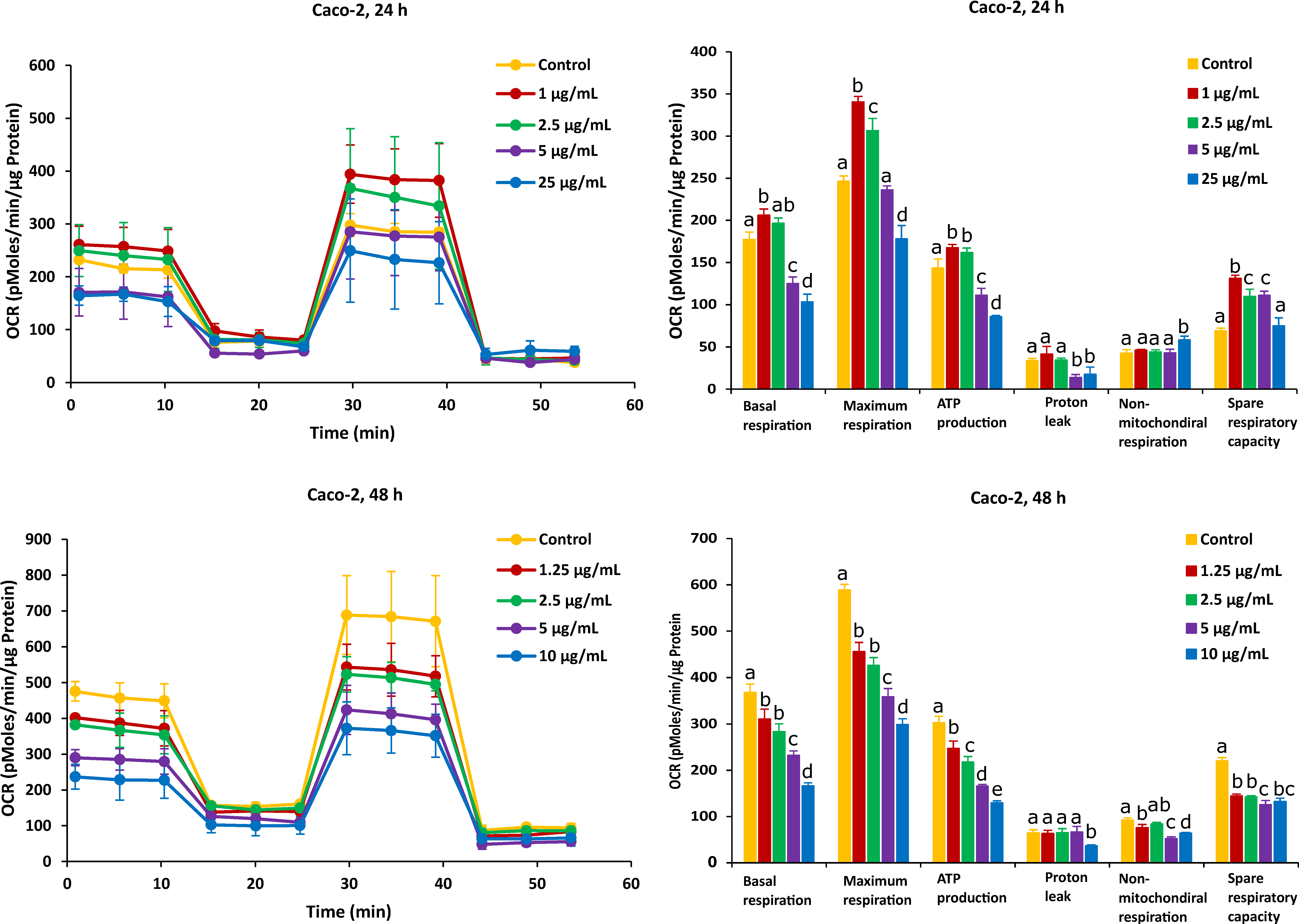
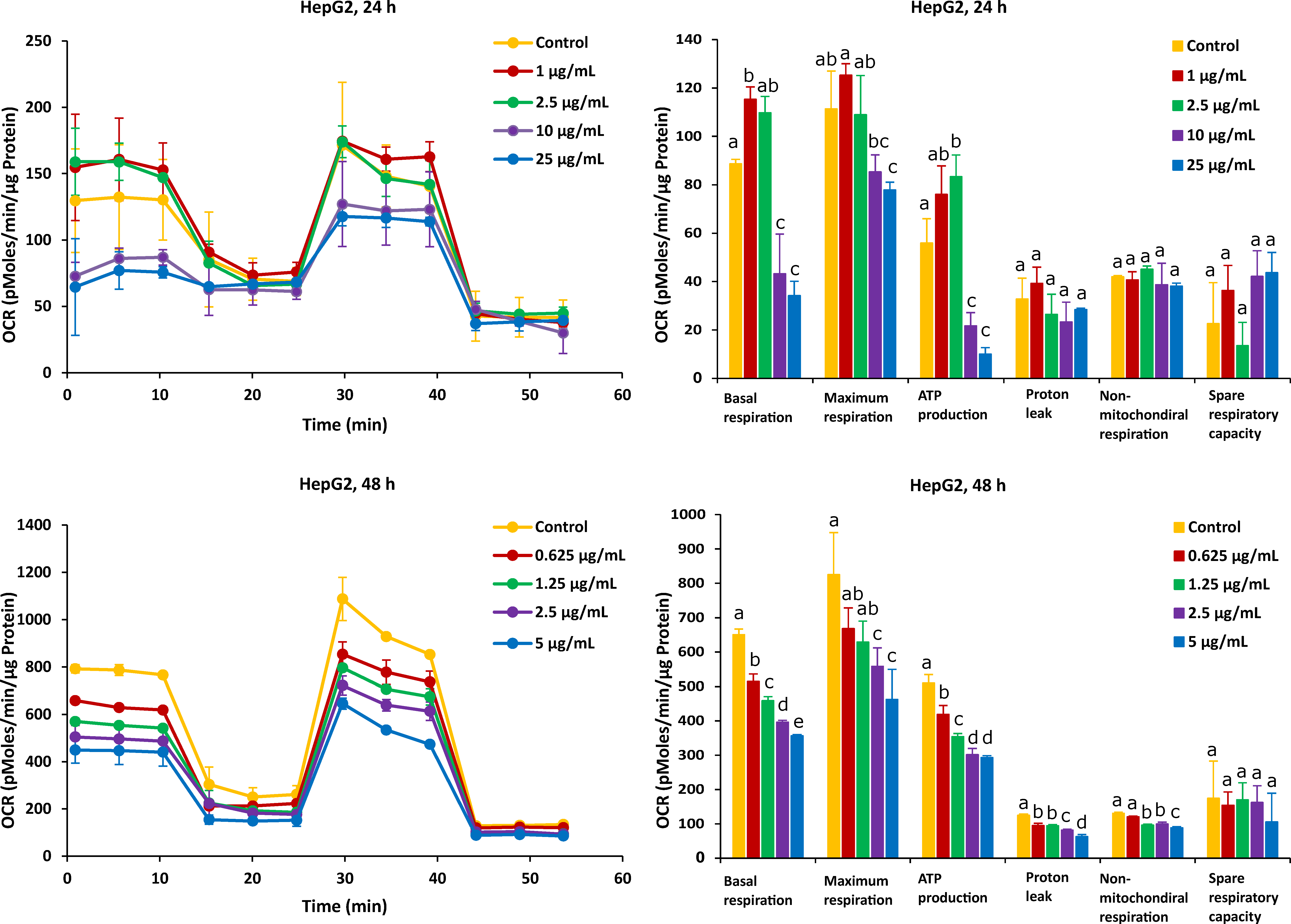
2.2. Different Time Effects of FB1 and AFB1 on Mitochondrial Respiration of Caco-2 and HepG2 Cells
2.3. ROS Production and MMP
3. Discussion
4. Materials and Methods
4.1. Chemical Reagents
4.2. Cell Culture Materials
4.3. Cell Viability and Protein Content Assays
4.4. Seahorse Extra-Cellular Flux Analysis of Mitochondrial Respiration
4.5. Cytotoxicity Endpoint Measurement
4.6. Data Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Alshannaq, A.; Yu, J.H. Occurrence, Toxicity, and Analysis of Major Mycotoxins in Food. Int. J. Environ. Res. Public Health 2017, 14, 632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moretti, A.; Logrieco, A.F.; Susca, A. Mycotoxins: An Underhand Food Problem. Methods Mol. Biol. 2017, 1542, 3–12. [Google Scholar] [PubMed]
- Eskola, M.; Kos, G.; Elliott, C.T.; Hajšlová, J.; Mayar, S.; Krska, R. Worldwide Contamination of Food-Crops with Mycotoxins: Validity of the Widely Cited ‘FAO Estimate’ of 25%. Crit. Rev. Food Sci. Nutr. 2020, 60, 2773–2789. [Google Scholar] [CrossRef] [PubMed]
- Ji, F.; He, D.; Olaniran, A.O.; Mokoena, M.P.; Xu, J.; Shi, J. Occurrence, Toxicity, Production and Detection of Fusarium Mycotoxin: A Review. Food Prod. Process. Nutr. 2019, 1, 6. [Google Scholar] [CrossRef]
- Rheeder, J.P.; Marasas, W.F.O.; Vismer, H.F. Production of Fumonisin Analogs by Fusarium Species. Appl. Environ. Microbiol. 2002, 68, 2101–2105. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Landschoot, S.; Detavernier, C.; De Saeger, S.; Rajkovic, A.; Audenaert, K. Cross-Talk between Fusarium Verticillioides and Aspergillus Flavus in Vitro and in Planta. Mycotoxin Res. 2021, 37, 229–240. [Google Scholar] [CrossRef]
- Amaike, S.; Keller, N.P. Aspergillus Flavus. Annu. Rev. Phytopathol. 2011, 49, 107–133. [Google Scholar] [CrossRef]
- Scott, P.M. Recent Research on Fumonisins: A Review. Food Addit. Contam. Part A Chem. Anal. Control. Expo. Risk Assess. 2012, 29, 242–248. [Google Scholar] [CrossRef]
- Stockmann-Juvala, H.; Savolainen, K. A Review of the Toxic Effects and Mechanisms of Action of Fumonisin B 1. Hum. Exp. Toxicol. 2008, 27, 799–809. [Google Scholar] [CrossRef]
- Abrar, M.; Anjum, F.M.; Butt, M.S.; Pasha, I.; Randhawa, M.A.; Saeed, F.; Waqas, K. Aflatoxins: Biosynthesis, Occurrence, Toxicity, and Remedies. Crit. Rev. Food Sci. Nutr. 2013, 53, 862–874. [Google Scholar] [CrossRef]
- Ismail, A.; Gonçalves, B.L.; de Neeff, D.V.; Ponzilacqua, B.; Coppa, C.F.S.C.; Hintzsche, H.; Sajid, M.; Cruz, A.G.; Corassin, C.H.; Oliveira, C.A.F. Aflatoxin in Foodstuffs: Occurrence and Recent Advances in Decontamination. Food Res. Int. 2018, 113, 74–85. [Google Scholar] [CrossRef] [PubMed]
- Demissie, N. A Review of Aflatoxin: Occurrence, Prevention, and Gaps in Both Food and Feed Safety. Nov. Tech. Nutr. Food Sci. 2018, 1, 1–35. [Google Scholar] [CrossRef]
- Lumsangkul, C.; Chiang, H.I.; Lo, N.W.; Fan, Y.K.; Ju, J.C. Developmental Toxicity of Mycotoxin Fumonisin B 1 in Animal Embryogenesis: An Overview. Toxins 2019, 11, 114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voss, K.A.; Smith, G.W.; Haschek, W.M. Fumonisins: Toxicokinetics, Mechanism of Action and Toxicity. Anim. Feed Sci. Technol. 2007, 137, 299–325. [Google Scholar] [CrossRef]
- Chen, J.; Wen, J.; Tang, Y.; Shi, J.; Mu, G.; Yan, R.; Cai, J.; Long, M. Research Progress on Fumonisin B1 Contamination and Toxicity: A Review. Molecules 2021, 26, 5238. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Safety Evaluation of Certain Food Additives and Contaminants: Prepared by the Seventy Fourth Meeting of the Joint FAO/WHO Expert Committee on Food Additives (JECFA); FAO: Rome, Italy, 2012; pp. 605–684. [Google Scholar]
- Abdul, N.S.; Chuturgoon, A.A. Fumonisin B1 Regulates LDL Receptor and ABCA1 Expression in an LXR Dependent Mechanism in Liver (HepG2) Cells. Toxicon 2021, 190, 58–64. [Google Scholar] [CrossRef]
- Kouadio, J.H.; Mobio, T.A.; Baudrimont, I.; Moukha, S.; Dano, S.D.; Creppy, E.E. Comparative Study of Cytotoxicity and Oxidative Stress Induced by Deoxynivalenol, Zearalenone or Fumonisin B1 in Human Intestinal Cell Line Caco-2. Toxicology 2005, 213, 56–65. [Google Scholar] [CrossRef]
- Arumugam, T.; Pillay, Y.; Ghazi, T.; Nagiah, S.; Abdul, N.S.; Chuturgoon, A.A. Fumonisin B 1 -Induced Oxidative Stress Triggers Nrf2-Mediated Antioxidant Response in Human Hepatocellular Carcinoma (HepG2) Cells. Mycotoxin Res. 2019, 35, 99–109. [Google Scholar] [CrossRef]
- Singh, M.P.; Kang, S.C. Endoplasmic Reticulum Stress-Mediated Autophagy Activation Attenuates Fumonisin B1 Induced Hepatotoxicity in Vitro and in Vivo. Food Chem. Toxicol. 2017, 110, 371–382. [Google Scholar] [CrossRef]
- Stockmann-Juvala, H.; Mikkola, J.; Naarala, J.; Loikkanen, J.; Elovaara, E.; Savolainen, K. Oxidative Stress Induced by Fumonisin B1 in Continuous Human and Rodent Neural Cell Cultures. Free Radic. Res. 2004, 38, 933–942. [Google Scholar] [CrossRef]
- Domijan, A.M. Fumonisin B1: A Neurotoxic Mycotoxin. Arh. Hig. Rada Toksikol. 2012, 63, 531–544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marí, M.; Morales, A.; Colell, A.; García-Ruiz, C.; Fernández-Checa, J.C. Mitochondrial Glutathione, a Key Survival Antioxidant. Antioxid. Redox Signal. 2009, 11, 2685–2700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Domijan, A.M.; Abramov, A.Y. Fumonisin B1 Inhibits Mitochondrial Respiration and Deregulates Calcium Homeostasis—Implication to Mechanism of Cell Toxicity. Int. J. Biochem. Cell Biol. 2011, 43, 897–904. [Google Scholar] [CrossRef] [PubMed]
- Mary, V.S.; Theumer, M.G.; Arias, S.L.; Rubinstein, H.R. Reactive Oxygen Species Sources and Biomolecular Oxidative Damage Induced by Aflatoxin B1 and Fumonisin B1 in Rat Spleen Mononuclear Cells. Toxicology 2012, 302, 299–307. [Google Scholar] [CrossRef] [PubMed]
- Antonissen, G.; Devreese, M.; De Baere, S.; Martel, A.; Van Immerseel, F.; Croubels, S. Impact of Fusarium Mycotoxins on Hepatic and Intestinal MRNA Expression of Cytochrome P450 Enzymes and Drug Transporters, and on the Pharmacokinetics of Oral Enrofloxacin in Broiler Chickens. Food Chem. Toxicol. 2017, 101, 75–83. [Google Scholar] [CrossRef]
- Kim, S.H.; Singh, M.P.; Sharma, C.; Kang, S.C. Fumonisin B1 Actuates Oxidative Stress-Associated Colonic Damage via Apoptosis and Autophagy Activation in Murine Model. J. Biochem. Mol. Toxicol. 2018, 32, e22161. [Google Scholar] [CrossRef]
- Meissonnier, G.M.; Pinton, P.; Laffitte, J.; Cossalter, A.M.; Gong, Y.Y.; Wild, C.P.; Bertin, G.; Galtier, P.; Oswald, I.P. Immunotoxicity of Aflatoxin B1: Impairment of the Cell-Mediated Response to Vaccine Antigen and Modulation of Cytokine Expression. Toxicol. Appl. Pharmacol. 2008, 231, 142–149. [Google Scholar] [CrossRef]
- Desaulniers, D.; Cummings-Lorbetskie, C.; Leingartner, K.; Xiao, G.H.; Zhou, G.; Parfett, C. Effects of Vanadium (Sodium Metavanadate) and Aflatoxin-B1 on Cytochrome P450 Activities, DNA Damage and DNA Methylation in Human Liver Cell Lines. Toxicol. In Vitro 2021, 70, 105036. [Google Scholar] [CrossRef]
- Zhang, J.; Zheng, N.; Liu, J.; Li, F.D.; Li, S.L.; Wang, J.Q. Aflatoxin B1 and Aflatoxin M1 Induced Cytotoxicity and DNA Damage in Differentiated and Undifferentiated Caco-2 Cells. Food Chem. Toxicol. 2015, 83, 54–60. [Google Scholar] [CrossRef]
- Erdélyi, M.; Balogh, K.; Pelyhe, C.; Kövesi, B.; Nakade, M.; Zándoki, E.; Mézes, M.; Kovács, B. Changes in the Regulation and Activity of Glutathione Redox System, and Lipid Peroxidation Processes in Short-Term Aflatoxin B1 Exposure in Liver of Laying Hens. J. Anim. Physiol. Anim. Nutr. 2018, 102, 947–952. [Google Scholar] [CrossRef]
- Schieber, M.; Chandel, N.S. ROS Function in Redox Signaling and Oxidative Stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.; Gan, Z.; Zhuo, R.; Zhang, L.; Wang, T.; Zhong, X. Resveratrol Attenuates Aflatoxin B1-Induced Ros Formation and Increase of M6a Rna Methylation. Animals 2020, 10, 677. [Google Scholar] [CrossRef] [PubMed]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial ROS-Induced ROS Release: An Update and Review. Biochim. Biophys. Acta Bioenerg. 2006, 1757, 509–517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Wang, W. Aflatoxin B1 Impairs Mitochondrial Functions, Activates ROS Generation, Induces Apoptosis and Involves Nrf2 Signal Pathway in Primary Broiler Hepatocytes. Anim. Sci. J. 2016, 87, 1490–1500. [Google Scholar] [CrossRef] [PubMed]
- Ge, J.; Yu, H.; Li, J.; Lian, Z.; Zhang, H.; Fang, H.; Qian, L. Assessment of Aflatoxin B1 Myocardial Toxicity in Rats: Mitochondrial Damage and Cellular Apoptosis in Cardiomyocytes Induced by Aflatoxin B1. J. Int. Med. Res. 2017, 45, 1015–1023. [Google Scholar] [CrossRef] [Green Version]
- Ganan, M.; Collins, M.; Rastall, R.; Hotchkiss, A.T.; Chau, H.K.; Carrascosa, A.V.; Martinez-Rodriguez, A.J. Inhibition by Pectic Oligosaccharides of the Invasion of Undifferentiated and Differentiated Caco-2 Cells by Campylobacter Jejuni. Int. J. Food Microbiol. 2010, 137, 181–185. [Google Scholar] [CrossRef]
- Ude, V.C.; Brown, D.M.; Viale, L.; Kanase, N.; Stone, V.; Johnston, H.J. Impact of Copper Oxide Nanomaterials on Differentiated and Undifferentiated Caco-2 Intestinal Epithelial Cells; Assessment of Cytotoxicity, Barrier Integrity, Cytokine Production and Nanomaterial Penetration. Part. Fibre Toxicol. 2017, 14, 31. [Google Scholar] [CrossRef]
- Lenaerts, K.; Bouwman, F.K.; Lamers, W.H.; Renes, J.; Mariman, E.C. Comparative Proteomic Analysis of Cell Lines and Scrapings of the Human Intestinal Epithelium. BMC Genom. 2007, 8, 91. [Google Scholar] [CrossRef] [Green Version]
- Haselsberger, K.; Peterson, D.C.; Thomas, D.; Darling, J.L. Assay of Anticancer Drugs in Tissue Culture: Comparison of a Tetrazolium-Based Assay and a Protein Binding Dye Assay in Short-Term Cultures Derived from Human Malignant Glioma. Anticancer Drugs 1996, 7, 331–338. [Google Scholar] [CrossRef]
- Wentzel, J.F.; Lombard, M.J.; Du Plessis, L.H.; Zandberg, L. Evaluation of the Cytotoxic Properties, Gene Expression Profiles and Secondary Signalling Responses of Cultured Cells Exposed to Fumonisin B1, Deoxynivalenol and Zearalenone Mycotoxins. Arch. Toxicol. 2017, 91, 2265–2282. [Google Scholar] [CrossRef]
- Cetin, Y.; Bullerman, L.B. Cytotoxicity of Fusarium Mycotoxins to Mammalian Cell Cultures as Determined by the MTT Bioassay. Food Chem. Toxicol. 2005, 43, 755–764. [Google Scholar] [CrossRef] [PubMed]
- Du, M.; Liu, Y.; Zhang, G. Interaction of Aflatoxin B1 and Fumonisin B1 in HepG2 Cell Apoptosis. Food Biosci. 2017, 20, 131–140. [Google Scholar] [CrossRef]
- Ji, J.; Wang, Q.; Wu, H.; Xia, S.; Guo, H.; Blaženović, I.; Zhang, Y.; Sun, X. Insights into Cellular Metabolic Pathways of the Combined Toxicity Responses of Caco-2 Cells Exposed to Deoxynivalenol, Zearalenone and Aflatoxin B1. Food Chem. Toxicol. 2019, 126, 106–112. [Google Scholar] [CrossRef] [PubMed]
- Corcuera, L.A.; Arbillaga, L.; Vettorazzi, A.; Azqueta, A.; López de Cerain, A. Ochratoxin A Reduces Aflatoxin B1 Induced DNA Damage Detected by the Comet Assay in Hep G2 Cells. Food Chem. Toxicol. 2011, 49, 2883–2889. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Du, M.; Zhang, G. Proapoptotic Activity of Aflatoxin B1 and Sterigmatocystin in HepG2 Cells. Toxicol. Rep. 2014, 1, 1076–1086. [Google Scholar] [CrossRef] [Green Version]
- Fulda, S.; Gorman, A.M.; Hori, O.; Samali, A. Cellular Stress Responses: Cell Survival and Cell Death. Int. J. Cell Biol. 2010, 2010, 214074. [Google Scholar] [CrossRef] [Green Version]
- Vakifahmetoglu-Norberg, H.; Ouchida, A.T.; Norberg, E. The Role of Mitochondria in Metabolism and Cell Death. Biochem. Biophys. Res. Commun. 2017, 482, 426–431. [Google Scholar] [CrossRef]
- Bionda, C.; Portoukalian, J.; Schmitt, D.; Rodriguez-Lafrasse, C.; Ardail, D. Subcellular Compartmentalization of Ceramide Metabolism: MAM (Mitochondria-Associated Membrane) and/or Mitochondria? Biochem. J. 2004, 382, 527–533. [Google Scholar] [CrossRef] [Green Version]
- Dmitriev, R.I.; Papkovsky, D.B. Optical Probes and Techniques for O2 Measurement in Live Cells and Tissue. Cell. Mol. Life Sci. 2012, 69, 2025–2039. [Google Scholar] [CrossRef] [Green Version]
- Brand, M.D.; Nicholls, D.G. Assessing Mitochondrial Dysfunction in Cells. Biochem. J. 2011, 435, 297–312. [Google Scholar] [CrossRef] [Green Version]
- Decleer, M.; Jovanovic, J.; Vakula, A.; Udovicki, B.; Agoua, R.-S.S.E.K.; Madder, A.; De Saeger, S.; Rajkovic, A. Oxygen Consumption Rate Analysis of Mitochondrial Dysfunction Caused by Bacillus Cereus Cereulide in Caco-2 and HepG2 Cells. Toxins 2018, 10, 266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loiseau, D.; Morvan, D.; Chevrollier, A.; Demidem, A.; Douay, O.; Reynier, P.; Stepien, G. Mitochondrial Bioenergetic Background Confers a Survival Advantage to HepG2 Cells in Response to Chemotherapy. Mol. Carcinog. 2009, 48, 733–741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jastroch, M.; Divakaruni, A.S.; Mookerjee, S.; Treberg, J.R.; Brand, M.D. Mitochondrial Proton and Electron Leaks. Essays Biochem. 2010, 47, 53–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marchetti, P.; Fovez, Q.; Germain, N.; Khamari, R.; Kluza, J. Mitochondrial Spare Respiratory Capacity: Mechanisms, Regulation, and Significance in Non-Transformed and Cancer Cells. FASEB J. 2020, 34, 13106–13124. [Google Scholar] [CrossRef]
- Wan, X.L.; Li, N.; Chen, Y.J.; Chen, X.S.; Yang, Z.; Xu, L.; Yang, H.M.; Wang, Z.Y. Protective Effects of Lycopene on Mitochondrial Oxidative Injury and Dysfunction in the Liver of Aflatoxin B1-Exposed Broilers. Poult. Sci. 2021, 100, 101441. [Google Scholar] [CrossRef]
- Shi, D.; Guo, S.; Liao, S.; Su, R.; Guo, M.; Liu, N.; Li, P.; Tang, Z. Protection of Selenium on Hepatic Mitochondrial Respiratory Control Ratio and Respiratory Chain Complex Activities in Ducklings Intoxicated with Aflatoxin B1. Biol. Trace Elem. Res. 2012, 145, 312–317. [Google Scholar] [CrossRef]
- Xu, F.; Li, Y.; Cao, Z.; Zhang, J.; Huang, W. AFB1-Induced Mice Liver Injury Involves Mitochondrial Dysfunction Mediated by Mitochondrial Biogenesis Inhibition. Ecotoxicol. Environ. Saf. 2021, 216, 112213. [Google Scholar] [CrossRef]
- Guerra, M.C.; Galvano, F.; Bonsi, L.; Speroni, E.; Costa, S.; Renzulli, C.; Cervellati, R. Cyanidin-3- O -β-Glucopyranoside, a Natural Free-Radical Scavenger against Aflatoxin B1- and Ochratoxin A-Induced Cell Damage in a Human Hepatoma Cell Line (Hep G2) and a Human Colonic Adenocarcinoma Cell Line (CaCo-2). Br. J. Nutr. 2005, 94, 211–220. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Maki, C.R.; Deng, Y.; Tian, Y.; Phillips, T.D. Development of High Capacity Enterosorbents for Aflatoxin B1 and Other Hazardous Chemicals. Chem. Res. Toxicol. 2017, 30, 1694–1701. [Google Scholar] [CrossRef]
- Ramsey, J.J.; Hagopian, K.; Kenny, T.M.; Koomson, E.K.; Bevilacqua, L.; Weindruch, R.; Harper, M.E. Proton Leak and Hydrogen Peroxide Production in Liver Mitochondria from Energy-Restricted Rats. Am. J. Physiol. Endocrinol. Metab. 2004, 286, 31–40. [Google Scholar] [CrossRef] [Green Version]
- Hill, B.G.; Benavides, G.A.; Lancaster, J.J.R.; Ballinger, S.; Dell’Italia, L.; Zhang, J.; Darley-Usmar, V.M. Integration of Cellular Bioenergetics with Mitochondrial Quality Control and Autophagy. Biol. Chem. 2012, 393, 1485–1512. [Google Scholar] [CrossRef] [Green Version]
- Marin, D.E.; Taranu, I. Overview on Aflatoxins and Oxidative Stress. Toxin Rev. 2012, 31, 32–43. [Google Scholar] [CrossRef]
- Zorova, L.D.; Popkov, V.A.; Plotnikov, E.Y.; Silachev, D.N.; Pevzner, I.B.; Jankauskas, S.S.; Babenko, V.A.; Zorov, S.D.; Balakireva, A.V.; Juhaszova, M.; et al. Mitochondrial Membrane Potential. Anal. Biochem. 2018, 552, 50–59. [Google Scholar] [CrossRef] [PubMed]
- Bayir, H. Reactive Oxygen Species. Crit. Care Med. 2005, 33, S498–S501. [Google Scholar] [CrossRef] [PubMed]
- Feissner, R.F.; Skalska, J.; Gaum, W.E.; Sheu, S.S. Crosstalk Signaling between Mitochondrial Ca2+ and ROS. Front. Biosci. 2009, 14, 1197–1218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheik Abdul, N.; Marnewick, J.L. Fumonisin B1-Induced Mitochondrial Toxicity and Hepatoprotective Potential of Rooibos: An Update. J. Appl. Toxicol. 2020, 40, 1602–1613. [Google Scholar] [CrossRef]
- Nicholls, D.G. Mitochondrial Membrane Potential and Aging. Aging Cell 2004, 3, 35–40. [Google Scholar] [CrossRef]
- Ly, J.D.; Grubb, D.R.; Lawen, A. The Mitochondrial Membrane Potential (Δψm) in Apoptosis; an Update. Apoptosis 2003, 8, 115–128. [Google Scholar] [CrossRef]
- Brookes, P.S.; Yoon, Y.; Robotham, J.L.; Anders, M.W.; Sheu, S.S. Calcium, ATP, and ROS: A Mitochondrial Love-Hate Triangle. Am. J. Physiol. Cell Physiol. 2004, 287, C817–C833. [Google Scholar] [CrossRef]
- Twarużek, M.; Zastempowska, E.; Soszczyńska, E.; Ałtyn, I. The Use of in Vitro Assays for the Assessment of Cytotoxicity on the Example of MTT Test. Folia Biol. Oecol. 2018, 14, 23–32. [Google Scholar] [CrossRef] [Green Version]
- Orellana, E.; Kasinski, A. Sulforhodamine B (SRB) Assay in Cell Culture to Investigate Cell Proliferation. Bio-Protocol 2016, 6, e1984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vissenaekens, H.; Grootaert, C.; Rajkovic, A.; Van De Wiele, T.; Calatayud, M. The Response of Five Intestinal Cell Lines to Anoxic Conditions in Vitro. Biol. Cell 2019, 111, 232–244. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mycotoxin | Cell | Exposure Time | MTT_IC50 (µg/mL) | SRB_IC50 (µg/mL) |
|---|---|---|---|---|
| Fumonisin B1 | Caco-2 | 24 h | 589.7 ± 23.4 | 369.2 ± 12.1 |
| 48 h | 158.7 ± 38.4 | 37.9 ± 11.8 | ||
| 72 h | 52.9 ± 13.6 | 19.0 ± 11.19 | ||
| HepG2 | 24 h | 156.2 ± 16.8 | 33.9 ± 11.2 | |
| 48 h | 14.9 ± 15.7 | 3.5 ± 0.81 | ||
| 72 h | 9.1 ± 10.1 | 3.0 ± 1.1 | ||
| Aflatoxin B1 | Caco-2 | 24 h | 48.0 ± 3.2 | 119.2 ± 11.0 |
| 48 h | 52.3 ± 3.3 | 58.0 ± 7.7 | ||
| 72 h | 59.3 ± 5.7 | 22.8 ± 11.7 | ||
| HepG2 | 24 h | 63.6 ± 23.3 | 23.5 ± 14.7 | |
| 48 h | 3.6 ± 3.9 | 1.6 ± 0.4 | ||
| 72 h | 2.0 ± 2.2 | 0.7 ± 0.1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, X.; Abdallah, M.F.; Grootaert, C.; Rajkovic, A. Bioenergetic Status of the Intestinal and Hepatic Cells after Short Term Exposure to Fumonisin B1 and Aflatoxin B1. Int. J. Mol. Sci. 2022, 23, 6945. https://doi.org/10.3390/ijms23136945
Chen X, Abdallah MF, Grootaert C, Rajkovic A. Bioenergetic Status of the Intestinal and Hepatic Cells after Short Term Exposure to Fumonisin B1 and Aflatoxin B1. International Journal of Molecular Sciences. 2022; 23(13):6945. https://doi.org/10.3390/ijms23136945
Chicago/Turabian StyleChen, Xiangrong, Mohamed F. Abdallah, Charlotte Grootaert, and Andreja Rajkovic. 2022. "Bioenergetic Status of the Intestinal and Hepatic Cells after Short Term Exposure to Fumonisin B1 and Aflatoxin B1" International Journal of Molecular Sciences 23, no. 13: 6945. https://doi.org/10.3390/ijms23136945
APA StyleChen, X., Abdallah, M. F., Grootaert, C., & Rajkovic, A. (2022). Bioenergetic Status of the Intestinal and Hepatic Cells after Short Term Exposure to Fumonisin B1 and Aflatoxin B1. International Journal of Molecular Sciences, 23(13), 6945. https://doi.org/10.3390/ijms23136945