
The Neonatal Fc Receptor Is Elevated in Monocyte-Derived Immune Cells in Pancreatic Cancer

 , , , and
, , , and
Abstract
:1. Introduction
2. Results
2.1. PDAC Tumor-Bearing Mice Exhibit Altered FcRn Expression among Monocytic Immune Cell Populations
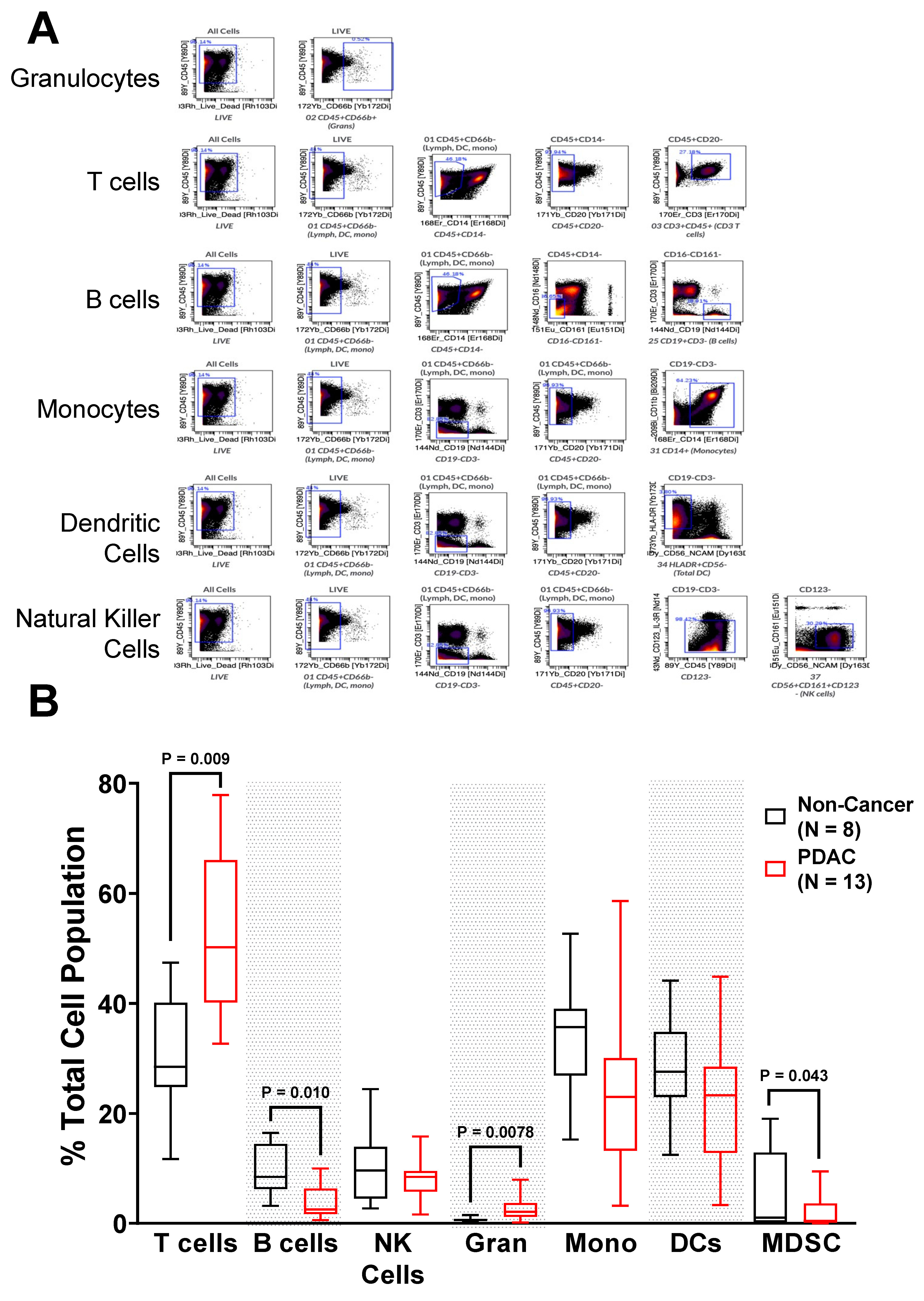
2.2. Immunophenotyping of Circulating PBMC from PDAC Patients Shows Alterations in Immune Populations
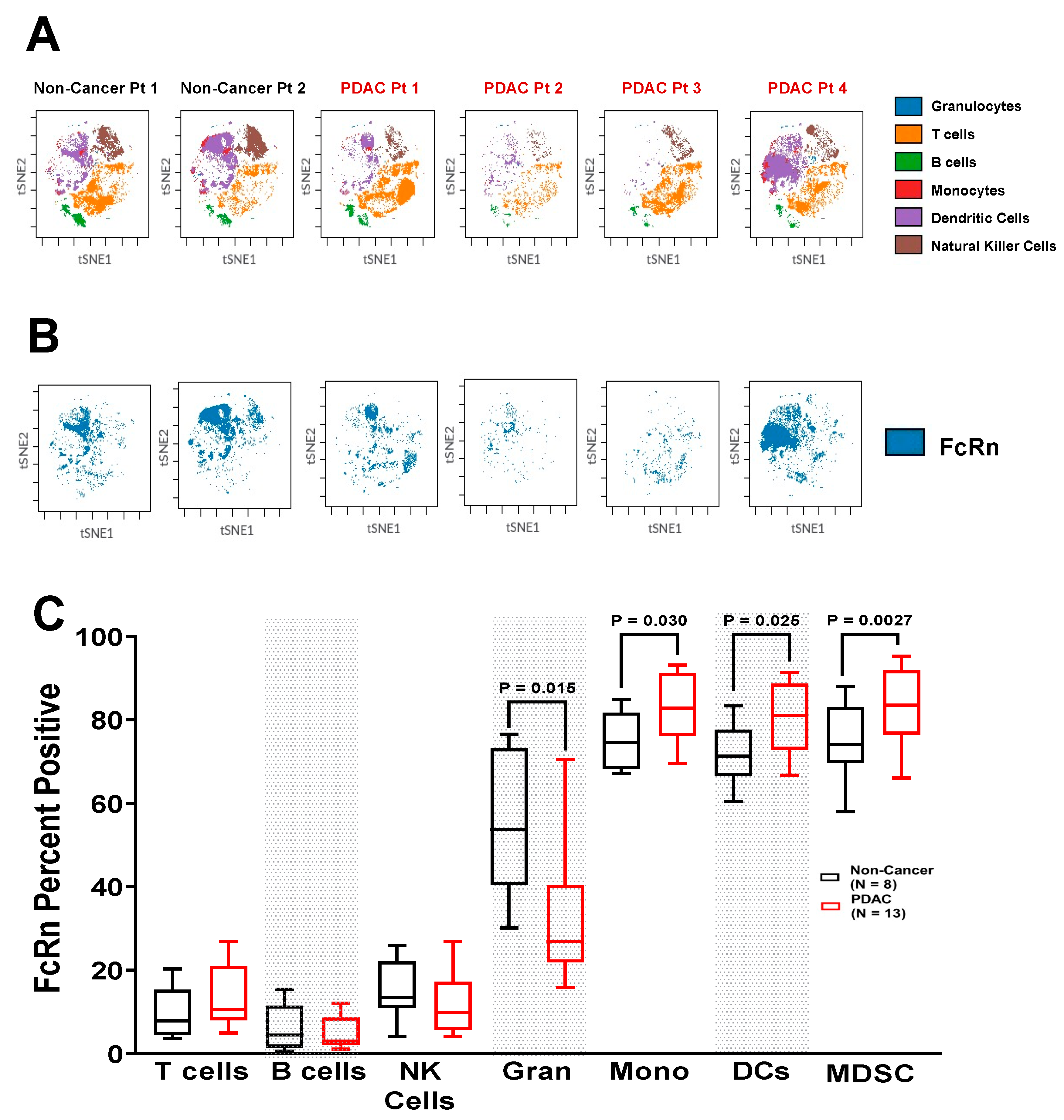
2.3. FcRn Is Localized to Myeloid-Derived Antigen-Presenting Cell Populations
2.4. Increased Expression of FcRn in PDAC Patient Immune Populations
2.5. FcRn Is Localized to Monocyte-Derived Subpopulations
3. Materials and Methods
3.1. Murine Model of Pancreatic Cancer and Splenocyte Isolation
3.2. Flow Cytometry Surface Marker Staining
3.3. PBMC Staining and Mass Cytometry (CyTOF)
3.4. Statistics
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Nimmerjahn, F.; Ravetch, J.V. Fc-receptors as regulators of immunity. Adv. Immunol. 2007, 96, 179–204. [Google Scholar] [PubMed]
- Nimmerjahn, F.; Ravetch, J.V. Fcgamma receptors: Old friends and new family members. Immunity 2006, 24, 19–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brambell, F.W. The transmission of immunity from mother to young and the catabolism of immunoglobulins. Lancet 1966, 2, 1087–1093. [Google Scholar] [CrossRef]
- Pyzik, M.; Sand, K.M.K.; Hubbard, J.J.; Andersen, J.T.; Sandlie, I.; Blumberg, R.S. The Neonatal Fc Receptor (FcRn): A Misnomer? Front. Immunol. 2019, 10, 1540. [Google Scholar] [CrossRef]
- Kontermann, R. Therapeutic Proteins: Strategies to Modulate Their Plasma Half-Life; John Wiley & Sons: Weinheim, Portland, OR, USA, 2011; p. 1. Available online: https://public.ebookcentral.proquest.com/choice/publicfullrecord.aspx?p=851166 (accessed on 10 May 2022).
- Andersen, J.T.; Daba, M.B.; Berntzen, G.; Michaelsen, T.E.; Sandlie, I. Cross-species binding analyses of mouse and human neonatal Fc receptor show dramatic differences in immunoglobulin G and albumin binding. J. Biol. Chem. 2010, 285, 4826–4836. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, M.; Kobayashi, K.; Kuo, T.T.; Bry, L.; Glickman, J.N.; Claypool, S.M.; Kaser, A.; Nagaishi, T.; Higgins, D.E.; Mizoguchi, E.; et al. Neonatal Fc receptor for IgG regulates mucosal immune responses to luminal bacteria. J. Clin. Investig. 2006, 116, 2142–2151. [Google Scholar] [CrossRef]
- Mi, W.; Wanjie, S.; Lo, S.T.; Gan, Z.; Pickl-Herk, B.; Ober, R.J.; Ward, E.S. Targeting the neonatal fc receptor for antigen delivery using engineered fc fragments. J. Immunol. 2008, 181, 7550–7561. [Google Scholar] [CrossRef] [Green Version]
- Dall’Acqua, W.F.; Kiener, P.A.; Wu, H. Properties of human IgG1s engineered for enhanced binding to the neonatal Fc receptor (FcRn). J. Biol. Chem. 2006, 281, 23514–23524. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Sun, M.; Liu, Z.; Kong, C.; Kong, W.; Ye, J.; Gong, J.; Huang, D.C.S.; Qian, F. KRAS-enhanced macropinocytosis and reduced FcRn-mediated recycling sensitize pancreatic cancer to albumin-conjugated drugs. J. Control. Release 2019, 296, 40–53. [Google Scholar] [CrossRef]
- Yardley, D.A. nab-Paclitaxel mechanisms of action and delivery. J. Control Release 2013, 170, 365–372. [Google Scholar] [CrossRef]
- Qi, T.; Cao, Y. In Translation: FcRn across the Therapeutic Spectrum. Int. J. Mol. Sci. 2021, 22, 3048. [Google Scholar] [CrossRef] [PubMed]
- Ward, E.S.; Devanaboyina, S.C.; Ober, R.J. Targeting FcRn for the modulation of antibody dynamics. Mol. Immunol. 2015, 67, 131–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goumas, F.A.; Holmer, R.; Egberts, J.H.; Gontarewicz, A.; Heneweer, C.; Geisen, U.; Hauser, C.; Mende, M.M.; Legler, K.; Rocken, C.; et al. Inhibition of IL-6 signaling significantly reduces primary tumor growth and recurrencies in orthotopic xenograft models of pancreatic cancer. Int. J. Cancer 2015, 137, 1035–1046. [Google Scholar] [CrossRef] [PubMed]
- Baker, K.; Qiao, S.W.; Kuo, T.T.; Aveson, V.G.; Platzer, B.; Andersen, J.T.; Sandlie, I.; Chen, Z.; de Haar, C.; Lencer, W.I.; et al. Neonatal Fc receptor for IgG (FcRn) regulates cross-presentation of IgG immune complexes by CD8-CD11b+ dendritic cells. Proc. Natl. Acad. Sci. USA 2011, 108, 9927–9932. [Google Scholar] [CrossRef] [Green Version]
- Baker, K.; Rath, T.; Flak, M.B.; Arthur, J.C.; Chen, Z.; Glickman, J.N.; Zlobec, I.; Karamitopoulou, E.; Stachler, M.D.; Odze, R.D.; et al. Neonatal Fc receptor expression in dendritic cells mediates protective immunity against colorectal cancer. Immunity 2013, 39, 1095–1107. [Google Scholar] [CrossRef] [Green Version]
- Kotsias, F.; Cebrian, I.; Alloatti, A. Antigen processing and presentation. Int. Rev. Cell Mol. Biol. 2019, 348, 69–121. [Google Scholar]
- Macri, C.; Morgan, H.; Villadangos, J.A.; Mintern, J.D. Regulation of dendritic cell function by Fc-gamma-receptors and the neonatal Fc receptor. Mol. Immunol. 2021, 139, 193–201. [Google Scholar] [CrossRef]
- Golay, J.; Semenzato, G.; Rambaldi, A.; Foa, R.; Gaidano, G.; Gamba, E.; Pane, F.; Pinto, A.; Specchia, G.; Zaja, F.; et al. Lessons for the clinic from rituximab pharmacokinetics and pharmacodynamics. MAbs 2013, 5, 826–837. [Google Scholar] [CrossRef]
- Cadena Castaneda, D.; Brachet, G.; Goupille, C.; Ouldamer, L.; Gouilleux-Gruart, V. The neonatal Fc receptor in cancer FcRn in cancer. Cancer Med. 2020, 9, 4736–4742. [Google Scholar] [CrossRef]
- Jansen, M.P.; Foekens, J.A.; van Staveren, I.L.; Dirkzwager-Kiel, M.M.; Ritstier, K.; Look, M.P.; Meijer-van Gelder, M.E.; Sieuwerts, A.M.; Portengen, H.; Dorssers, L.C.; et al. Molecular classification of tamoxifen-resistant breast carcinomas by gene expression profiling. J. Clin. Oncol. 2005, 23, 732–740. [Google Scholar] [CrossRef]
- Shi, L.; Zhang, W.; Zou, F.; Mei, L.; Wu, G.; Teng, Y. KLHL21, a novel gene that contributes to the progression of hepatocellular carcinoma. BMC Cancer 2016, 16, 815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dalloneau, E.; Baroukh, N.; Mavridis, K.; Maillet, A.; Gueugnon, F.; Courty, Y.; Petit, A.; Kryza, T.; Del Rio, M.; Guyetant, S.; et al. Downregulation of the neonatal Fc receptor expression in non-small cell lung cancer tissue is associated with a poor prognosis. Oncotarget 2016, 7, 54415–54429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larsen, M.T.; Kuhlmann, M.; Hvam, M.L.; Howard, K.A. Albumin-based drug delivery: Harnessing nature to cure disease. Mol. Cell Ther. 2016, 4, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2022. CA Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef]
- Perri, G.; Prakash, L.; Qiao, W.; Varadhachary, G.R.; Wolff, R.; Fogelman, D.; Overman, M.; Pant, S.; Javle, M.; Koay, E.J.; et al. Response and Survival Associated with First-line FOLFIRINOX vs Gemcitabine and nab-Paclitaxel Chemotherapy for Localized Pancreatic Ductal Adenocarcinoma. JAMA Surg. 2020, 155, 832–839. [Google Scholar] [CrossRef]
- Oberstein, P.E.; Olive, K.P. Pancreatic cancer: Why is it so hard to treat? Ther. Adv. Gastroenterol. 2013, 6, 321–337. [Google Scholar] [CrossRef] [Green Version]
- Kabacaoglu, D.; Ciecielski, K.J.; Ruess, D.A.; Algul, H. Immune Checkpoint Inhibition for Pancreatic Ductal Adenocarcinoma: Current Limitations and Future Options. Front. Immunol. 2018, 9, 1878. [Google Scholar] [CrossRef]
- Gabitass, R.F.; Annels, N.E.; Stocken, D.D.; Pandha, H.A.; Middleton, G.W. Elevated myeloid-derived suppressor cells in pancreatic, esophageal and gastric cancer are an independent prognostic factor and are associated with significant elevation of the Th2 cytokine interleukin-13. Cancer Immunol. Immunother. 2011, 60, 1419–1430. [Google Scholar] [CrossRef] [Green Version]
- Tzeng, A.; Diaz-Montero, C.M.; Rayman, P.A.; Kim, J.S.; Pavicic, P.G., Jr.; Finke, J.H.; Barata, P.C.; Lamenza, M.; Devonshire, S.; Schach, K.; et al. Immunological Correlates of Response to Immune Checkpoint Inhibitors in Metastatic Urothelial Carcinoma. Target Oncol. 2018, 13, 599–609. [Google Scholar] [CrossRef]
- Du, J.; Gu, J.; Li, J. Mechanisms of drug resistance of pancreatic ductal adenocarcinoma at different levels. Biosci. Rep. 2020, 40, BSR20200401. [Google Scholar] [CrossRef]
- Carbone, C.; Piro, G.; Agostini, A.; Delfino, P.; De Sanctis, F.; Nasca, V.; Spallotta, F.; Sette, C.; Martini, M.; Ugel, S.; et al. Intratumoral injection of TLR9 agonist promotes an immunopermissive microenvironment transition and causes cooperative antitumor activity in combination with anti-PD1 in pancreatic cancer. J. Immunother. Cancer 2021, 9, e002876. [Google Scholar] [CrossRef] [PubMed]
- Choueiry, F.; Torok, M.; Shakya, R.; Agrawal, K.; Deems, A.; Benner, B.; Hinton, A.; Shaffer, J.; Blaser, B.W.; Noonan, A.M.; et al. CD200 promotes immunosuppression in the pancreatic tumor microenvironment. J. Immunother. Cancer 2020, 8, e000189. [Google Scholar] [CrossRef] [PubMed]
- Mace, T.A.; Shakya, R.; Pitarresi, J.R.; Swanson, B.; McQuinn, C.W.; Loftus, S.; Nordquist, E.; Cruz-Monserrate, Z.; Yu, L.; Young, G.; et al. IL-6 and PD-L1 antibody blockade combination therapy reduces tumour progression in murine models of pancreatic cancer. Gut 2018, 67, 320–332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merad, M.; Sathe, P.; Helft, J.; Miller, J.; Mortha, A. The dendritic cell lineage: Ontogeny and function of dendritic cells and their subsets in the steady state and the inflamed setting. Annu. Rev. Immunol. 2013, 31, 563–604. [Google Scholar] [CrossRef] [Green Version]
- Motulsky, H.J.; Brown, R.E. Detecting outliers when fitting data with nonlinear regression—A new method based on robust nonlinear regression and the false discovery rate. BMC Bioinform. 2006, 7, 123. [Google Scholar] [CrossRef] [Green Version]
- Rahib, L.; Smith, B.D.; Aizenberg, R.; Rosenzweig, A.B.; Fleshman, J.M.; Matrisian, L.M. Projecting cancer incidence and deaths to 2030: The unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014, 74, 2913–2921. [Google Scholar] [CrossRef] [Green Version]
- McGuigan, A.; Kelly, P.; Turkington, R.C.; Jones, C.; Coleman, H.G.; McCain, R.S. Pancreatic cancer: A review of clinical diagnosis, epidemiology, treatment and outcomes. World J. Gastroenterol. 2018, 24, 4846–4861. [Google Scholar] [CrossRef]
- Robert, C. A decade of immune-checkpoint inhibitors in cancer therapy. Nat. Commun. 2020, 11, 3801. [Google Scholar] [CrossRef]
- Vaddepally, R.K.; Kharel, P.; Pandey, R.; Garje, R.; Chandra, A.B. Review of Indications of FDA-Approved Immune Checkpoint Inhibitors per NCCN Guidelines with the Level of Evidence. Cancers 2020, 12, 738. [Google Scholar] [CrossRef] [Green Version]
- He, M.; Henderson, M.; Muth, S.; Murphy, A.; Zheng, L. Preclinical mouse models for immunotherapeutic and non-immunotherapeutic drug development for pancreatic ductal adenocarcinoma. Ann. Pancreat. Cancer 2020, 3, 7. [Google Scholar] [CrossRef]
- Baker, K.; Rath, T.; Pyzik, M.; Blumberg, R.S. The Role of FcRn in Antigen Presentation. Front. Immunol. 2014, 5, 408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vidarsson, G.; Dekkers, G.; Rispens, T. IgG subclasses and allotypes: From structure to effector functions. Front. Immunol. 2014, 5, 520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Byrne, K.T.; Yan, F.; Yamazoe, T.; Chen, Z.; Baslan, T.; Richman, L.P.; Lin, J.H.; Sun, Y.H.; Rech, A.J.; et al. Tumor Cell-Intrinsic Factors Underlie Heterogeneity of Immune Cell Infiltration and Response to Immunotherapy. Immunity 2018, 49, 178–193.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Borresen-Dale, A.L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [Green Version]
- Bern, M.; Sand, K.M.; Nilsen, J.; Sandlie, I.; Andersen, J.T. The role of albumin receptors in regulation of albumin homeostasis: Implications for drug delivery. J. Control. Release 2015, 211, 144–162. [Google Scholar] [CrossRef]
- Yousefpour, P.; Ahn, L.; Tewksbury, J.; Saha, S.; Costa, S.A.; Bellucci, J.J.; Li, X.; Chilkoti, A. Conjugate of Doxorubicin to Albumin-Binding Peptide Outperforms Aldoxorubicin. Small 2019, 15, e1804452. [Google Scholar] [CrossRef]
- Fong, Z.V.; Ferrone, C.R. Surgery After Response to Chemotherapy for Locally Advanced Pancreatic Ductal Adenocarcinoma: A Guide for Management. J. Natl. Compr. Cancer Netw. 2021, 19, 459–467. [Google Scholar] [CrossRef]
- Cullis, J.; Siolas, D.; Avanzi, A.; Barui, S.; Maitra, A.; Bar-Sagi, D. Macropinocytosis of Nab-paclitaxel Drives Macrophage Activation in Pancreatic Cancer. Cancer Immunol. Res. 2017, 5, 182–190. [Google Scholar] [CrossRef] [Green Version]
- Macri, C.; Pang, E.S.; Patton, T.; O’Keeffe, M. Dendritic cell subsets. Semin. Cell Dev. Biol. 2018, 84, 11–21. [Google Scholar] [CrossRef]
- Sichien, D.; Lambrecht, B.N.; Guilliams, M.; Scott, C.L. Development of conventional dendritic cells: From common bone marrow progenitors to multiple subsets in peripheral tissues. Mucosal Immunol. 2017, 10, 831–844. [Google Scholar] [CrossRef]
- Tcyganov, E.; Mastio, J.; Chen, E.; Gabrilovich, D.I. Plasticity of myeloid-derived suppressor cells in cancer. Curr. Opin. Immunol. 2018, 51, 76–82. [Google Scholar] [CrossRef]
- Groth, C.; Hu, X.; Weber, R.; Fleming, V.; Altevogt, P.; Utikal, J.; Umansky, V. Immunosuppression mediated by myeloid-derived suppressor cells (MDSCs) during tumour progression. Br. J. Cancer 2019, 120, 16–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marabelle, A.; Le, D.T.; Ascierto, P.A.; Di Giacomo, A.M.; De Jesus-Acosta, A.; Delord, J.P.; Geva, R.; Gottfried, M.; Penel, N.; Hansen, A.R.; et al. Efficacy of Pembrolizumab in Patients with Noncolorectal High Microsatellite Instability/Mismatch Repair-Deficient Cancer: Results from the Phase II KEYNOTE-158 Study. J. Clin. Oncol. 2020, 38, 1–10. [Google Scholar] [CrossRef] [PubMed]
- O’Reilly, E.M.; Oh, D.Y.; Dhani, N.; Renouf, D.J.; Lee, M.A.; Sun, W.; Fisher, G.; Hezel, A.; Chang, S.C.; Vlahovic, G.; et al. Durvalumab with or without Tremelimumab for Patients with Metastatic Pancreatic Ductal Adenocarcinoma: A Phase 2 Randomized Clinical Trial. JAMA Oncol. 2019, 5, 1431–1438. [Google Scholar] [CrossRef] [PubMed]
- Bergtold, A.; Desai, D.D.; Gavhane, A.; Clynes, R. Cell surface recycling of internalized antigen permits dendritic cell priming of B cells. Immunity 2005, 23, 503–514. [Google Scholar] [CrossRef] [Green Version]
- Bruhns, P. Properties of mouse and human IgG receptors and their contribution to disease models. Blood 2012, 119, 5640–5649. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sex | PDAC | Non-Cancer | |
|---|---|---|---|
| Male | 6 | 5 | |
| Female | 7 | 3 | |
| Race | |||
| White | 11 | N/A | |
| Black/African American | 1 | N/A | |
| Initial Diagnosis | |||
| PDAC | 10 | N/A | |
| Pancreatic Mass | 3 | N/A | |
| Neoadjuvant Treatment | |||
| Gemcitabine/Abraxane | 1 | N/A | |
| FOLFIRINOX | 9 | N/A | |
| Height (in.) | Median (Range) | 65.98 (58.4–72.6) | 65.47 (61.0–72.8) |
| Weight (lbs.) | Median (Range) | 152.9 (107.58–198.1) | 221.01 (173.94–261.25) |
| BMI | Median (Range) | 25.14 (17.38–32.39) | 32.57 (30.29–43.28) |
| Serum Pre-albumin | Median (Range) | 18.5 (9–29) | N/A |
| Serum Albumin | Median (Range) | 3.8 (3.3–4.4) | N/A |
| Serum Glucose | Median (Range) | 105 (87–207) | 88.5 (76–100) |
| Cell Type | Mouse FcRn Positivity | Human FcRn Positivity | Mouse FcRn MFI | Human FcRn MMI |
|---|---|---|---|---|
| Monocytic DC | No Change | Elevated | No Change | Elevated |
| Plasmacytoid DC | No Change | No Change | No Change | No Change |
| Classical DC | No Change | Elevated | Elevated | Elevated |
| Macrophages | No Change | No Change | No Change | No Change |
| Granulocytic MDSC | No Change | Elevated | No Change | No Change |
| Monocytic MDSC | Elevated | Elevated | Elevated | Elevated |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thomas, J.; Torok, M.A.; Agrawal, K.; Pfau, T.; Vu, T.T.; Lyberger, J.; Chang, H.; Castillo, A.M.M.; Chen, M.; Remaily, B.; et al. The Neonatal Fc Receptor Is Elevated in Monocyte-Derived Immune Cells in Pancreatic Cancer. Int. J. Mol. Sci. 2022, 23, 7066. https://doi.org/10.3390/ijms23137066
Thomas J, Torok MA, Agrawal K, Pfau T, Vu TT, Lyberger J, Chang H, Castillo AMM, Chen M, Remaily B, et al. The Neonatal Fc Receptor Is Elevated in Monocyte-Derived Immune Cells in Pancreatic Cancer. International Journal of Molecular Sciences. 2022; 23(13):7066. https://doi.org/10.3390/ijms23137066
Chicago/Turabian StyleThomas, Justin, Molly A. Torok, Kriti Agrawal, Timothy Pfau, Trang T. Vu, Justin Lyberger, Hsiaochi Chang, Alyssa Marie M. Castillo, Min Chen, Bryan Remaily, and et al. 2022. "The Neonatal Fc Receptor Is Elevated in Monocyte-Derived Immune Cells in Pancreatic Cancer" International Journal of Molecular Sciences 23, no. 13: 7066. https://doi.org/10.3390/ijms23137066
APA StyleThomas, J., Torok, M. A., Agrawal, K., Pfau, T., Vu, T. T., Lyberger, J., Chang, H., Castillo, A. M. M., Chen, M., Remaily, B., Kim, K., Xie, Z., Dillhoff, M. E., Kulp, S. K., Behbehani, G. K., Cruz-Monserrate, Z., Ganesan, L. P., Owen, D. H., Phelps, M. A., ... Mace, T. A. (2022). The Neonatal Fc Receptor Is Elevated in Monocyte-Derived Immune Cells in Pancreatic Cancer. International Journal of Molecular Sciences, 23(13), 7066. https://doi.org/10.3390/ijms23137066