
Synthesis of Indole-Coupled KYNA Derivatives via C–N Bond Cleavage of Mannich Bases




Abstract
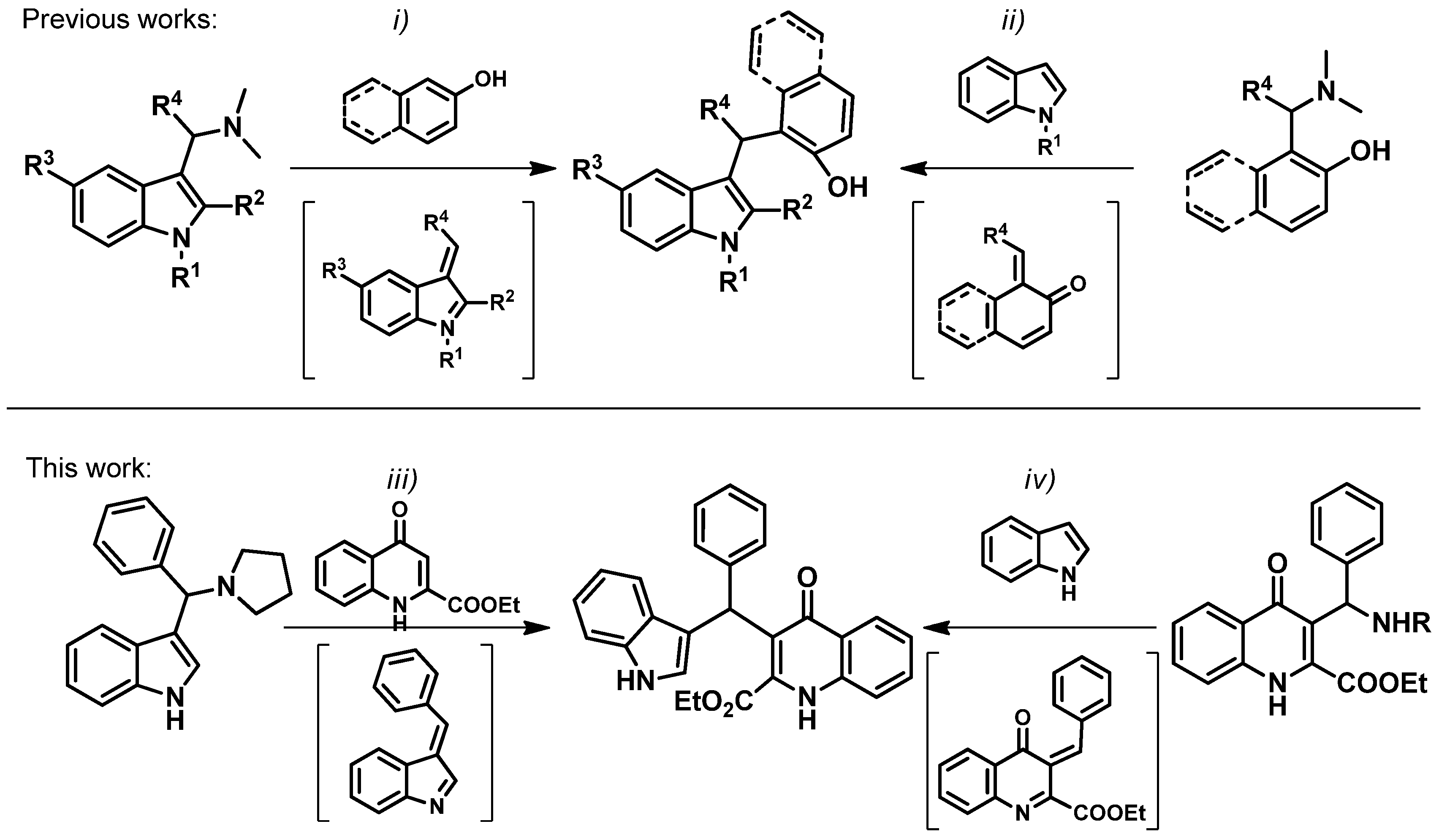
:1. Introduction
2. Materials and Methods
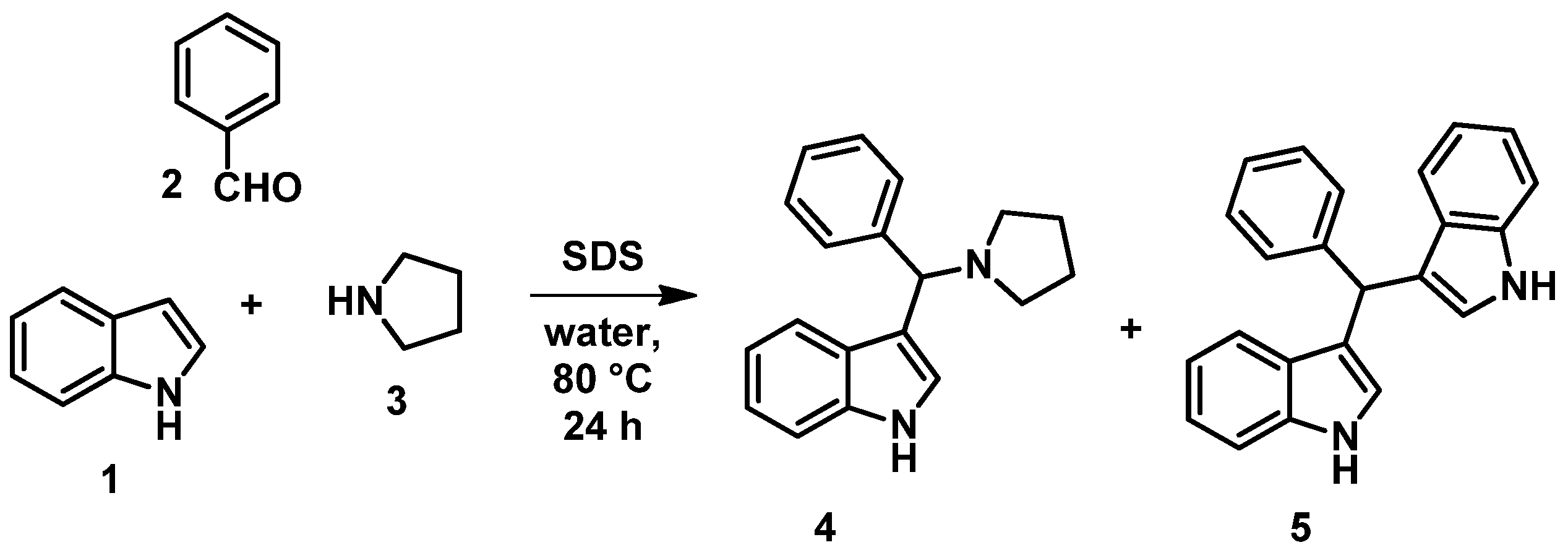
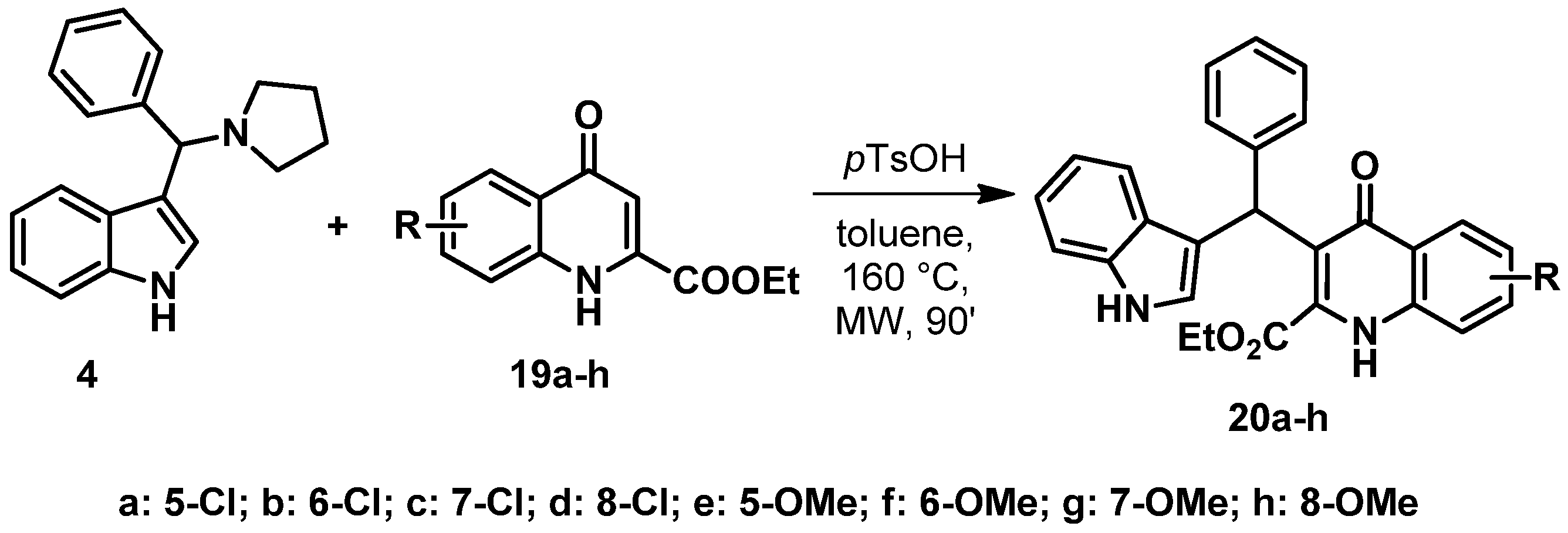
2.1. General Procedure for the Synthesis of 7, 20a–h
- (A)
- 4-Oxo-1,4-dihydroquinoline-2-carboxylic acid ethyl ester (6) or its derivatives (19a–h, 0.1 mmol) and 4 (0.15 mmol) were placed in a pressure-resistant 10 mL vessel with toluene (5 mL). The mixture was kept at 160 °C for 3 h in a CEM Discover SP microwave reactor (300 W). Work-up is similar to method B) but lower conversions and yields have been achieved with method A).
- (B)
- 4-Oxo-1,4-dihydroquinoline-2-carboxylic acid ethyl ester (6) or its derivatives (19a–h, 0.5 mmol) and 4 (0.75 mmol) were placed in a 50 mL round-bottom flask. The mixture was heated at reflux temperature in toluene (25 mL) for 1.5–18 h. Work-up is described separately.
2.1.1. Ethyl 3-((1H-indol-3-yl)(phenyl)methyl)-4-oxo-1,4-dihydroquinoline-2-carboxylate (7)
2.1.2. Ethyl 3-((1H-indol-3-yl)(phenyl)methyl)-5-chloro-4-oxo-1,4-dihydroquinoline-2-carboxylate (20a)
2.1.3. Ethyl 3-((1H-indol-3-yl)(phenyl)methyl)-6-chloro-4-oxo-1,4-dihydroquinoline-2-carboxylate (20b)
2.1.4. Ethyl 3-((1H-indol-3-yl)(phenyl)methyl)-7-chloro-4-oxo-1,4-dihydroquinoline-2-carboxylate (20c)
2.1.5. Ethyl 3-((1H-indol-3-yl)(phenyl)methyl)-8-chloro-4-oxo-1,4-dihydroquinoline-2-carboxylate (20d)
2.1.6. Ethyl 3-((1H-indol-3-yl)(phenyl)methyl)-5-methoxy-4-oxo-1,4-dihydroquinoline-2-carboxylate (20e)
2.1.7. Ethyl 3-((1H-indol-3-yl)(phenyl)methyl)-6-methoxy-4-oxo-1,4-dihydroquinoline-2-carboxylate (20f)
2.1.8. Ethyl 3-((1H-indol-3-yl)(phenyl)methyl)-7-methoxy-4-oxo-1,4-dihydroquinoline-2-carboxylate (20g)
2.1.9. Ethyl 3-((1H-indol-3-yl)(phenyl)methyl)-8-methoxy-4-oxo-1,4-dihydroquinoline-2-carboxylate (20h)
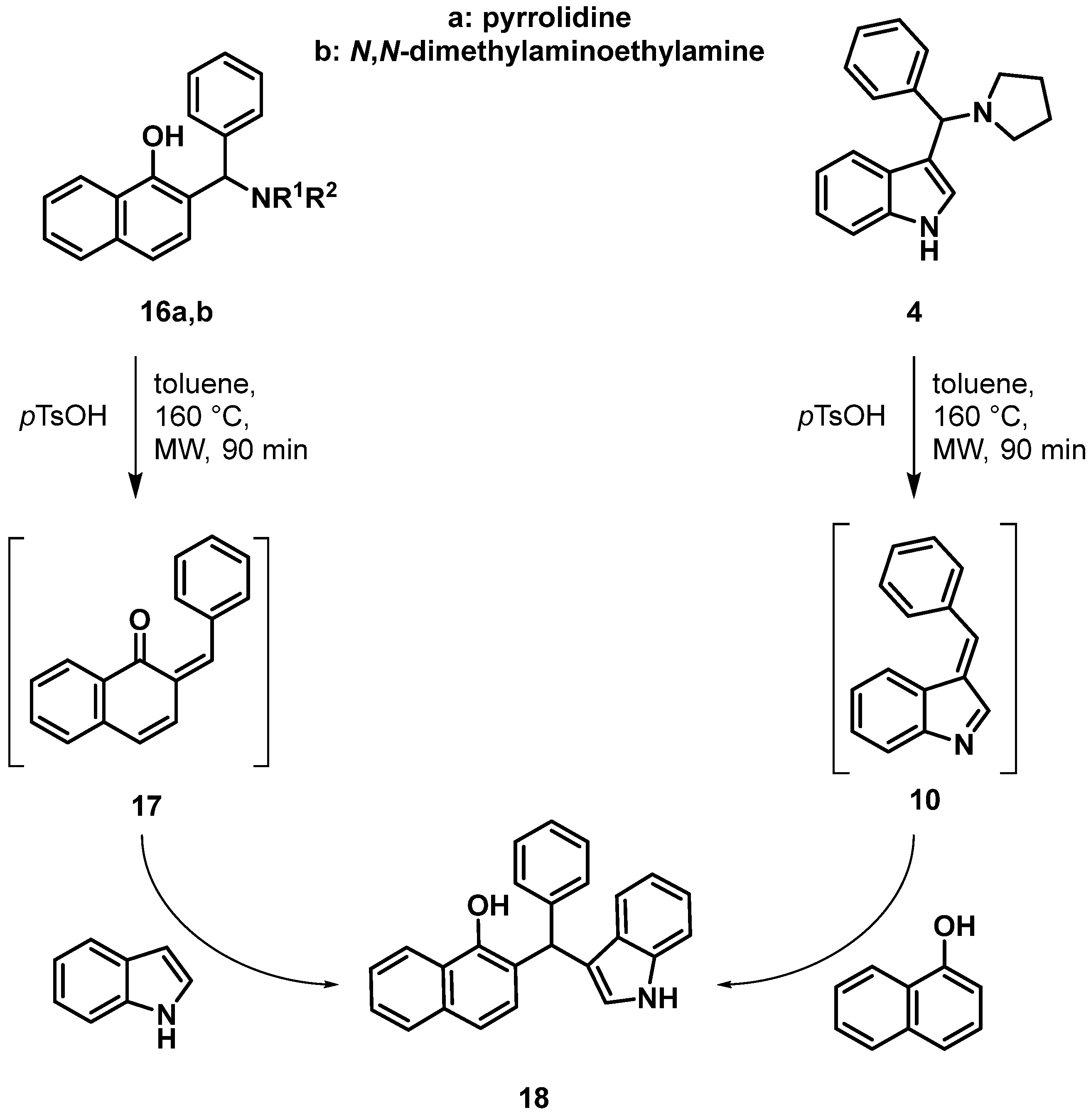
2.2. Procedure for the Synthesis of 2-(((2-(dimethylamino)ethyl)amino)(phenyl)methyl)naphthalen-1-ol (16b)
2.3. Procedures for the synthesis of 2-((1H-indol-3-yl)(phenyl)methyl)naphthalen-1-ol (18)
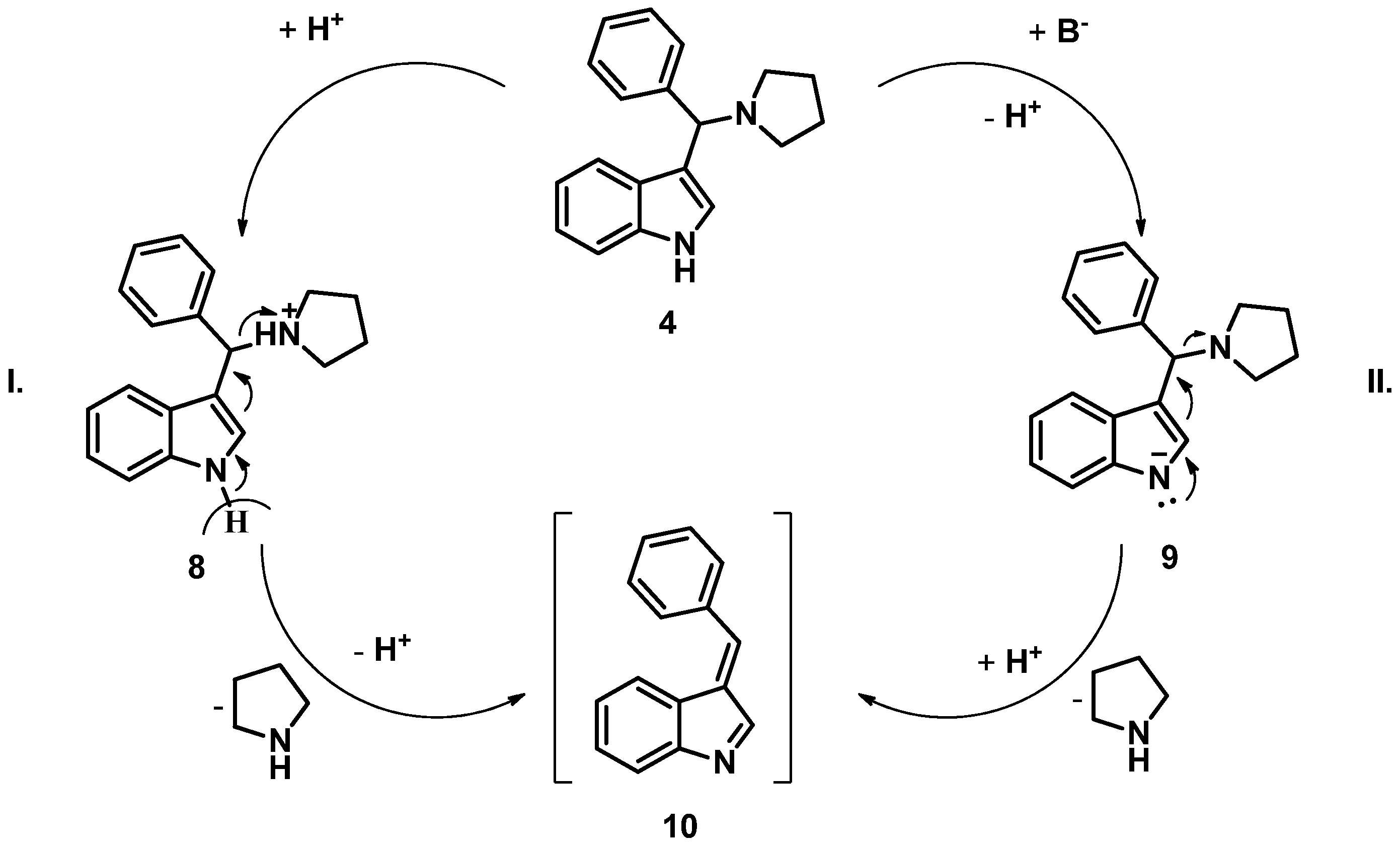
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sundberg, R.J. Indoles; Academic Press: London, UK, 1996. [Google Scholar]
- Gribble, G.W. Heterocyclic Chemistry II; Springer: Berlin, Germany, 2010; Volume 26. [Google Scholar]
- Yang, T.; Lu, H.; Shu, Y.; Ou, Y.; Hong, L.; Au, C.T.; Qiu, R. CF3SO2Na-Mediated, UV-Light-Induced Friedel–Crafts Alkylation of Indoles with Ketones/Aldehydes and Bioactivities of Products. Org. Lett. 2020, 22, 827–831. [Google Scholar] [CrossRef]
- Humphrey, G.R.; Kuethe, J.T. Practical methodologies for the synthesis of indoles. Chem. Rev. 2006, 106, 2875–2911. [Google Scholar] [CrossRef]
- Cacchi, S.; Fabrizi, G. Synthesis and Functionalization of Indoles Through Palladium-catalyzed Reactions. Chem. Rev. 2005, 105, 2873–2920. [Google Scholar] [CrossRef]
- Bartoli, G.; Bencivenni, G.; Dalpozzo, R. Organocatalytic strategies for the asymmetric functionalization of indoles. Chem. Soc. Rev. 2010, 39, 4449–4465. [Google Scholar] [CrossRef]
- Bandini, M.; Eichholzer, A. Catalytic Functionalization of Indoles in a New Dimension. Angew. Chem. Int. Ed. 2009, 48, 9608–9644. [Google Scholar] [CrossRef]
- Shiri, M. Indoles in Multicomponent Processes (MCPs). Chem. Rev. 2012, 112, 3508–3549. [Google Scholar] [CrossRef]
- Szatmári, I.; Sas, J.; Fülöp, F. Catalyst-free coupling of indole derivatives with 3,4-dihydroisoquinoline and related compounds. Tetrahedron Lett. 2013, 54, 5069–5071. [Google Scholar] [CrossRef]
- Takeda, S.; Yamamoto, A.; Okada, T.; Matsumura, E.; Nose, E.; Kogure, K.; Kojima, S.; Haga, T. Identification of surrogate ligands for orphan G protein-coupled receptors. Life Sci. 2003, 74, 367–377. [Google Scholar] [CrossRef]
- Nikaido, Y.; Koyama, Y.; Yoshikawa, Y.; Furuya, T.; Takeda, S. Mutation analysis and molecular modeling for the investigation of ligand-binding modes of GPR84. J. Biochem. 2015, 157, 311–320. [Google Scholar] [CrossRef]
- Fares, F. The anti-carcinogenic effect of indole-3-carbinol and 3, 3′-diindolylmethane and their mechanism of action. Med. Chem. 2014, S1, 1–8. [Google Scholar] [CrossRef]
- Kiselev, V.I.; Drukh, V.M.; Muyzhnek, E.L.; Kuznetsov, I.N.; Pchelintseva, O.I.; Paltsev, M.A. Preclinical antitumor activity of the diindolylmethane formulation in xenograft mouse model of prostate cancer. Exp. Oncol. 2014, 36, 90–93. [Google Scholar] [PubMed]
- Pillaiyar, T.; Köse, M.; Sylvester, K.; Weighardt, H.; Thimm, D.; Borges, G.; Förster, I.; Kügelgen, I.; Müller, C.E. Diindolylmethane Derivatives: Potent Agonists of the Immunostimulatory Orphan G Protein-Coupled Receptor GPR84. J. Med. Chem. 2017, 60, 3636–3655. [Google Scholar] [CrossRef] [PubMed]
- Safe, S.; Papineni, S.; Chintharlapalli, S. Cancer chemotherapy with indole-3-carbinol, bis(3′-indolyl)methane and synthetic analogs. Cancer Lett. 2008, 269, 326–338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kramer, H.-J.; Podobinska, M.; Bartsch, A.; Battmann, A.; Thoma, W.; Bernd, A.; Kummer, W.; Irlinger, B.; Steglich, W.; Mayser, P. Malassezin, a novel agonist of the aryl hydrocarbon receptor from the yeast Malassezia furfur, induces apoptosis in primary human melanocytes. ChemBioChem 2005, 6, 860–865. [Google Scholar] [CrossRef] [PubMed]
- Queiroz, M.M.F.; Queiroz, E.F.; Zeraik, M.L.; Ebrahimi, S.N.; Marcourt, L.; Cuendet, M.; Castro-Gamboa, I.; Hamburger, M.; Bolzani, V.S.; Wolfender, J.-L. Chemical composition of the bark of Tetrapterys mucronata and identification of acetylcholinesterase inhibitory constituents. J. Nat. Prod. 2014, 77, 650–656. [Google Scholar] [CrossRef]
- Liu, X.; Ma, S.; Toy, P.H. Halogen Bond-Catalyzed Friedel-Crafts Reactions of Aldehydes and Ketones Using a Bidentate Halogen Bond Donor Catalyst: Synthesis of Symmetrical Bis-(indolyl)methanes. Org. Lett. 2019, 21, 9212–9216. [Google Scholar] [CrossRef]
- Pillaiyar, T.; Gorska, E.; Schnakenburg, G.; Müller, C.E. General Synthesis of Unsymmetrical 3,3′-(Aza)diindolylmethane Derivatives. J. Org. Chem. 2018, 83, 9902–9913. [Google Scholar] [CrossRef]
- Xiao, J.; Wen, H.; Wang, L.; Xu, L.B.; Hao, Z.H.; Shao, C.-L.; Wang, C.-Y. Catalyst-free dehydrative SN1-type reaction of indolyl alcohols with diverse nucleophiles on water. Green Chem. 2016, 18, 1032–1037. [Google Scholar] [CrossRef]
- Huo, C.D.; Sun, C.G.; Wang, C.; Jia, X.D.; Chang, W.J. Triphenylphosphine-m-sulfonate/Carbon Tetrabromide as an Efficient and Easily Recoverable Catalyst System for Friedel–Crafts Alkylation of Indoles with Carbonyl Compounds or Acetals. ACS Sustain. Chem. Eng. 2013, 1, 549–553. [Google Scholar] [CrossRef]
- Bayindir, S.; Saracoglu, N. A facile one-pot method to synthesise 2-alkylated indole and 2,2′-bis(indolyl)methane derivatives using ketones as electrophiles and their anion sensing ability. RSC Adv. 2016, 6, 72959–72967. [Google Scholar] [CrossRef]
- Sas, K.; Robotka, H.; Toldi, J.; Vécsei, L. Mitochondria, metabolic disturbances, oxidative stress and the kynurenine system, with focus on neurodegenerative disorders. J. Neurol. Sci. 2007, 257, 221–239. [Google Scholar] [CrossRef] [PubMed]
- Gigler, G.; Szénási, G.; Simó, A.; Lévay, G.; Hársing, L.G., Jr.; Sas, K.; Vécsei, L.; Toldi, J. Neuroprotective effect of L-kynurenine sulfate administered before focal cerebral ischemia in mice and global cerebral ischemia in gerbils. Eur. J. Pharmacol. 2007, 564, 116–122. [Google Scholar] [CrossRef] [PubMed]
- Luchowska, E.; Luchowski, P.; Sarnowska, A.; Wielosz, M.; Turski, W.A.; Urbańska, E.M. Endogenous level of kynurenic acid and activities of kynurenine aminotransferases following transient global ischemia in the gerbil hippocampus. Pol. J. Pharmacol. 2003, 55, 443–447. [Google Scholar] [PubMed]
- Harrison, B.L.; Baron, B.M.; Cousino, D.M.; McDonald, I.A. 4-[(Carboxymethyl)oxy]- and 4-[(carboxymethyl)amino]-5,7-dichloroquinoline-2-carboxylic acid: New antagonists of the strychnine-insensitive glycine binding site on the N-methyl-D-aspartate receptor complex. J. Med. Chem. 1990, 12, 3130–3132. [Google Scholar] [CrossRef]
- Edmont, D.; Rocher, R.; Plisson, C.; Chenault, J. Synthesis and evaluation of quinoline carboxyguanidines as antidiabetic agents. Bioorg. Med. Chem. Lett. 2000, 16, 1831–1834. [Google Scholar] [CrossRef]
- Bonina, F.P.; Arenare, L.; Ippolito, R.; Boatto, G.; Battaglia, G.; Bruno, V.; de Caprariis, P. Synthesis, pharmacokinetics and anticonvulsant activity of 7-chlorokynurenic acid prodrugs. Int. J. Pharm. 2000, 202, 79–88. [Google Scholar] [CrossRef]
- Manfredini, S.; Pavan, B.; Vertuani, S.; Scaglianti, M.; Compagnone, D.; Biondi, C.; Scatturin, A.; Tanganelli, S.; Ferraro, L.; Prasad, P.; et al. Design, synthesis and activity of ascorbic acid prodrugs of nipecotic, kynurenic and diclophenamic acids, liable to increase neurotropic activity. J. Med. Chem. 2002, 45, 559–562. [Google Scholar] [CrossRef]
- Manfredini, S.; Vertuani, S.; Pavan, B.; Vitali, F.; Scaglianti, M.; Bortolotti, F.; Biondi, C.; Scatturin, A.; Prasad, P.; Dalpiaz, A. Design, synthesis and in vitro evaluation on HRPE cells of ascorbic and 6-bromoascorbic acid conjugates with neuroactive molecules. Bioorgan. Med. Chem. 2004, 12, 5453–5463. [Google Scholar] [CrossRef]
- Stone, T.W. Development and therapeutic potential of kynurenic acid and kynurenine derivatives for neuroprotection. Trends Pharmacol. Sci. 2000, 21, 149–154. [Google Scholar] [CrossRef]
- Stone, T.W. Inhibitors of the kynurenine pathway. Eur. J. Med. Chem. 2000, 35, 179–186. [Google Scholar] [CrossRef]
- Nichols, A.C.; Yielding, K.L. Anticonvulsant activity of 4-urea-5,7-dichlorokynurenic acid derivatives that are antagonists at the NMDA-associated glycine binding site. Mol. Chem. Neuropathol. 1998, 35, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Füvesi, J.; Somlai, C.; Németh, H.; Varga, H.; Kis, Z.; Farkas, T.; Károly, N.; Dobszay, M.; Penke, Z.; Penke, B.; et al. Comparative study on the effects of kynurenic acid and glucosamine-kynurenic acid. Pharmacol. Biochem. Behav. 2004, 77, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Sun, F.; Li, Y.; Sun, X.; Liu, X.; Huang, Y.; Zhang, L.H.; Ye, X.S.; Xiao, J. Rapid synthesis of iminosugar derivatives for cell-based in situ screening: Discovery of “hit” compounds with anticancer activity. ChemMedChem. 2007, 2, 1594–1597. [Google Scholar] [CrossRef] [PubMed]
- Brik, A.; Lin, Y.C.; Elder, J.; Wong, C.H. A quick diversity-oriented amide-forming reaction to optimise P-subsite residues of HIV protease inhibitors. Chem. Biol. 2002, 9, 891–896. [Google Scholar] [CrossRef] [Green Version]
- Tossi, A.; Benedetti, F.; Norbedo, S.; Skrbec, D.; Berti, F.; Romeo, D. Small hydroxyethylene-based peptidomimetics inhibiting both HIV-1 and C. albicans aspartic proteases. Bioorg. Med. Chem. 2003, 11, 4719–4727. [Google Scholar] [CrossRef] [PubMed]
- Knyihár-Csillik, E.; Mihály, A.; Krisztin-Péva, B.; Robotka, H.; Szatmári, I.; Fülöp, F.; Toldi, J.; Csillik, B.; Vécsei, L. The kynurenate analog SZR-72 prevents the nitroglycerol-induced increase of c-Fos immunoreactivity in the rat caudal trigeminal nucleus: Comparative studies of the effects of SZR-72 and kynurenic acid. Neurosci. Res. 2008, 61, 429–432. [Google Scholar] [CrossRef] [Green Version]
- Fülöp, F.; Szatmári, I.; Vámos, E.; Zádori, D.; Toldi, J.; Vécsei, L. Syntheses, transformations and pharmaceutical applications of kynurenic acid derivatives. Curr. Med. Chem. 2009, 16, 4828–4842. [Google Scholar] [CrossRef] [Green Version]
- Barta, P.; Fülöp, F.; Szatmári, I. Mannich base-connected syntheses mediated by ortho-quinone methides. Beilstein J. Org. Chem. 2018, 14, 560–575. [Google Scholar] [CrossRef] [Green Version]
- Lőrinczi, B.; Csámpai, A.; Fülöp, F.; Szatmári, I. Synthesis of New C-3 Substituted Kynurenic Acid Derivatives. Molecules 2020, 25, 937. [Google Scholar] [CrossRef] [Green Version]
- Deb, M.L.; Das, C.; Deka, B.; Saikla, P.J.; Baruah, P.K. Hydrogen-Bond-Catalyzed Arylation of 3-(Aminoalkyl)indoles via C–N Bond Cleavage with Thiourea under Microwave Irradiation: An Approach to 3-(α,α-Diarylmethyl)indoles. Synlett 2016, 27, 2788–2794. [Google Scholar] [CrossRef] [Green Version]
- Deb, M.L.; Pegu, C.D.; Deka, B.; Dutta, P.; Kotmale, A.S.; Baruah, P.K. Brønsted-Acid-Mediated Divergent Reactions of Betti Bases with Indoles: An Approach to Chromeno[2,3-b]indoles through Intramolecular Dehydrogenative C2-Alkoxylation of Indole. Eur. J. Org. Chem. 2016, 2016, 3441–3448. [Google Scholar] [CrossRef]
- Pegu, C.D.; Nasrin, S.B.; Deb, M.L.; Das, D.J.; Saikia, K.K.; Baruah, P.K. CAN-Catalyzed microwave promoted reaction of indole with betti bases under solvent-free condition and evaluation of antibacterial activity of the products. Synth. Commun. 2017, 47, 2007–2014. [Google Scholar] [CrossRef]
- Saidi, M.R.; Azizi, N.; Naimi-Jamal, M.R. Lithium perchlorate assisted one-pot three-component aminoalkylation of electron-rich aromatic compounds. Tetrahedron Lett. 2001, 42, 8111–8113. [Google Scholar] [CrossRef]
- Zhu, P.; Yi, Y.; Liu, Y.; Peng, Y. Asymmetric Addition of α-Diazomethylphosphonate to Alkylideneindolenine Catalyzed by a Trifunctional BINAP-Based Monophosphonium Salt. Org. Lett. 2022, 8, 1657–1661. [Google Scholar] [CrossRef] [PubMed]
- Aghaalikhani, S.; Behbahani, F.K. Three-Component Synthesis of 3-Aminoalkylindoles using Iron(III) Phosphate. ChemistrySelect 2016, 1, 5530–5532. [Google Scholar] [CrossRef]
- Depa, N.; Erothu, H. One-Pot Three-Component Synthesis of 3-Aminoalkyl Indoles Catalyzed by Molecular Iodine. ChemistrySelect 2019, 4, 9722–9725. [Google Scholar] [CrossRef]
- Cao, J.-F.; Chen, Y.-L.; Guan, Z.; He, Y.-H. Catalyst-free, Solvent-promoted and Scalable Multicomponent Synthesis of 3-Aminoalkylated Indoles via a Mannich-type Reaction. Z. Nat. B 2014, 69, 721–727. [Google Scholar] [CrossRef]
- Rajesh, C.; Kholiya, R.; Pavan, V.S.; Rawat, D.S. Catalyst-free, ethylene glycol promoted one-pot three component synthesis of 3-amino alkylated indoles via Mannich-type reaction. Tetrahedron Lett. 2014, 55, 2977–2981. [Google Scholar] [CrossRef]
- Kumar, A.; Gupta, M.K.; Kumar, M. L-Proline catalysed multicomponent synthesis of 3-amino alkylated indoles via a Mannich-type reaction under solvent-free conditions. Green Chem. 2012, 14, 290–295. [Google Scholar] [CrossRef]
- Deb, M.L.; Baruah, P.K. Deamination of Indole Mannich Bases: An Efficient Route to 3-Benzyl/Alkylindoles via a Metal-Free Transfer Hydrogenation Under Microwave Irradiation. Curr. Organocatal. 2016, 3, 84–89. [Google Scholar] [CrossRef]
- Kumar, A.; Gupta, M.K.; Kumar, M.; Saxena, D. Micelle promoted multicomponent synthesis of 3-amino alkylated indoles via a Mannich-type reaction in water. RSC Adv. 2013, 3, 1673–1678. [Google Scholar] [CrossRef]
- Lőrinczi, B.; Csámpai, A.; Fülöp, F.; Szatmári, I. Synthetic- and DFT modelling studies on regioselective modified Mannich reactions of hydroxy-KYNA derivatives. RSC Adv. 2021, 11, 543–554. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Solvent | Catalyst | Temperature (°C) | Time (min.) | Conversion (%) a |
|---|---|---|---|---|---|
| MeCN | thiourea | 100 | 10 | 5 |
| toluene | pTsOH | 100 | 180 | 20 |
| toluene | pTsOH | 130 | 180 | 60 |
| toluene | pTsOH | 160 | 90 | 80 b |
| toluene | - | 160 | 90 | 5 |
| toluene | thiourea | 160 | 90 | 40 |
| toluene | L-proline | 160 | 90 | 65 |
| toluene | TEA | 160 | 90 | 70 |
| 1,2-dichlorobenzene | pTsOH | 160 | 90 | 0 |
| MeCN | pTsOH | 160 | 90 | 10 |
| anisole | pTsOH | 160 | 90 | 5 |
| EtOH | pTsOH | 160 | 90 | 0 |
| water | SDS | 160 | 60 | 0 |
| Starting Material | Temperature | Time (h) | Conversion (%) a | Yield (%) |
|---|---|---|---|---|---|
| 19a | 160 | 3 | 60 | 44 |
| reflux | 8 | 99 | 91 | |
| 19b | 160 | 3 | 60 | 49 |
| reflux | 8 | 90 | 83 | |
| 19c | 160 | 3 | 70 | 62 |
| reflux | 8 | 85 | 76 | |
| 19d | 160 | 3 | 60 | 53 |
| reflux | 8 | 50 | 38 | |
| 19e | 160 | 3 | 30 | 18 |
| reflux | 8 | 90 | 81 | |
| 19f | 160 | 3 | 45 | 34 |
| reflux | 8 | 70 | 58 | |
| 19g | 160 | 3 | 60 | 48 |
| reflux | 5 | 99 | 95 | |
| 19h | 160 | 3 | 10 | 5 |
| reflux | 18 | 20 | 10 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lőrinczi, B.; Simon, P.; Szatmári, I. Synthesis of Indole-Coupled KYNA Derivatives via C–N Bond Cleavage of Mannich Bases. Int. J. Mol. Sci. 2022, 23, 7152. https://doi.org/10.3390/ijms23137152
Lőrinczi B, Simon P, Szatmári I. Synthesis of Indole-Coupled KYNA Derivatives via C–N Bond Cleavage of Mannich Bases. International Journal of Molecular Sciences. 2022; 23(13):7152. https://doi.org/10.3390/ijms23137152
Chicago/Turabian StyleLőrinczi, Bálint, Péter Simon, and István Szatmári. 2022. "Synthesis of Indole-Coupled KYNA Derivatives via C–N Bond Cleavage of Mannich Bases" International Journal of Molecular Sciences 23, no. 13: 7152. https://doi.org/10.3390/ijms23137152
APA StyleLőrinczi, B., Simon, P., & Szatmári, I. (2022). Synthesis of Indole-Coupled KYNA Derivatives via C–N Bond Cleavage of Mannich Bases. International Journal of Molecular Sciences, 23(13), 7152. https://doi.org/10.3390/ijms23137152