
Glioblastoma and Methionine Addiction

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
1.1. Methionine Addiction Is a Metabolic Vulnerability in Cancer
1.2. Traditional Methods for In Vitro Cell Culture Do Not Reflect the Tumor Microenvironment
1.3. The Accumulation of 11C-Methionine by Glioblastoma Tumor Cells Is Diagnostic and Reveals Important Aspects of Tumor Biology
1.4. Is Dietary Methionine Restriction a Potential Treatment Strategy?
1.5. Why Does Glioblastoma Need Exogenous Methionine?
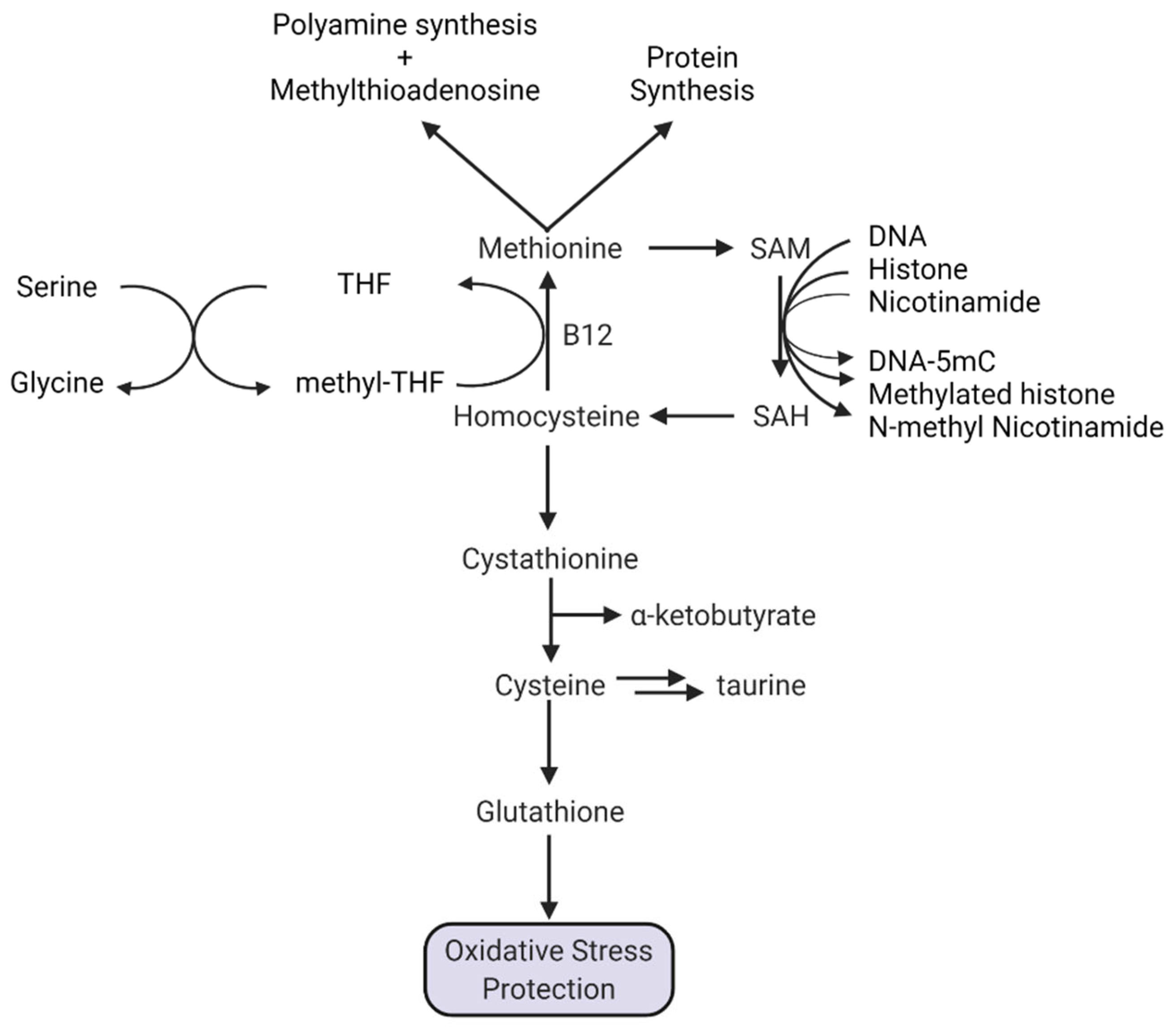
1.5.1. Methionine and Protein Synthesis
1.5.2. Methionine and Polyamine Synthesis
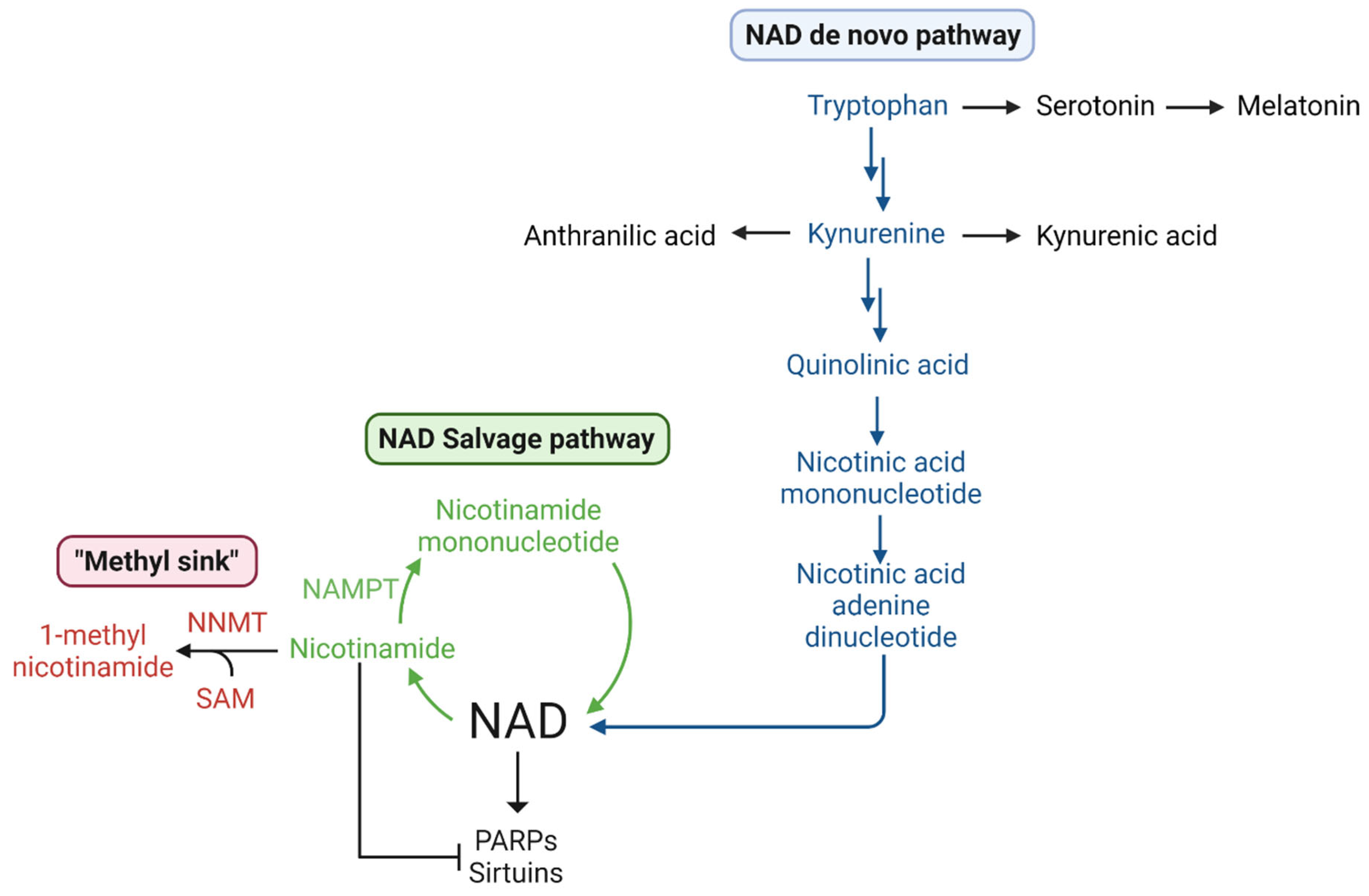
1.5.3. Methionine, SAM, Transmethylation, and NAD+
1.5.4. A Special Role for Methionine in DNA Methylation
1.6. Potential Consequences of Methionine Restriction in Glioblastoma Patients
2. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chello, P.L.; Bertino, J.R. Dependence of 5 Methyltetrahydrofolate Utilization by L5178Y Murine Leukemia Cells in vitro on the Presence of Hydroxycobalamin and Transcobalamin II. Cancer Res. 1973, 33, 1898–1904. [Google Scholar] [PubMed]
- Halpern, B.C.; Clark, B.R.; Hardy, D.N.; Halpern, R.M.; Smith, R.A. The Effect of Replacement of Methionine by Homocystine on Survival of Malignant and Normal Adult Mammalian Cells in Culture. Proc. Natl. Acad. Sci. USA 1974, 71, 1133–1136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoffman, R.M.; Erbe, R.W. High in vivo Rates of Methionine Biosynthesis in Transformed Human and Malignant Rat Cells Auxotrophic for Methionine. Proc. Natl. Acad. Sci. USA 1976, 73, 1523–1527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Judde, J.G.; Frost, P. Patterns of Methionine Auxotrophy in Normal and Neoplastic Cells: The Methionine Independence of Lymphocyte Mitogenesis and Low Frequency of the Methionine-Dependent Phenotype in Human Tumors. Cancer Res. 1988, 48, 6775–6779. [Google Scholar]
- Guo, H.-Y.; Herrera, H.; Groce, A.; Hoffman, R.M. Expression of the Biochemical Defect of Methionine Dependence in Fresh Patient Tumors in Primary Histoculture. Cancer Res. 1993, 53, 2479–2483. [Google Scholar]
- Fiskerstrand, T.; Christensen, B.; Tvsnes, O.B.; Ueland, P.M.; Refsum, H. Development and Reversion of Methionine Dependence in a Human Glioma Cell Line: Relation to Homocysteine Remethylation and Cobalamin Status. Cancer Res. 1994, 54, 4899–4906. [Google Scholar]
- Zhang, K.; Xu, P.; Sowers, J.L.; Machuca, D.F.; Mirfattah, B.; Herring, J.; Tang, H.; Chen, Y.; Tian, B.; Brasier, A.R.; et al. Proteome Analysis of Hypoxic Glioblastoma Cells Reveals Sequential Metabolic Adaptation of One-Carbon Metabolic Pathways. Mol. Cell. Proteom. 2017, 16, 1906–1921. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Yip, L.Y.; Lee, J.H.J.; Wu, Z.; Chew, H.Y.; Chong, P.K.W.; Teo, C.C.; Ang, H.Y.K.; Peh, K.L.E.; Yuan, J.; et al. Methionine Is a Metabolic Dependency of Tumor-Initiating Cells. Nat. Med. 2019, 25, 825–837. [Google Scholar] [CrossRef]
- Klein, S.G.; Alsolami, S.M.; Steckbauer, A.; Arossa, S.; Parry, A.J.; Ramos Mandujano, G.; Alsayegh, K.; Izpisua Belmonte, J.C.; Li, M.; Duarte, C.M. A Prevalent Neglect of Environmental Control in Mammalian Cell Culture Calls for Best Practices. Nat. Biomed. Eng. 2021, 5, 787–792. [Google Scholar] [CrossRef]
- Mally, J.; Szalai, G.; Stone, T.W. Changes in the Concentration of Amino Acids in Serum and Cerebrospinal Fluid of Patients with Parkinson’s Disease. J. Neurol. Sci. 1997, 151, 159–162. [Google Scholar] [CrossRef]
- Hladky, S.B.; Barrand, M.A. Elimination of Substances from the Brain Parenchyma: Efflux via Perivascular Pathways and via the Blood-Brain Barrier 11 Medical and Health Sciences 1109 Neurosciences. Fluids Barriers CNS 2018, 15, 30. [Google Scholar] [CrossRef] [Green Version]
- Cantor, J.R.; Abu-Remaileh, M.; Kanarek, N.; Freinkman, E.; Gao, X.; Louissaint, A.; Lewis, C.A.; Sabatini, D.M. Physiologic Medium Rewires Cellular Metabolism and Reveals Uric Acid as an Endogenous Inhibitor of UMP Synthase. Cell 2017, 169, 258–272.e17. [Google Scholar] [CrossRef] [Green Version]
- Hultberg, B.; Andersson, A.; Isaksson, A. Metabolism of Homocysteine, Its Relation to the Other Cellular Thiols and Its Mechanism of Cell Damage in a Cell Culture Line (Human Histiocytic Cell Line U-937). BBA-Mol. Cell Res. 1995, 1269, 6–12. [Google Scholar] [CrossRef] [Green Version]
- Verbeek, M.M.; Leen, W.G.; Willemsen, M.A.; Slats, D.; Claassen, J.A. Hourly Analysis of Cerebrospinal Fluid Glucose Shows Large Diurnal Fluctuations. J. Cereb. Blood Flow Metab. 2015, 36, 899–902. [Google Scholar] [CrossRef]
- Muir, A.; Vander Heiden, M.G. The Nutrient Environment Affects Therapy. Science 2018, 360, 962–963. [Google Scholar] [CrossRef]
- Vande Voorde, J.; Ackermann, T.; Pfetzer, N.; Sumpton, D.; Mackay, G.; Kalna, G.; Nixon, C.; Blyth, K.; Gottlieb, E.; Tardito, S. Improving the Metabolic Fidelity of Cancer Models with a Physiological Cell Culture Medium. Sci. Adv. 2019, 5, eaau7314. [Google Scholar] [CrossRef] [Green Version]
- Ackermann, T.; Tardito, S. Cell Culture Medium Formulation and Its Implications in Cancer Metabolism. Trends Cancer 2019, 5, 329–332. [Google Scholar] [CrossRef]
- Tardito, S.; Oudin, A.; Ahmed, S.U.; Fack, F.; Keunen, O.; Zheng, L.; Miletic, H.; Sakariassen, P.Ø.; Weinstock, A.; Wagner, A.; et al. Glutamine Synthetase Activity Fuels Nucleotide Biosynthesis and Supports Growth of Glutamine-Restricted Glioblastoma. Nat. Cell Biol. 2015, 17, 1556–1568. [Google Scholar] [CrossRef] [Green Version]
- DeBerardinis, R.J.; Mancuso, A.; Daikhin, E.; Nissim, I.; Yudkoff, M.; Wehrli, S.; Thompson, C.B. Beyond Aerobic Glycolysis: Transformed Cells Can Engage in Glutamine Metabolism That Exceeds the Requirement for Protein and Nucleotide Synthesis. Proc. Natl. Acad. Sci. USA 2007, 104, 19345–19350. [Google Scholar] [CrossRef] [Green Version]
- Lilja, A.; Bergström, K.; Hartvig, P.; Spännare, B.; Halldin, C.; Lundqvist, H.; Långstrom, B. Dynamic Study of Supratentorial Gliomas with L-Methyl-11C-Methionine and Positron Emission Tomography Anders. AJNR Am. J. Neuroradiol. 1985, 6, 505–514. [Google Scholar]
- Kaschten, B.; Stevenaert, A.; Sadzot, B.; Deprez, M.; Degueldre, C.; Del Fiore, G.; Luxen, A.; Reznik, M. Preoperative Evaluation of 54 Gliomas by PET with Fluorine-18- Fluorodeoxyglucose and/or Carbon-11-Methionine. J. Nucl. Med. 1998, 39, 778–785. [Google Scholar]
- Kläsner, B.D.; Krause, B.J.; Beer, A.J.; Drzezga, A. PET Imaging of Gliomas Using Novel Tracers: A Sleeping Beauty Waiting to Be Kissed. Expert Rev. Anticancer Ther. 2010, 10, 609–613. [Google Scholar] [CrossRef]
- Nawashiro, H.; Otani, N.; Uozumi, Y.; Ooigawa, H.; Toyooka, T.; Suzuki, T.; Katoh, H.; Tsuzuki, N.; Ohnuki, A.; Shima, K.; et al. High Expression of L-Type Amino Acid Transporter 1 in Infiltrating Glioma Cells. Brain Tumor Pathol. 2005, 22, 89–91. [Google Scholar] [CrossRef]
- Nozaki, S.; Nakatani, Y.; Mawatari, A.; Hume, W.E.; Wada, Y.; Ishii, A.; Tanaka, M.; Tsuyuguchi, N.; Doi, H.; Watanabe, Y. First-in-Human Assessment of the Novel LAT1 Targeting PET Probe 18F-FIMP. Biochem. Biophys. Res. Commun. 2022, 596, 83–87. [Google Scholar] [CrossRef]
- Van Dijken, B.R.J.; Schuuring, B.; Jeltema, H.R.; van Laar, P.J.; Enting, R.H.; Dierckx, R.A.J.O.; Stormezand, G.N.; van der Hoorn, A. Ventricle Contact May Be Associated with Higher 11C Methionine PET Uptake in Glioblastoma. Neuroradiology 2022, 64, 247–252. [Google Scholar] [CrossRef]
- Steed, T.C.; Treiber, J.M.; Taha, B.; Engin, H.B.; Carter, H.; Patel, K.S.; Dale, A.M.; Carter, B.S.; Chen, C.C. Glioblastomas Located in Proximity to the Subventricular Zone (SVZ) Exhibited Enrichment of Gene Expression Profiles Associated with the Cancer Stem Cell State. J. Neurooncol. 2020, 148, 455–462. [Google Scholar] [CrossRef]
- Yamaki, T.; Shibahra, I.; Matsuda, K.I.; Kanemura, Y.; Konta, T.; Kanamori, M.; Yamakawa, M.; Tominaga, T.; Sonoda, Y. Relationships between Recurrence Patterns and Subventricular Zone Involvement or CD133 Expression in Glioblastoma. J. Neurooncol. 2020, 146, 489–499. [Google Scholar] [CrossRef]
- Fontán-Lozano, Á.; Morcuende, S.; Davis-López de Carrizosa, M.A.; Benítez-Temiño, B.; Mejías, R.; Matarredona, E.R. To Become or Not to Become Tumorigenic: Subventricular Zone Versus Hippocampal Neural Stem Cells. Front. Oncol. 2020, 10, 602217. [Google Scholar] [CrossRef]
- Bender, K.; Träger, M.; Wahner, H.; Onken, J.; Scheel, M.; Beck, M.; Ehret, F.; Budach, V.; Kaul, D. What Is the Role of the Subventricular Zone in Radiotherapy of Glioblastoma Patients? Radiother. Oncol. 2021, 158, 138–145. [Google Scholar] [CrossRef]
- Brockman, A.A.; Mobley, B.C.; Ihrie, R.A. Histological Studies of the Ventricular–Subventricular Zone as Neural Stem Cell and Glioma Stem Cell Niche. J. Histochem. Cytochem. 2021, 69, 819–834. [Google Scholar] [CrossRef]
- Comas, S.; Luguera, E.; Molero, J.; Balaña, C.; Estival, A.; Castañer, S.; Carrato, C.; Hostalot, C.; Teixidor, P.; Villà, S. Influence of Glioblastoma Contact with the Subventricular Zone on Survival and Recurrence Patterns. Clin. Transl. Oncol. 2021, 23, 554–564. [Google Scholar] [CrossRef] [PubMed]
- Ripari, L.B.; Norton, E.S.; Bodoque-Villar, R.; Jeanneret, S.; Lara-Velazquez, M.; Carrano, A.; Zarco, N.; Vazquez-Ramos, C.A.; Quiñones-Hinojosa, A.; de la Rosa-Prieto, C.; et al. Glioblastoma Proximity to the Lateral Ventricle Alters Neurogenic Cell Populations of the Subventricular Zone. Front. Oncol. 2021, 11, 650316. [Google Scholar] [CrossRef] [PubMed]
- Pala, A.; Reske, S.N.; Eberhardt, N.; Scheuerle, A.; König, R.; Schmitz, B.; Beer, A.J.; Wirtz, C.R.; Coburger, J. Diagnostic Accuracy of Intraoperative Perfusion-Weighted MRI and 5-Aminolevulinic Acid in Relation to Contrast-Enhanced Intraoperative MRI and 11 C-Methionine Positron Emission Tomography in Resection of Glioblastoma: A Prospective Study. Neurosurg. Rev. 2019, 42, 471–479. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.K.; Jung, T.Y.; Jung, S.; Kim, I.Y.; Jang, W.Y.; Moon, K.S.; Kim, S.K.; Kim, J.H.; Lee, K.H. Relationship between Tumor Cell Infiltration and 5-Aminolevulinic Acid Fluorescence Signals after Resection of MR-Enhancing Lesions and Its Prognostic Significance in Glioblastoma. Clin. Transl. Oncol. 2021, 23, 459–467. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, K.; Tamura, K.; Hara, S.; Inaji, M.; Tanaka, Y.; Kobayashi, D.; Sugawara, T.; Wakimoto, H.; Nariai, T.; Ishii, K.; et al. Correlation of Intraoperative 5-ALA-Induced Fluorescence Intensity and Preoperative 11C-Methionine PET Uptake in Glioma Surgery. Cancers 2022, 14, 1449. [Google Scholar] [CrossRef]
- Inoue, A.; Nishikawa, M.; Ohnishi, T.; Yano, H.; Kanemura, Y.; Ohtsuka, Y.; Ozaki, S.; Nakamura, Y.; Matsumoto, S.; Suehiro, S.; et al. Prediction of Glioma Stemlike Cell Infiltration in the Non–Contrast-Enhancing Area by Quantitative Measurement of Lactate on Magnetic Resonance Spectroscopy in Glioblastoma. World Neurosurg. 2021, 153, e76–e95. [Google Scholar] [CrossRef]
- Yang, C.; Tian, G.; Dajac, M.; Doty, A.; Wang, S.; Lee, J.H.; Rahman, M.; Huang, J.; Reynolds, B.A.; Sarkisian, M.R.; et al. Slow-Cycling Cells in Glioblastoma: A Specific Population in the Cellular Mosaic of Cancer Stem Cells. Cancers 2022, 14, 1126. [Google Scholar] [CrossRef]
- Nakajo, K.; Uda, T.; Kawashima, T.; Terakawa, Y.; Ishibashi, K.; Tsuyuguchi, N.; Tanoue, Y.; Nagahama, A.; Uda, H.; Koh, S.; et al. Maximum 11C-Methionine PET Uptake as a Prognostic Imaging Biomarker for Newly Diagnosed and Untreated Astrocytic Glioma. Sci. Rep. 2022, 12, 546. [Google Scholar] [CrossRef]
- Maeda, Y.; Yamamoto, Y.; Norikane, T.; Mitamura, K.; Hatakeyama, T.; Miyake, K.; Nishiyama, Y.; Kudomi, N. Fractal Analysis of 11C-Methionine PET in Patients with Newly Diagnosed Glioma. EJNMMI Phys. 2021, 8, 76. [Google Scholar] [CrossRef]
- Sugimura, T.; Birnbaum, S.M.; Winitz, M.; Greenstein, J.P. Quantitative Nutritional Studies with Water-Soluble, Chemically Defined Diets. VIII. The Forced Feeding of Diets Each Lacking in One Essential Amino Acid. Arch. Biochem. Biophys. 1959, 81, 448–455. [Google Scholar] [CrossRef]
- Cavuoto, P.; Fenech, M.F. A Review of Methionine Dependency and the Role of Methionine Restriction in Cancer Growth Control and Life-Span Extension. Cancer Treat. Rev. 2012, 38, 726–736. [Google Scholar] [CrossRef]
- Chaturvedi, S.; Hoffman, R.M.; Bertino, J.R. Exploiting Methionine Restriction for Cancer Treatment. Biochem. Pharmacol. 2018, 154, 170–173. [Google Scholar] [CrossRef]
- Guéant, J.L.; Oussalah, A.; Zgheib, R.; Siblini, Y.; Hsu, S.B.; Namour, F. Genetic, Epigenetic and Genomic Mechanisms of Methionine Dependency of Cancer and Tumor-Initiating Cells: What Could We Learn from Folate and Methionine Cycles. Biochimie 2020, 173, 123–128. [Google Scholar] [CrossRef]
- Wanders, D.; Hobson, K.; Ji, X. Methionine Restriction and Cancer Biology. Nutrients 2020, 12, 684. [Google Scholar] [CrossRef] [Green Version]
- Goseki, N.; Yamazaki, S.; Shimojyu, K.; Kando, F.; Maruyama, M.; Endo, M.; Koike, M.; Takahashi, H. Synergistic Effect of Methionine-depleting Total Parenteral Nutrition with 5-Fluorouracil on Human Gastric Cancer: A Randomized, Prospective Clinical Trial. Jpn. J. Cancer Res. 1995, 86, 484–489. [Google Scholar] [CrossRef]
- Epner, D.E.; Morrow, S.; Wilcox, M.; Houghton, J.L. Nutrient Intake and Nutritional Indexes in Adults with Metastatic Cancer on a Phase I Clinical Trial of Dietary Methionine Restriction. Nutr. Cancer 2002, 42, 158–166. [Google Scholar] [CrossRef]
- Thivat, E.; Farges, M.C.; Bacin, F.; D’Incan, M.; Mouret-Reynier, M.A.; Cellarier, E.; Madelmont, J.C.; Vasson, M.P.; Chollet, P.; Durando, X. Phase II Trial of the Association of a Methionine-Free Diet with Cystemustine Therapy in Melanoma and Glioma. Anticancer Res. 2009, 29, 5235–5240. [Google Scholar]
- Tan, Y.; Zavala, J.S.; Xu, M.; Zavala, J.J.; Hoffman, R.M. Serum Methionine Depletion without Side Effects by Methioninase in Metastatic Breast Cancer Patients. Anticancer Res. 1996, 16, 3937–3942. [Google Scholar]
- Gay, F.; Aguera, K.; Sénéchal, K.; Tainturier, A.; Berlier, W.; Maucort-Boulch, D.; Honnorat, J.; Horand, F.; Godfrin, Y.; Bourgeaux, V. Methionine Tumor Starvation by Erythrocyte-Encapsulated Methionine Gamma-Lyase Activity Controlled with per Os Vitamin B6. Cancer Med. 2017, 6, 1437–1452. [Google Scholar] [CrossRef]
- Kearse, M.G.; Wilusz, J.E. Non-AUG Translation: A New Start for Protein Synthesis in Eukaryotes. Genes Dev. 2017, 31, 1717–1731. [Google Scholar] [CrossRef] [Green Version]
- Lin, M.; Zhang, X.; Jia, B.; Guan, S. Suppression of Glioblastoma Growth and Angiogenesis through Molecular Targeting of Methionine Aminopeptidase-2. J. Neurooncol. 2018, 136, 243–254. [Google Scholar] [CrossRef]
- Li, Q.Z.; Zuo, Z.W.; Zhou, Z.R.; Ji, Y. Polyamine Homeostasis-Based Strategies for Cancer: The Role of Combination Regimens. Eur. J. Pharmacol. 2021, 910, 174456. [Google Scholar] [CrossRef]
- Hansen, L.J.; Yang, R.; Roso, K.; Wang, W.; Chen, L.; Yang, Q.; Pirozzi, C.J.; He, Y. MTAP Loss Correlates with an Immunosuppressive Profile in GBM and Its Substrate MTA Stimulates Alternative Macrophage Polarization. Sci. Rep. 2022, 12, 4183. [Google Scholar] [CrossRef]
- Avila, M.A.; García-Trevijano, E.R.; Lu, S.C.; Corrales, F.J.; Mato, J.M. Methylthioadenosine. Int. J. Biochem. Cell Biol. 2004, 36, 2125–2130. [Google Scholar] [CrossRef]
- Marjon, K.; Cameron, M.J.; Quang, P.; Clasquin, M.F.; Mandley, E.; Kunii, K.; McVay, M.; Choe, S.; Kernytsky, A.; Gross, S.; et al. MTAP Deletions in Cancer Create Vulnerability to Targeting of the MAT2A/PRMT5/RIOK1 Axis. Cell Rep. 2016, 15, 574–587. [Google Scholar] [CrossRef] [Green Version]
- Mavrakis, K.J.; Robert McDonald, E.; Schlabach, M.R.; Billy, E.; Hoffman, G.R.; DeWeck, A.; Ruddy, D.A.; Venkatesan, K.; Yu, J.; McAllister, G.; et al. Disordered Methionine Metabolism in MTAP/CDKN2A-Deleted Cancers Leads to Dependence on PRMT5. Science 2016, 351, 1208–1213. [Google Scholar] [CrossRef]
- Kryukov, G.V.; Wilson, F.H.; Ruth, J.R.; Paulk, J.; Tsherniak, A.; Marlow, S.E.; Vazquez, F.; Weir, B.A.; Fitzgerald, M.E.; Tanaka, M.; et al. MTAP Deletion Confers Enhanced Dependency on the PRMT5 Arginine Methyltransferase in Cancer Cells. Science 2016, 351, 1214–1218. [Google Scholar] [CrossRef] [Green Version]
- Sanderson, S.M.; Mikhael, P.G.; Ramesh, V.; Dai, Z.; Locasale, J.W. Nutrient Availability Shapes Methionine Metabolism in P16/MTAP-Deleted Cells. Sci. Adv. 2019, 5, eaav7769. [Google Scholar] [CrossRef] [Green Version]
- Barekatain, Y.; Ackroyd, J.J.; Yan, V.C.; Khadka, S.; Wang, L.; Chen, K.C.; Poral, A.H.; Tran, T.; Georgiou, D.K.; Arthur, K.; et al. Homozygous MTAP Deletion in Primary Human Glioblastoma Is Not Associated with Elevation of Methylthioadenosine. Nat. Commun. 2021, 12, 4228. [Google Scholar] [CrossRef] [PubMed]
- Locasale, J.W.; Melman, T.; Song, S.; Yang, X.; Swanson, K.D.; Cantley, L.C.; Wong, E.T.; Asara, J.M. Metabolomics of Human Cerebrospinal Fluid Identifies Signatures of Malignant Glioma. Mol. Cell. Proteomics 2012, 11, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Ulanovskaya, O.A.; Zuhl, A.M.; Cravatt, B.F. NNMT Promotes Epigenetic Remodeling in Cancer by Creating a Metabolic Methylation Sink. Nat. Chem. Biol. 2013, 9, 300–306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palanichamy, K.; Kanji, S.; Gordon, N.; Thirumoorthy, K.; Jacob, J.R.; Litzenberg, K.T.; Patel, D.; Chakravarti, A. NNMT Silencing Activates Tumor Suppressor PP2A, Inactivates Oncogenic STKs, and Inhibits Tumor Forming Ability. Clin. Cancer Res. 2017, 23, 2325–2334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eckert, M.A.; Coscia, F.; Chryplewicz, A.; Chang, J.W.; Hernandez, K.M.; Pan, S.; Tienda, S.M.; Nahotko, D.A.; Li, G.; Blaženović, I.; et al. Proteomics Reveals NNMT as a Master Metabolic Regulator of Cancer-Associated Fibroblasts. Nature 2019, 569, 723–728. [Google Scholar] [CrossRef] [PubMed]
- Bowman, R.L.; Wang, Q.; Carro, A.; Verhaak, R.G.W.; Squatrito, M. GlioVis Data Portal for Visualization and Analysis of Brain Tumor Expression Datasets. Neuro. Oncol. 2017, 19, 138–141. [Google Scholar] [CrossRef] [Green Version]
- Matta, E.; Kiribayeva, A.; Khassenov, B.; Matkarimov, B.T.; Ishchenko, A.A. Insight into DNA Substrate Specificity of PARP1-Catalysed DNA Poly(ADP-Ribosyl)Ation. Sci. Rep. 2020, 10, 3699. [Google Scholar] [CrossRef] [Green Version]
- Sim, H.W.; Galanis, E.; Khasraw, M. PARP Inhibitors in Glioma: A Review of Therapeutic Opportunities. Cancers 2022, 14, 1003. [Google Scholar] [CrossRef]
- Audrito, V.; Messana, V.G.; Deaglio, S. NAMPT and NAPRT: Two Metabolic Enzymes with Key Roles in Inflammation. Front. Oncol. 2020, 10, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Kujundžić, R.N.; Prpić, M.; Đaković, N.; Dabelić, N.; Tomljanović, M.; Mojzeš, A.; Fröbe, A.; Trošelj, K.G. Nicotinamide N-methyltransferase in Acquisition of Stem Cell Properties and Therapy Resistance in Cancer. Int. J. Mol. Sci. 2021, 22, 5681. [Google Scholar] [CrossRef]
- Lautrup, S.; Sinclair, D.A.; Mattson, M.P.; Fang, E.F. NAD+ in Brain Aging and Neurodegenerative Disorders. Cell Metab. 2019, 30, 630–655. [Google Scholar] [CrossRef]
- Bockwoldt, M.; Houry, D.; Niere, M.; Gossmann, T.I.; Reinartz, I.; Schug, A.; Ziegler, M.; Heiland, I. Identification of Evolutionary and Kinetic Drivers of NAD-Dependent Signaling. Proc. Natl. Acad. Sci. USA 2019, 116, 15957–15966. [Google Scholar] [CrossRef] [Green Version]
- Sharma, P.; Xu, J.; Williams, K.; Easley, M.; Elder, J.B.; Lonser, R.; Lang, F.F.; Lapalombella, R.; Sampath, D.; Puduvalli, V.K. Inhibition of Nicotinamide Phosphoribosyltransferase (NAMPT), the Rate-Limiting Enzyme of the Nicotinamide Adenine Dinucleotide (NAD) Salvage Pathway, to Target Glioma Heterogeneity through Mitochondrial Oxidative Stress. Neuro. Oncol. 2022, 24, 229–244. [Google Scholar] [CrossRef]
- Adams, S.; Teo, C.; McDonald, K.L.; Zinger, A.; Bustamante, S.; Lim, C.K.; Sundaram, G.; Braidy, N.; Brew, B.J.; Guillemin, G.J. Involvement of the Kynurenine Pathway in Human Glioma Pathophysiology. PLoS ONE 2014, 9, e112945. [Google Scholar] [CrossRef] [Green Version]
- Panitz, V.; Končarević, S.; Sadik, A.; Friedel, D.; Bausbacher, T.; Trump, S.; Farztdinov, V.; Schulz, S.; Sievers, P.; Schmidt, S.; et al. Tryptophan Metabolism Is Inversely Regulated in the Tumor and Blood of Patients with Glioblastoma. Theranostics 2021, 11, 9217–9233. [Google Scholar] [CrossRef]
- Guastella, A.R.; Michelhaugh, S.K.; Klinger, N.V.; Fadel, H.A.; Kiousis, S.; Ali-Fehmi, R.; Kupsky, W.J.; Juhász, C.; Mittal, S. Investigation of the Aryl Hydrocarbon Receptor and the Intrinsic Tumoral Component of the Kynurenine Pathway of Tryptophan Metabolism in Primary Brain Tumors. J. Neurooncol. 2018, 139, 239–249. [Google Scholar] [CrossRef]
- Zaragoza-Ojeda, M.; Apatiga-Vega, E.; Arenas-Huertero, F. Role of Aryl Hydrocarbon Receptor in Central Nervous System Tumors: Biological and Therapeutic Implications (Review). Oncol. Lett. 2021, 21, 1–12. [Google Scholar] [CrossRef]
- Jin, U.H.; Karki, K.; Cheng, Y.; Michelhaugh, S.K.; Mittal, S.; Safe, S. The Aryl Hydrocarbon Receptor Is a Tumor Suppressor-like Gene in Glioblastoma. J. Biol. Chem. 2019, 294, 11342–11353. [Google Scholar] [CrossRef]
- Herring, J.L.; Rogstad, D.K.; Sowers, L.C. Enzymatic Methylation of DNA in Cultured Human Cells Studied by Stable Isotope Incorporation and Mass Spectrometry. Chem Res Toxicol. 2009, 22, 1060–1068. [Google Scholar] [CrossRef] [Green Version]
- Robertson, K.D.; Jones, P.A. DNA Methylation: Past, Present and Future Directions. Carcinogenesis 2000, 21, 461–467. [Google Scholar] [CrossRef] [Green Version]
- Weng, J.Y.; Salazar, N. DNA Methylation Analysis Identifies Patterns in Progressive Glioma Grades to Predict Patient Survival. Int. J. Mol. Sci. 2021, 22, 1020. [Google Scholar] [CrossRef]
- Tornaletti, S.; Pfeifer, G.P. Complete and Tissue-Independent Methylation of CpG Sites in the P53 Gene: Implications for Mutations in Human Cancers. Oncogene 1995, 10, 1493–1499. [Google Scholar]
- Buchmuller, B.C.; Kosel, B.; Summerer, D. Complete Profiling of Methyl-CpG-Binding Domains for Combinations of Cytosine Modifications at CpG Dinucleotides Reveals Differential Read-out in Normal and Rett-Associated States. Sci. Rep. 2020, 10, 4053. [Google Scholar] [CrossRef]
- Ohgaki, H.; Kleihues, P. Epidemiology and Etiology of Gliomas. Acta Neuropathol. 2005, 109, 93–108. [Google Scholar] [CrossRef]
- Sowers, J.L.; Johnson, K.M.; Conrad, C.; Patterson, J.T.; Sowers, L.C. The Role of Inflammation in Brain Cancer. Adv Exp Med Biol. 2014, 816, 75–105. [Google Scholar] [CrossRef]
- Zhang, M.; Yang, D.; Gold, B. Origin of Mutations in Genes Associated with Human Glioblastoma Multiform Cancer: Random Polymerase Errors versus Deamination. Heliyon 2019, 5, e01265. [Google Scholar] [CrossRef] [Green Version]
- Costa, G.; Barra, V.; Lentini, L.; Cilluffo, D.; Di Leonardo, A. DNA Demethylation Caused By 5-Aza-2’-Deoxycytidine Induces Mitotic Alterations and Aneuploidy. Oncotarget 2016, 7, 3726–3739. [Google Scholar] [CrossRef] [Green Version]
- González, B.; Navarro-jim, M.; Gennaro, A.; Jansen, S.M.; Granada, I.; Perucho, M.; Alonso, S. Somatic Hypomethylation of Pericentromeric SST1 Repeats And Tetraploidization in Human Colorectal Cancer Cells. Cancers 2021, 13, 5353. [Google Scholar] [CrossRef]
- Kanai, Y. Amino Acid Transporter LAT1 (SLC7A5) as a Molecular Target for Cancer Diagnosis and Therapeutics. Pharmacol. Ther. 2022, 230, 107964. [Google Scholar] [CrossRef]
- Zhen, H.; Kawai, N.; Okada, M.; Okubo, S.; Tamiya, T.; Zhang, X.; Liu, W.; Huo, J.; Fei, Z. Relation of 4F2hc Expression to Pathological Grade Proliferation and Angiogenesis in Human Brain Gliomas. Chin. J. Clin. Oncol. 2012, 39, 1161–1164. [Google Scholar] [CrossRef]
- Shen, J.; Song, R.; Hodges, T.R.; Heimberger, A.B.; Zhao, H. Identification of Metabolites in Plasma for Predicting Survival in Glioblastoma. Mol. Carcinog. 2018, 57, 1078–1084. [Google Scholar] [CrossRef]
- Phillips, H.S.; Kharbanda, S.; Chen, R.; Forrest, W.F.; Soriano, R.H.; Wu, T.D.; Misra, A.; Nigro, J.M.; Colman, H.; Soroceanu, L.; et al. Molecular Subclasses of High-Grade Glioma Predict Prognosis, Delineate a Pattern of Disease Progression, and Resemble Stages in Neurogenesis. Cancer Cell 2006, 9, 157–173. [Google Scholar] [CrossRef] [Green Version]
- Segerman, A.; Niklasson, M.; Haglund, C.; Bergström, T.; Jarvius, M.; Xie, Y.; Westermark, A.; Sönmez, D.; Hermansson, A.; Kastemar, M.; et al. Clonal Variation in Drug and Radiation Response among Glioma-Initiating Cells Is Linked to Proneural-Mesenchymal Transition. Cell Rep. 2016, 17, 2994–3009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kling, T.; Ferrarese, R.; Ó hAilín, D.; Johansson, P.; Heiland, D.H.; Dai, F.; Vasilikos, I.; Weyerbrock, A.; Jörnsten, R.; Carro, M.S.; et al. Integrative Modeling Reveals Annexin A2-Mediated Epigenetic Control of Mesenchymal Glioblastoma. EBioMedicine 2016, 12, 72–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minniti, G.; Salvati, M.; Arcella, A.; Buttarelli, F.; D’Elia, A.; Lanzetta, G.; Esposito, V.; Scarpino, S.; Maurizi Enrici, R.; Giangaspero, F. Correlation between O6-Methylguanine-DNA Methyltransferase and Survival in Elderly Patients with Glioblastoma Treated with Radiotherapy plus Concomitant and Adjuvant Temozolomide. J. Neurooncol. 2011, 102, 311–316. [Google Scholar] [CrossRef] [PubMed]
- Binabaj, M.M.; Bahrami, A.; ShahidSales, S.; Joodi, M.; Joudi Mashhad, M.; Hassanian, S.M.; Anvari, K.; Avan, A. The Prognostic Value of MGMT Promoter Methylation in Glioblastoma: A Meta-Analysis of Clinical Trials. J. Cell. Physiol. 2018, 233, 378–386. [Google Scholar] [CrossRef]
- Paz, M.F.; Fraga, M.F.; Avila, S.; Guo, M.; Pollan, M.; Herman, J.G.; Esteller, M. A Systematic Profile of DNA Methylation in Human Cancer Cell Lines. Cancer Res. 2003, 63, 1114–1121. [Google Scholar]
- Karpf, A.R.; Matsui, S.I. Genetic Disruption of Cytosine DNA Methyltransferase Enzymes Induces Chromosomal Instability in Human Cancer Cells. Cancer Res. 2005, 65, 8635–8639. [Google Scholar] [CrossRef] [Green Version]
- Barra, V.; Schillaci, T.; Lentini, L.; Costa, G.; Di Leonardo, A. Bypass of Cell Cycle Arrest Induced by Transient DNMT1 Post-Transcriptional Silencing Triggers Aneuploidy in Human Cells. Cell Div. 2012, 7, 2. [Google Scholar] [CrossRef] [Green Version]
- Bergo, A.; Strollo, M.; Gai, M.; Barbiero, I.; Stefanelli, G.; Sertic, S.; Gigli, C.C.; Di Cunto, F.; Kilstrup-Nielsen, C.; Landsberger, N. Methyl-CpG Binding Protein 2 (MeCP2) Localizes at the Centrosome and Is Required for Proper Mitotic Spindle Organization. J. Biol. Chem. 2015, 290, 3223–3237. [Google Scholar] [CrossRef] [Green Version]
- Roussel-Gervais, A.; Naciri, I.; Kirsh, O.; Kasprzyk, L.; Velasco, G.; Grillo, G.; Dubus, P.; Defossez, P.A. Loss of the Methyl-CpG-Binding Protein ZBTB4 Alters Mitotic Checkpoint, Increases Aneuploidy, and Promotes Tumorigenesis. Cancer Res. 2017, 77, 62–73. [Google Scholar] [CrossRef] [Green Version]
- Hervouet, E.; Debien, E.; Campion, L.; Charbord, J.; Menanteau, J.; Vallette, F.M.; Cartron, P.F. Folate Supplementation Limits the Aggressiveness of Glioma via the Remethylation of DNA Repeats Element and Genes Governing Apoptosis and Proliferation. Clin. Cancer Res. 2009, 15, 3519–3529. [Google Scholar] [CrossRef] [Green Version]
- Cartron, P.F.; Hervouet, E.; Debien, E.; Olivier, C.; Pouliquen, D.; Menanteau, J.; Loussouarn, D.; Martin, S.A.; Campone, M.; Vallette, F.M. Folate Supplementation Limits the Tumourigenesis in Rodent Models of Gliomagenesis. Eur. J. Cancer 2012, 48, 2431–2441. [Google Scholar] [CrossRef]
- Jung, J.; Kim, L.J.Y.; Wang, X.; Sanvoranart, T.; Hubert, C.G.; Prager, B.C.; Wu, Q.; Wallace, L.C.; Jin, X.; Mack, S.C.; et al. Nicotinamide Metabolism Regulates Glioblastoma Stem Cell Maintenance. JCI Insight 2017, 2, e90019. [Google Scholar] [CrossRef] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sowers, M.L.; Sowers, L.C. Glioblastoma and Methionine Addiction. Int. J. Mol. Sci. 2022, 23, 7156. https://doi.org/10.3390/ijms23137156
Sowers ML, Sowers LC. Glioblastoma and Methionine Addiction. International Journal of Molecular Sciences. 2022; 23(13):7156. https://doi.org/10.3390/ijms23137156
Chicago/Turabian StyleSowers, Mark L., and Lawrence C. Sowers. 2022. "Glioblastoma and Methionine Addiction" International Journal of Molecular Sciences 23, no. 13: 7156. https://doi.org/10.3390/ijms23137156
APA StyleSowers, M. L., & Sowers, L. C. (2022). Glioblastoma and Methionine Addiction. International Journal of Molecular Sciences, 23(13), 7156. https://doi.org/10.3390/ijms23137156