Abstract
Huntington’s Disease (HD) is a fatal autosomal dominant neurodegenerative disease manifested through motor dysfunction and cognitive deficits. Decreased fertility is also observed in HD animal models and HD male patients, due to altered spermatogenesis and sperm function, thus resulting in reduced fertilization potential. Although some pharmaceuticals are currently utilized to mitigate HD symptoms, an effective treatment that remedies the pathogenesis of the disease is yet to be approved by the FDA. Identification of genes and relevant diagnostic biomarkers and therapeutic target pathways including glycolysis and mitochondrial complex-I-dependent respiration may be advantageous for early diagnosis, management, and treatment of the disease. This review addresses the HD pathway in neuronal and sperm metabolism, including relevant gene and protein expression in both neurons and spermatozoa, indicated in the pathogenesis of HD. Furthermore, zinc-containing and zinc-interacting proteins regulate and/or are regulated by zinc ion homeostasis in both neurons and spermatozoa. Therefore, this review also aims to explore the comparative role of zinc in both neuronal and sperm function. Ongoing studies aim to characterize the products of genes implicated in HD pathogenesis that are expressed in both neurons and spermatozoa to facilitate studies of future treatment avenues in HD and HD-related male infertility. The emerging link between zinc homeostasis and the HD pathway could lead to new treatments and diagnostic methods linking genetic sperm defects with somatic comorbidities.
1. Introduction
Huntington’s disease (HD) is an autosomal dominant, late-onset neurodegenerative disease caused by CAG repeat expansion in exon 1 of the Huntingtin gene on chromosome 4. It is characterized by disordered movement and cognitive decline in both humans and animal models [1]. The worldwide prevalence of HD in the population of European descent is ~12 per 100,000 individuals [2] and is less common in African, Chinese, and Japanese descent [3]. The age of onset of the motor symptoms can occur from childhood to old age, with the mean age of onset ~45 years [4]. A CAG repeat of 36 or more is pathogenic, with 36–39 repeats conferring reduced penetrance, while 40 and more repeats include complete penetrance with full clinical expression [5,6]. A premotor onset of HD has been suggested by recent evidence [5]. Wild-type Huntingtin (HTT) is a large (350 kDa), ubiquitously expressed protein made up of polyQ N-terminus (as the result of CAG repeats) and several consensus areas called HEAT repeats [7]. HTT is highly expressed in striatal neurons of the central nervous system (CNS), shuttling between cytoplasm and nucleus [8,9,10]. Known functions of HTT are: (i) early embryonic development [11,12] and neurogenesis [13]; (ii) protein scaffolding [14], intracellular trafficking [15], and controlling spindle orientation in neuronal cells [16]; (iii) transcriptional regulator [17,18]; (iv) it is required for proper function of cortical and striatal excitatory synapses [19], and (v) autophagy [20]. A more descriptive overview of HTT functions can be found in [21]. Mutant Huntingtin (mHTT), caused by a minimum of 36 CAG trinucleotide repeats in the Huntingtin gene, results in a loss of efferent medium spiny neurons (MSNs) due to degeneration of striata [22]. The elongated polyglutamine tract at the amino terminus of mHTT affects posttranslational modification of HTT, resulting in altered subcellular distribution, stability, cleavage, and its function [23]. The presence of mHTT disrupts transcription in the brain as well as in peripheral tissues such as muscles and blood [24].
To date, there are numerous cellular (in vitro), invertebrate, and small animal models to study HD including human cellular models [25], C. elegans [26], D. melanogaster [27], and widely used mouse models [28]. Despite providing valuable knowledge on HD pathology, these models do not always faithfully recapitulate human disease pathology and/or adequately predict clinical response to treatment. Large animal models of HD, spanning the gap between small animal models and humans have been developed in the last decade and currently include sheep [29], minipig [30,31], and macaque [32,33]. The strengths and weaknesses of each said large animal model are discussed in [34]. The subject of the present review, i.e., the unexpected link between the HD pathway and male reproductive function, emerged from our ongoing research on sperm capacitation—a complex signaling and cell structure remodeling process that endows spermatozoa traveling within the female oviduct with fertilizing ability. In our continuous efforts to fully understand the process of sperm capacitation and particularly the role of zinc ions in its regulation, we recently performed a comparative proteomic study of zinc-containing/binding proteins (further zincoproteins) between ejaculated and capacitated spermatozoa [35]. To our surprise, we found that the Huntington’s and Parkinson’s disease pathways were the most represented in the sperm zincoproteome, making the spermatozoon a candidate model for studying neurodegenerative diseases.
This review aims to provide a background on HD pathogenesis, explore the comparative role of zinc in both neuronal function and sperm capacitation, and discuss sperm proteins encoded by genes implicated in HD to facilitate future treatment avenues.
2. Mechanisms of HD Pathogenesis
This section aims to provide concise background on HD pathogenesis rather than to give a detailed review, which is provided elsewhere [1,5,6,36,37,38]. Due to the immense complexity of its pathogenesis, implications of HD within striatal neurons are still under investigation; however, there are currently seven proposed mechanisms of pathogenesis, as depicted in Figure 1.
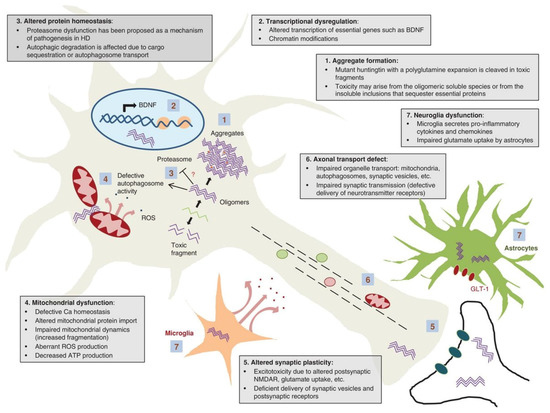
Figure 1.
Selected mechanisms of HD Pathogenesis. (1) Aggregation of mutant huntingtin, (2) Transcriptional dysfunction, (3) Proteasomal and autophagy dysfunction, (4) Mitochondrial dysfunction, (5) Alteration of synaptic plasticity, (6) Defective axonal transport, (7) Microglial dysfunction. The nuclear transport failure is not depicted. The figure was adapted from Jimenez-Sanchez et al. (2017) [37] and is replicated with permission from Cold Spring Harbor Laboratory Press, obtained on 15 December 2021.
The hallmark of HD is the presence of protein aggregates (1). These are initiated by N-terminal proteolytic cleavage of mHTT into N-terminal polyglutamine fragments that form amyloid fibrils and large inclusions. The cytotoxicity of N-terminal polyglutamine fragments is a question of debate; however, evidence suggests that N-terminal mHTT oligomers are toxic and the toxicity is mitigated by their aggregation into inclusions [39,40]. mHTT aggregates are found in both nuclei and perinuclei (cytoplasm) of a neuron, and it seems that nuclear aggregates are relatively benign when compared to cytoplasmic mHTT aggregates [41].
Binding and inactivation of a number of polyglutamine-containing transcription factors result in altered gene transcription (2), influencing a wide spectrum of cellular functions on the level of transcriptional networks. For instance, mHTT actively transports repressor peptide REST into the nucleus, where it forms a repressor complex on the BDNF gene, and reduces levels of BDNF protein—a striatal neuronal pro-survival factor, thus increasing the susceptibility of the striatum to HD [42]. Other transcription-related molecules that mHHT interacts with are: (i) transcription factors: CREB, CBP, SP1, NF-κB, NEUROD1, TP53, PCG-1α; (ii) transcription activators/repressors: TAFII130, CA150, NCOR, REST/NRSF; and (iii) nuclear receptors: LXRα, PPARγ, TRα1 [36]. MHHT interferes with gene expression on the epigenetic level by inducing an imbalance between histone acetylation and deacetylation, DNA methylation and demethylation, and non-coding RNA levels [43].
Another consequence of polyglutamine-expanded mHTT expression is compromised protein degradation systems (3). Impairment of ubiquitin-proteasome system (UPS) is explained by sequestration of UPS components into inclusions or interaction between (and saturation of) proteasomes and aggregation-resistant mHTT [44]. The impaired proteasomal activity is initially compensated by the lysosomal autophagy system that eventually becomes compromised with time in chronic mHTT expression [44,45]. The impairment of mHTT recognition by the cell, endoplasmic reticulum dysfunction in its interaction with N-terminal polyglutamine fragments, as well as disturbance of autophagy regulation by mHTT, ultimately leads to a systemic failure of proteostasis in neurons [46].
Mitochondrial dysfunction (4) is illustrated through decreased transcription of mitochondrial genes due to repression of PGC-1α (peroxisome proliferator-activating receptor coactivator-1α), which regulates the expression of genes mediating mitochondrial biogenesis and respiration, leading to increased susceptibility to oxidative stress and neuronal degeneration [47,48]. Furthermore, mHTT induces an imbalance in mitochondrial dynamics by increasing the transcription of DRP1, a fission-mediating GTPase, resulting in the accumulation of the impaired mitochondria in the cytoplasm [49,50]. In addition, mHTT interacts with both the outer and inner mitochondrial membrane, resulting in defective Ca2+ regulation and buffering capacity, and altering the import of mitochondrial proteins due to inhibition of TIM23, respectively [5,37]. The results of such interactions are failures in complex II and IV of the respiratory chain; an influx of Ca2+ into mitochondria, promoting the release of cytochrome C; increased ROS production; and ATP synthesis defect [47]. On the macromolecular level, mHTT inhibits mitophagy by affecting the formation of mitophagy initiation protein complexes [51] and impedes axonal transport of mitochondria [52].
Alteration of synaptic plasticity (5) is likely due to mHTT aggregates blocking axons, sequestration of motor proteins, or wild-type HTT loss of function. Mutant Huntingtin impedes the delivery of GABA and AMPA receptors to neuronal membranes, therefore inhibiting synaptic excitability, as well as transport and release of BNDF or retrograde transport of its receptor TrkB, necessary to promote survival signals in the neuronal cell body [50,53]. Excitotoxicity is a detrimental consequence of altered NMDAR signaling and glutamate uptake due to mHTT expression [50,53]. Excitotoxicity is instigated through excess extracellular glutamate via increased glutamate release or impaired uptake due to downregulation of GLT1 [37,54]. This leads to constituent stimulation of NMDAR and ultimately the death of striatal neurons. Excitotoxicity is central to other neurological disorders including Alzheimer’s disease, Parkinson’s disease, and amyotrophic lateral sclerosis (ALS/Lou Gehrig’s disease) [54].
Fast axonal transport of organelles (6) including mitochondria, autophagosomes, and synaptic vesicles is disrupted by mHTT. Impaired transport of organelles occurs either by the direct interference of mHTT with motor protein function of dynein, kinesin, and actin; and/or indirect interaction of mHTT with Huntingtin-Associated Protein 1 (HAP1) through the disintegration of the HAP1/DCTN1 complex followed by its detachment from the microtubules resulting in suppression of the rapid axonal transport [36].
Microglial and astrocyte function (7) is dysregulated by mHTT by inducing the release of proinflammatory cytokine IL-6 [55]. As mentioned earlier, mHTT positively regulates the NF-κB signaling pathway resulting in the release of pro-inflammatory cytokines and chemokines such as IL-6 and IL-8, respectively [56]. Cytokine profile of HD patients had increased levels of IL-4, IL-10, and TNF-α with the disease progression when compared to the control group [57]. Immune and inflammatory changes also occur in peripheral organs during HD development and progression [58].
Last but not least, mHTT disrupts the nuclear transport cycle by sequestering RanGAP1, NUP62, and NUP88 in mHTT aggregates [59].
3. Treatments of HD
The same as in the previous section, the purpose of this section is to give a concise overview of currently available treatments, with an in-depth review available elsewhere [1,6,37,60,61]. Current pharmaceutical treatments approved by the FDA in the United States remain symptomatic, only targeting motor, cognitive, and psychiatric symptoms. No disease-modifying treatments are currently available [1].
3.1. Symptomatic Treatment Approaches
Symptomatic treatment management aims to improve HD patients’ quality of life. Two examples include Tetrabenazine and Deutetrabenazine, which aid in the management of chorea symptoms caused by neurodegeneration in HD. Chorea, which occurs in early-onset and worsens over time, is characterized by jerky, involuntary movements caused by generalized muscle contraction. Active ingredients in Tetrabenazine and Deutetrabenazine decrease uptake of monoamines (dopamine, serotonin, norepinephrine) in synaptic vesicles by binding and inhibiting Vesicular Monoamine Transporter type-2 (VMAT2) receptors, leading to depletion of monoamines from nerve terminals [62,63]. Tetrabenazine is often characterized by potential adverse effects including hypotension, hyperprolactinemia, suicidal thoughts, and depression [63]. Deutetrabenazine, a derivation of Tetrabenazine, is notably safer than Tetrabenazine as indicated by the mitigated prevalence of adverse events including depression, somnolence, and insomnia [62]. In addition, it is comparably effective at lower dosages.
Cognitive symptoms including decreased processing speed, poor attention, poor problem-solving abilities, and poor memory retrieval can manifest decades before the onset of motor symptoms in HD patients. To date, there is no effective therapy for HD-associated dementia. Metamatine that is used to treat Alzheimer’s dementia seems promising with a potential neuroprotective effect with long-term use [64].
The most commonly reported psychiatric symptom in HD patients is major depressive disorder [65]. Although there is no guide to pharmacotherapy of HD depression, selective serotonin reuptake inhibitors have been shown to be beneficial by HD patients [66].
3.2. Clinical Trials
As stated previously, no treatment for HD is currently available that would target the disease itself. However, the potential efficacy of numerous treatment approaches for HD is currently being tested through clinical trials. These are also available at ClinicalTrials.gov (accessed on 16 March 2021). A summary of selected interventions and their mechanisms are discussed below.
3.2.1. Genetic Manipulation
Various approaches, as listed in Table 1, aim to destroy or inactivate HTT/mHTT translation through gene silencing and are currently undergoing clinical trials. Both allele-specific and non-specific approaches have proven effective in mouse models. Allele-specific and non-specific mouse models show a significant correlation between reduced mHTT expression and decreased atrophy, improved motor function, and improved cognition [61]. Although such methods prove effective, drug developers may ultimately need to combine gene silencing with immunomodulatory drugs to complement immune activation. The anti-semaphorin 4D antibody VX15/2503 is a monoclonal antibody against semaphorin 4D, a transmembrane protein modulating microglial activation, oligodendrocyte viability, and permeability of the blood/brain barrier [67].

Table 1.
Huntingtin lowering therapies targeting DNA and RNA.
Allele-specific, WVE-120101, and WVE-120102 are mHTT-specific lowering antisense drugs that target SNP variants linked to CAG expansions, leading to pre-mRNA degradation [61,67]. mHTT-specific drugs by Imperial College London and Sangamo Therapeutics under preclinical trials integrate zinc-finger domains into adeno-associated viruses (AAVs) and lentiviral vectors to modify effector elements and repress transcription of mHTT. In mouse models, a 78% reduction of mRNA was achieved, resulting in reduced mHTT aggregate levels if administered early in disease onset [61].
Allele-non-specific, HTT-lowering miRNA-based gene silencing approaches include AMT-130 and VY-HTT01. Both harness a particular RNAi pathway to silence both HTT and mHTT expression through the use of viral vectors [61,67]. RNA-induced silencing complexes target mRNA for degradation, yielding reduced protein expression [61]. After an exact dose of AMT-130 to achieve knockout was identified, the FDA granted fast-track designation to the first AAV gene therapy approved for human trials. In monkey models, VT-HTT01 has shown 50% improvement in motor function [61]. Direct injection of AMT-130 and VY-HTT01 AAVs or lentiviral vectors could provide lifelong silencing of mHTT [61,67]. An additional non-specific drug, RG6042, aids in the prevention of toxic fragment buildup, as it silences the expression of all HTT forms to inhibit the synthesis of toxic byproducts and prevents neurodegeneration [67]. This method has proven effective through the significant correlation between lowered mHTT levels and function, cognitive, and motor scores [61]. For a more in-depth review of mHTT lowering therapies, see [68].
3.2.2. Mitochondrial Function and Biogenesis
Three clinical trials are pending with relevance to increase mitochondrial function: (i) Fenofibrate is a peroxisome proliferator-activated receptor (PPAR) agonist that may induce activation of PGC-1α and aid in mitochondrial biogenesis [69]; (ii) Triheptanoin is a C7 fatty acid oil that is hydrolyzed into acetyl-CoA and propionyl-CoA and may provide substrates to the Krebs cycle, thus increasing ATP availability and correcting the bioenergetic profile in the HD brain [70]; and (iii) Metforminis an AMP-activated protein kinase (AMPK) activator commonly used to treat type 2 diabetes that has been shown to be protective of polyQ-expressing models [71,72], increasing the lifespan of HD transgenic mice [73] and increasing cognitive function in HD patients [74].
3.2.3. Modulation of BDNF Levels
A clinical trial of Pridopidine, a first-in-class sigma-1 receptor (S1R) agonist and dopamine receptor antagonist [75], is underway. S1R plays a key role in neuroprotection through the increased production of BDNF, a striatal neuronal pro-survival factor, levels.
3.2.4. Synaptic Modulation
A clinical trial of Neflamapimod, a small molecule that can penetrate the brain and inhibit the enzyme P38α (MAPK14), is underway. P38α is typically involved in regulating inflammation, and its chronic activation negatively affects nerve cell communication due to excessive inflammation [76].
3.2.5. Stem Cell Therapies
Considerable potential for the treatment of neurodegenerative diseases is represented by stem cell therapies with neuronal progenitor cells derived from induced pluripotent stem cells (iPST) [77]. Studies have shown that transplanted iPST can differentiate into functional neurons and improve partial striatal function and metabolism after implantation in a rat HD model [78,79]. Three clinical trials are pending to assess the dose, safety, and efficacy of intravenous delivery of stem cells in human HD patients [1].
4. Zinc Homeostasis, Reproduction and Huntington’s Disease
Both the brain and the testes contain high zinc ion (Zn2+) concentrations: brain ~70 µg·g−1 dry weight [80] and testes ~74.6 µg·g−1 dry weight [81]. An increased concentration leads to neurotoxicity [82]/reprotoxicity [83], while decreased concentration leads to impaired brain functioning [84] and impaired spermatogenesis [85], respectively. Therefore, zinc homeostasis needs to be tightly regulated in order for both the central nervous system and reproduction to function properly. In this section, similarities in neuronal and sperm zinc homeostasis will be discussed.
4.1. Neuronal Zinc Homeostasis
Zinc is one of the most prevalent and essential elements involved in brain function, tightly regulated for its proper function, and a potent neurotoxin when dysregulated. Zinc plays several roles in brain function including neurotransmission and sensory processing, and activation of both pro-survival and pro-death neuronal signaling pathways [86]. Zinc homeostasis in the brain is maintained by three groups of proteins: (i) zinc importers (ZIPs), transporting Zn2+ into the cytosol; (ii) zinc transporters (ZnT), transporting Zn2+ out of cytosol into both membranous organelles and to the extracellular environment; and (iii) metallothioneins (MTs), actively increasing/decreasing concentration of free/unbound Zn2+ in the cytosol (Figure 2) [87,88]. Expression of zinc transporter 3 (ZnT3) localized on the membrane of synaptic vesicles is required for the transport of zinc from the cytosol into presynaptic vesicles. The concentration of vesicular zinc (10–15% of total brain zinc) is dependent on the abundance of ZnT3 transporters [89]. In excitotoxic settings and neurodegeneration, MTs metal-binding redox-sensitive proteins mobilize large amounts of Zn2+ to re-establish vesicular neuronal zinc homeostasis [90]. PrPc (cellular prion protein) promotes zinc uptake as PrPc binds the ion via octapeptide repeats to facilitate the transport of the metal across the membrane through the AMPA receptor channel [91]. PrPc contributes to neuronal zinc homeostasis in three ways: by acting as a zinc sink, prompting zinc uptake via endocytosis into the postsynaptic neuron, and serving as a zinc sensor, monitoring levels of the ion in the synaptic cleft [91]. If extracellular zinc concentration rises above a certain threshold, a signaling cascade is triggered to increase the transcription of metal transporter genes. Excess zinc must be cleared from the synaptic cleft to prevent neuronal damage [91]. In this way, PrPc acts as a zinc sensor in coordination with AMPA receptors to maintain healthy levels of zinc in the synaptic cleft.
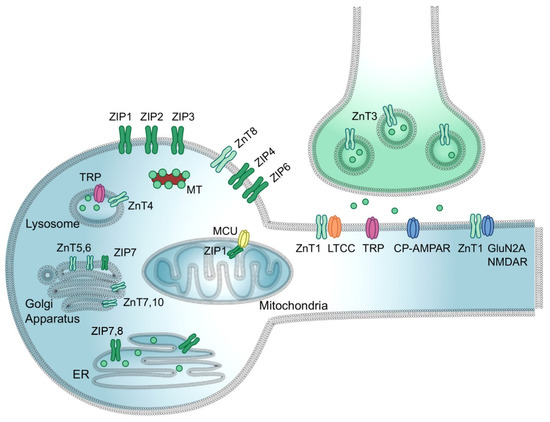
Figure 2.
Zinc transport in neurons. Zn2+ is loaded into presynaptic vesicles via ZnT3, released into the synaptic cleft, and conducted to the cytosol of a postsynaptic neuron via L-type calcium channels (LTCC), transient receptor potential channels (TRP), NMDA receptors, and calcium-permeable AMPA receptor (CP-AMPAR). Within the neuron, Zn2+ uptake to/release from specific organelles is mediated by organelle-specific ZIPs and ZnTs. Zn2+ import into mitochondrial matrix is mediated by mitochondrial Ca2+/cation uniporter (MCU) coupled with ZIP1. Free Zn2+ concertation within the cytosol is buffered by metallothioneins (MT). The figure was adapted from Krall et al. [86] and replicated with permission from Elsevier Ltd., license #5203200109216, obtained on 6 December 2021.
Vesicular zinc serves as a neuromodulator in glutamatergic neurons for glutamate signaling and cognitive ability [89]. In its role as a neuromodulator, Zn2+ is released during synaptic transmission and binds to pre- or postsynaptic membranes, essentially translocating from presynaptic terminals to postsynaptic neurons (Figure 2) [92]. When released into the synaptic cleft with glutamate during excitatory neurotransmission, Zn2+ exerts downstream inhibitory effects on NMDAR and AMPAR [90]. In this way, Zn2+ protects neurons from excitotoxicity and attenuates excess pre-synaptic glutamate [89,90].
Zn2+ is critical for the activation state of matrix metalloproteinases (MMPs) that are involved in BDNF maturation from its precursor form proBDNF [93]. Pro-BDNF/BDNF balance is critical for neuronal function, as Pro-BDNF activates neuronal death-related pathways and BDNF promotes long-term memory processes and neuronal survival [90]. Zn2+ also activates BDNF receptor TrKB activating downstream signal transduction [94].
4.2. Zinc Dysregulation in HD
Zinc homeostasis is dysregulated in a wide range of neurological diseases including Alzheimer’s disease (AD), amyotrophic lateral sclerosis (ALS), traumatic brain injury (TBI), hypoglycemia-induced neuronal death, ischemia-induced neuronal death, schizophrenia (SCZ), major depressive disorder, and Parkinson’s disease (PD), as well as HD [90,92,95].
Zn2+ dysregulation is believed to contribute to HD in a condition characterized by the occurrence of a chronic glutamatergic overdrive [90]. In such a condition, the aberrant release of glutamate and Zn2+ from presynaptic vesicles occurs. Glutamate stimulates NMDAR that causes Ca2+ influx to postsynaptic neurons, while Zn2+ enters via respective channels (Figure 2). Increased cytosolic Ca2+ concentration triggers irreversible cytotoxic effects including mitochondrial dysfunction, oxidative stress, and reduced ZnT3 expression. Following Zn2+ uptake via mitochondrial Ca2+/cation uniporters (MCU), the function of electron transport chain complexes and α-ketoglutarate dehydrogenase is inhibited, leading to ROS production and metabolic failure. Mitochondrial uptake of Zn2+ also triggers activation of mitochondrial permeability transition pores, promoting neuronal apoptosis [90]. Cytosolic Zn2+ activates neuronal isoform of nitric oxide synthase (nNOS), promoting increased NO production, as well as NADPH oxidase resulting in overproduction of superoxide anion (). Free combined with NO yields peroxynitrite (ONOO−), a neurotoxic reactive nitrogen species (RNS) [90]. Zn2+-driven ROS and RNS promote further metal release from MTs, resulting in a positive feedback loop of dyshomeostasis. Motor neurons are especially vulnerable to oxidative stress and dysregulation because they express CP-AMPARS, which offers a rationale for the degeneration of motor neurons [90]. Other factors leading to neuronal failure include Zn2+-driven NADPH oxidase activation, NAD+ depletion, GAPDH inhibition, ATP breakdown, and outward K+ currents [90]. As a result, patients often express a mix of neurotoxic proteins including mutant prion proteins, tau, and synuclein [90].
In addition, studies indicate that vesicular zinc deficiency also causes neuronal apoptosis and memory deficit in HD. mHTT inhibits the binding of Sp2 to the ZnT3 promoter, downregulating ZnT3 expression in motor neurons [89]. Transgenic mice models with a loss of synaptic vesicular zinc and disruption of vesicular zinc homeostasis have shown a decrease in dendritic spines, defective presynaptic synaptosome-associated protein 25 (SNAP25) and postsynaptic PSD95, early synaptic dysfunction, neurodegeneration, and cognitive decline in HD [89]. In addition, a loss of vesicular zinc leads to glutamate-mediated excitotoxic neuronal death [89].
4.3. Role of Zinc in Sperm Maturation
An increasing level of attention has been given to the role of Zn2+ in male reproduction since our last review [96]. Zn2+ is essentially necessary for almost every step of sperm maturation. Similarly, as in neuronal cells, zinc trafficking is mediated by ZnTs and ZIPs during sperm maturation. In the human male reproductive organs, ZnTs are expressed in the testis (Sertoli cells, ZnT1) and epididymis (ZnT1,2), but no ZnT expression was reported in spermatozoa [97]. On the contrary, ZIP1, 5, 6, and 8 are expressed in human testicular and epididymal spermatozoa and the epithelial/stromal cells of the testis and epididymis.
Starting with spermatogenesis, the Zn2+ requirement has been shown for the maintenance of germ cells and progression of spermatogenesis in the Japanese eel [85], and spermiogenesis activation in C. elegans [98,99]. Further, Zn2+ is vital for the maintenance of normal spermatogenesis in rats [100,101,102], mice [103], and rams [104]; by maintaining male germ cell proliferation [100,101,102], by expression of testis germ cell-specific genes during the cell differentiation and spermatogenesis [104], and by regulating Leydig cell generation [100]. Zn2+ accumulates in spermatocytes with its presence in the nucleus and chromatin [105], where it participates in the histone for protamine substitution event [98]; is required for the proper functioning of zinc finger protein (ZFP) transcription factors, such as ZFP185 or ZFP318 [106,107]; and stabilization of sperm chromatin with zinc bridges [108]. Furthermore, Zn2+ stabilizes nascent outer dense fibers (ODF) of the sperm flagellum and protects them from premature oxidation [109,110]. Zinc deficiency has therefore an adverse effect on spermatogenesis and the general abnormalities include hypogonadism and Leydig cell damage resulting in a deficiency of sex hormone production and impaired spermatogenesis, as well as inflammation, antioxidant depletion, and sperm death leading to male infertility [111]. The main mechanism of zinc deficiency seems to be oxidative stress induced by ROS that causes depletion of antioxidants, sperm DNA fragmentation, lipid peroxidation, and apoptosis; and, consequently, poor sperm quality [111].
During the epididymal transit of rat spermatozoa, approximately 60% of Zn2+ content is lost from epididymal spermatozoa [112,113] and reabsorbed by the epididymal epithelium [114]. This maturation step allows for the oxidation of sulfhydryl groups thus stiffening the ODF, and is mandatory to obtain functional ODF, a prerequisite for progressive motility. A thorough study was performed on zinc trafficking in human spermatozoa during the passage through the human male reproductive tract [97]. The authors found that contrary to the aforementioned rat studies, the total human sperm zinc content increases as spermatozoa pass through the male reproductive tract (2.56, 12.58, and 40.48 ng Zn2+/106 sperm in testicular, epididymal, and ejaculated spermatozoa, respectively). The autometallography assessment of Zn2+ accumulation revealed that Zn2+ is preferentially accumulated within the residual cytoplasm of immature testicular and epididymal spermatozoa, while in the ejaculated spermatozoa Zn2+ is localized predominantly in the midpiece, connecting piece, and flagellum [97]. Thus, Zn2+ trafficking likely plays a role in post-testicular sperm maturation including cytoplasmic rearrangement and chromatin condensation as previously suggested [115]. For the ejaculated human spermatozoa, the same trend was observed by another collective of authors [116] where 93% of the total zinc content was allocated to the flagellum. For the porcine (Figure 3) and bull spermatozoa, we reported that zinc is present throughout the entire spermatozoa; however, we did not perform organelle- or compartment-specific quantification of zinc content [117]. Higher content of flagellar Zn2+ has been negatively correlated with motility [97,116] and therefore it is reasonable to believe that Zn2+ may regulate sperm motility via a direct or indirect mechanism. We have previously suggested a model for how this may be orchestrated [96] and this model was further developed by another group [118]. Further, incorporation of Zn2+ into spermatozoa during ejaculation was proposed to have sperm chromatin stabilization effects in humans [119,120,121] and delay acrosomal exocytosis in humans [122]. It may also maintain plasma membrane structure and function, as shown in a model membrane system as well as in erythrocytes of multiple species [123,124], and protect against free radicals in human and goat spermatozoa [125,126].
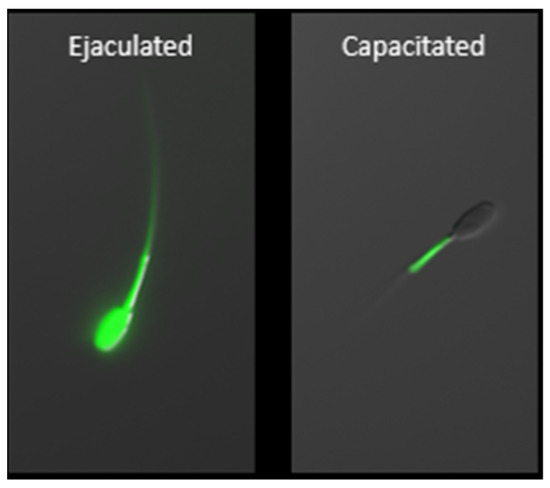
Figure 3.
Zinc localization in boar ejaculated and capacitated spermatozoa using fluorescent probe FluoZinTM-3, AM. Capacitation-related efflux of zinc ions from the sperm head and principal piece is a prerequisite for achieving capacitated, hyperactivated status. Following capacitation, zinc is localized exclusively within the mitochondrial sheath where it is functional, thus indicative of the importance of zinc homeostasis in the sperm mitochondria. The scale bar represents 10 µm.
During the last stage of sperm maturation, known as capacitation, that occurs in vivo within the oviductal epithelium sperm reservoir [127], Zn2+ is gradually released from spermatozoa (Figure 3), causing activation of signal transduction pathways resulting in hyperactivation, acrosomal and sperm surface remodeling, and increased affinity to zona pellucida. As reported previously, Zn2+ removal is an absolute necessity for spermatozoa in achieving full fertilization potential [96,97,116,117,118,128]. These zinc localization patterns have been termed the sperm zinc signature, with four patterns discovered initially in boar, bull, and man [117]; with subsequent follow-up, zinc localization to the sperm acrosome in bull [128]. These localization patterns are indicative of key stages of the sperm capacitation process. We have shown that the spermatozoa release from glycans, mimicking the oviductal epithelium reservoir in response to the oocyte-produced chemoattractant progesterone, is inhibited in the presence of biologically relevant millimolar Zn2+ concentrations, found in seminal plasma. This is notable because progesterone (P4) is contained in oocyte follicular fluid when released at the time of ovulation, thus guiding spermatozoa to the final site of fertilization. Further, we have shown that high, biologically relevant levels of Zn2+ directly inhibit the activity of matrix metalloproteinase (MMP) 2 and 9, as well as ACR and 26S proteasome [128]. Proteasome-inhibited bovine spermatozoa fail to undergo acrosomal exocytosis likely due to insufficient acrosomal remodeling [129]. It has been proposed that the inner acrosomal membrane localization of both MMP2 and ACR predestines these enzymes for a role in the ZP degradation at fertilization [130]; however, participation of MMP2 in the acrosomal remodeling also seems to be plausible and would be in support of previous findings [122]. Inhibition of said enzymes by millimolar Zn2+ concentration might be an evolutionarily conserved polyspermy defense mechanism, given that the sperm-activated oocytes of many species release billions of zinc ions at the time of fertilization during an event referred to as the zinc spark (Xenopus [131], mice [132,133], cattle [134], and human [133]).
5. HD Implications in Male Fertility
Similar membrane characteristics and features are shared between neurons and spermatozoa; many neuronal receptors are also present in spermatozoa which have been labeled as “neurons with tails” [135]. Spermatozoa share excitability functions with neurons, although they lack the synaptic mechanism [136]. Several neural receptors in spermatozoa have been linked with sperm function, such as acrosomal exocytosis mediated by dopamine and 5-HT receptors [137], or motility associated with purinergic [138], nicotinic [136,139], angiotensin II [140], cannabinoid [141], and olfactory receptors [142]. It is, therefore, no surprise that in idiopathic infertile men, asthenozoospermia was associated with polymorphism related to neurotransmission genes such as HTR2A, MAOA, and SLC18A1 [143].
The brain and testes share the highest similarity in HTT gene expression patterns [144], with both having the highest HTT expression among all body tissues [145]. Similarly, in HD individuals, the brain and testes have a high level of somatic mosaicism of CAG repeats in HTT. The CAG repeat track was shown to progressively increase with the age in sperm and lymphocytes of human patients, HD mice, and HD monkeys [146,147,148,149,150]. During spermatogenesis, HTT is localized in the cytoplasm of spermatogonia, while nuclear localization was observed in spermatids and spermatozoa [151]. Nuclear delocalization of HTT during meiosis suggests its role in it. Indeed, male transgenic mice expressing solely mHTT were characterized by infertility, testicular atrophy, aspermia, and massive apoptotic cell death in testes [152]. This was later confirmed in a conditional KO mouse with germ-line specific ablation of HTT which resulted in male infertility, with spermiogenesis arrested at step 3 of the Golgi phase and a significant testis protein profile alteration [153]. In humans, specific testicular pathology with reduced numbers of germ cells and abnormal seminiferous tubule pathology were reported in HD patients [154].
Some aspects of male reproduction were examined in the transgenic HD (TgHD) minipig model [30]. The TgHD boars gradually develop the neurodegenerative phenotype accompanied by testicular degeneration within 24 months of age [155]. After reaching one year of age, the TgHD boars had reduced fertility, fewer spermatozoa per unit in the ejaculate, and a significant decline in the number of WT oocytes penetrated by TgHD spermatozoa in vitro [30]. A follow-up report [156] confirmed previous mouse and human studies by showing that TgHD minipigs had testicular tubule atrophy as a result of mHTT accumulation. Multinucleated spermatogenic cells were frequently present and spermatogonia were shrunk with dilated ER, swollen mitochondria, and condensed chromatin, while spermatocytes and spermatids were rarely present. Further, some tubules contained Sertoli cells only, which were characterized by increased density and vacuolization of cytoplasm, dilation of ER, swollen mitochondria, and alterations of nuclear structure [156]. Sperm defects of TgHD minipig included deformation of the mitochondrial sheath, and folded or coiled flagella containing double or triple axoneme with observations of a fused mitochondrial sheet, while mHTT was detected along the whole sperm tail of TgHD boars [156]. In their later report [157], the same group explored whether the HD pathology was connected with a defect in mitochondrial metabolism. They incubated TgHD spermatozoa with different 14C substrates of the mitochondrial energy-generating system and monitored the production of 14CO2. The production of 14CO2 was significantly reduced in four incubations when compared to WT spermatozoa, implying a non-specific pathology in mitochondrial complex I of the oxidative phosphorylation system and/or in substrate-level phosphorylation of glycolysis, possibly linked with mHTT toxicity [157]. This finding has implications for HD progression monitoring and the development of an efficient strategy for targeted therapy.
5.1. Dysregulation of Neurodegenerative Pathway Genes Linked with Subfertility
In a study utilizing a buffalo model, transcriptomic profiling was conducted in low-fertile buffaloes and compared to high-fertile buffaloes [158]. The authors found 709 significantly dysregulated genes in total between low- and high-fertile buffaloes. Of the top 10 downregulated pathways, (i) AD pathway with 28 downregulated genes was the top one, (ii) HD pathway with 25 downregulated genes was the fifth (Table 2), and (iii) Parkinson’s disease with 22 downregulated genes was the eighth-most downregulated pathway. Even though the implications of genes of neurodegenerative pathways in sperm fertility are still unknown, these results offer an interesting avenue for a role exploration of the neurodegenerative pathway genes in sperm physiology.
Table 2.
HD pathway genes, significantly downregulated in low-fertile buffaloes as reported by Paul et al. [158]. Function(s) of the proteins were taken from the UniProtKB database (https://www.uniprot.org/), accessed on 3 May 2021.
The same group performed transcriptomic profiling of low-fertile bulls and compared their transcriptome profile to high-fertile bulls [159]. Surprisingly, or not so much at this point, Huntington’s and Parkinson’s disease pathway transcripts were uniquely present in high-fertile bulls, further supporting the hypothesis of the role of neurodegenerative pathways in male fertility.
5.2. Zinc-Interacting Proteins of Boar Spermatozoa and Neurodegenerative Pathways
Identification of zinc efflux during mammalian sperm capacitation [117] stimulated further research into characterizing sperm zinc-interacting/zinc-binding proteins (zincoproteins/zincoproteome). In our newest study [35], we identified 1752 zincoproteins with 102 changing significantly during sperm capacitation in vitro. After performing pathway analysis on identified zincoproteins, we made an observation that HD and PD pathways were the two most represented and AD was the tenth-most represented one. HD pathway represented forty-two zincoproteins (Table 3), of which five showed significantly different expression in boar spermatozoa following capacitation (DNAH7, DNAH8, DNAI2, HIP1, and TUBB4B). Their primary critical functions in spermatozoa include ATPase activity, microtubule motor activity, actin filament binding, and cytoskeleton organization. Spermatozoa and sperm capacitation, especially with these five zincoproteins, could be used as a model to explore the effects of HD on spermatozoan zinc homeostasis throughout disease progression.
Table 3.
Identified boar sperm zincoproteins of the HD pathway. Proteins were classified according to PANTHER classification system (http://pantherdb.org/) (accessed on 3 May 2021). Function(s) of the proteins were taken from the UniProtKB database (https://www.uniprot.org/), accessed on 3 May 2021.
6. Spermatozoon as a Potential Model for Studying HD
The spectrum of similarities between neurons and spermatozoa suggests a potential for developing a very easily manageable alternative cellular model system to study and diagnose neurological disorders such as HD, PD, and AD. Further, spermatozoa can be obtained in a non-invasive manner giving a possibility to screen and monitor the disease progression and/or regression during treatment. Studying these pathways in the male pig, which provides an abundant quantity of spermatozoa necessary for various study techniques, could be advantageous [160], especially since the pig is three times more genetically similar to the human than rodent models on the nucleotide level [161]. Likewise, given the non-invasive nature of semen collection, human spermatozoa could be used to study these neurological disorders as a model directly from a human. Studies on higher-order mammals such as the pig would undoubtedly identify pathways more similar to corresponding human pathways given the current number one model organism for HD is Drosophila [162].
In Vitro Spermatogenesis Model for Studying HD
Due to limited availability and ethical concerns, studying mechanisms involved in the pathogenesis of HD in human spermatogenesis has been challenging. However, recent development in the stem cell research field, especially induced pluripotent stem cell (iPSC) and directed differentiation helped to derive many tissue-specific cell types that can be used in research. Research on in vitro gametogenesis (IVG) is an active field of research, and the goal is to achieve complete oogenesis and spermatogenesis in culture. A complete in vitro spermatogenesis from pluripotent stem cells resulting in offspring has been reported in the mouse [163,164]. Recently, the first development of blastocyst was reported in non-human primates (NHP) followed by fertilization with in vitro derived spermatid from embryonic stem cells utilizing the same directed-differentiation protocol used in the human [164,165]. The differentiation protocol is being improved [166,167]. The successful generation of fertile male gametes requires a seamless orchestration between male germ cells and supporting cells including Sertoli, Leydig, and peritubular cells where spermatogenesis occurs in a highly organized tubular microenvironment, the seminiferous tubules, which constitute the mammalian testis. Besides generating haploid male gametes, the production of androgens through steroidogenesis is also an important function of the mammalian testis that regulates spermatogenesis as well as secondary male features.
The research on human in vitro spermatogenesis started in the 1960s from the organ culture [168]. In mice, reconstituting embryonic testicular cells has been used to derive and propagate spermatogonial stem cells from embryonic stem cells [169]. In vitro differentiation showed protracted and less efficient differentiation, and derived cells did not enter meiosis even in the prolonged culture [169]. Studies were conducted where both NHP and human pluripotent stem cells (hPSCs) were differentiated into primordial germ cell-like cells [170,171,172,173,174,175,176,177,178,179]. However, most of those studies failed to generate advanced spermatogenic past meiosis cells such as spermatid or spermatozoa. In later studies, hPSCs were differentiated into advanced spermatogenic cells and even haploid cells [164,174,175,180]. Even though haploid spermatid-like cells were derived from hPSCs, elongations have not been reported from 2D culture methods, and due to the ethical problems, fertilizations with in vitro-derived spermatid-like haploid cells have not been reported. However, in a recent study, elongation of spermatid-like cells, fertilization, and development of blastocyst with in vitro-derived spermatid-like cells have been reported in NHP [165]. Up-to-date, fully matured spermatozoa have only been reported by grafting primordial germ cell-like cells (PGCLCs) or spermatogonial stem cell-like cells (SSCLCs) in the testes or in ex vivo organ culture in rodents, NHPs, and humans. Self-organization of the testicular organoid was reported during extended culture with testicular cells that cannot be achieved in 2D culture conditions [181]. Most importantly, meiotic and post-meiotic germ cells were derived from undifferentiated spermatogonia isolated from prepubertal rhesus monkeys in a 3D soft agar culture [182]. A recent report on human 3D testicular organoids with the production of testosterone without the stimulation of hCG and the expression of protamine 1 (PRM1) and Acrosin in male germ cells suggests functional testicular organoids can be reconstructed in vitro by using testicular cells and extracellular matrix (ECM) [181,183]. The recent advancement of in vitro spermatogenesis and different schemes are summarized in Figure 4A.
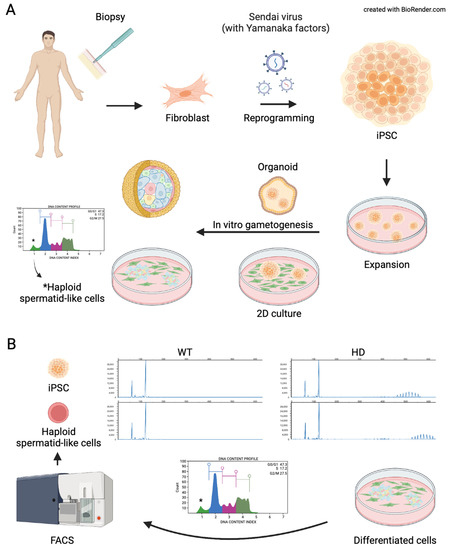
Figure 4.
Schematic representation of current models of in vitro spermatogenesis. (A) somatic cells can be collected from a patient where they can be reprogramed with reprogramming factors to derive induced pluripotent stem cells (iPSCs). The iPSCs can be preserved, expanded, and induced to produce advanced spermatogenic cells via 2D culture methods or 3D culture methods (3D). (B) A schematic representation of a recent study where differentiated spermatogenic cells were used to study developmental mechanisms involved in the pathogenesis of Huntington’s disease. BioRender.com, access on 20 April 2022.
Besides utilizations in regenerative reproductive medicine wherein in vitro derived advanced spermatogenic cells are used to fertilize oocytes or repopulate testes, a recent study utilized in vitro spermatogenesis to investigate the underlying mechanism of pathogenesis in HD. Trinucleotide repeat (TNR) expansion predominantly occurs through male lineage in HD. Unlike in rodents, where TNR expansion occurs in the post-meiotic stage [184,185], in humans, TNR expansion occurs in pre-meiotic and post-meiotic spermatogenic cells [186]. Those differences could be due to kinetic, biological, life span, and developmental process differences between rodents and humans [187,188]. Because NHP resembles biological and developmental processes during spermatogenesis, a recent study utilized in vitro spermatogenesis to study CAG repeat behavior in spermatid-like cells derived from NHP ESC [189]. The study showed the in vitro model resembles CAG repeat expansion observed in the sperm cells where repeat size is the main factor affecting the CAG repeat instability (Figure 4B) [189].
7. Conclusions
The goal of this review was to demonstrate the extensive parallels between neurons and spermatozoa. As “neurons with tails”, spermatozoa represent a unique, high-potential model for the study, diagnosis, and therapy/drug development targeting Huntington’s disease. Future studies should investigate the roles of neurodegenerative disease pathways in mammalian reproduction and how these are involved in regulating and sustaining male fertility. The feasibility and efficacy of this unique model need to be explored as well. In addition, targeting zinc dysregulation and exploitation of zincoproteome involved in spermatozoan capacitation will serve as an avenue for the exploration of potential therapeutic remedies to combat HD. Targeting Zn2+ dysregulation might serve as a potential therapeutic remedy to prevent the onset of disease, as altered neuronal zinc homeostasis and spermatozoan metabolism occur in the early stages of HD. Future clinical studies must evaluate the most efficient way of preserving zinc homeostasis and evaluate when, where, and how much Zn2+ is ideal for proper neural and reproductive function. In addition, quantification of mitochondrial metabolic biomarkers in spermatozoa, including those linked with HD pathology, could be useful for the diagnosis of early-onset HD. Moreover, in vitro spermatogenesis models can be used to study pathogenesis mechanisms involved during spermatogenesis in HD. Spermatozoa may be suitable noninvasive sampling material for monitoring HD disease progression and therapy. In addition, spermatozoa could be used to monitor the efficacy of preclinical therapeutic treatments.
Author Contributions
Conceptualization, M.L., M.Z. and P.S.; writing—original draft preparation, M.L., M.Z. and I.K.C.; writing—review and editing, K.K., C.A.E.IV and P.S.; visualization, M.L. and M.Z.; supervision, C.A.E.IV and P.S.; project administration, C.A.E.IV and P.S.; funding acquisition, K.K. and P.S. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by USDA National Institute of Food and Agriculture, Agriculture and Food Research Initiative Competitive Grants no. grants 2020-67015-31017 (PS), 2021-67015-33404 (PS), 2022-67015-36298 (KK), 2019-67012-29714 (KK), NIH/OD1 R01 OD028223-01 (IKC, CAE), and the CAFNR Joy of Discovery Seed Grant award (PS).
Institutional Review Board Statement
Studies reviewed here were conducted according to the guidelines of the Animal Care and Use protocol, approved by the Animal Care and Use Committee (ACUC) of the University of Missouri (Animal Welfare Assurance number/ACUC protocol # 9500—National Swine Resource and Research Center Procedures for Surgical Recovery of Oocytes, Embryos, and Fetuses, Surgical Embryo Transfer, and Cesarean Delivery, reviewed annually).
Informed Consent Statement
This article does not contain any studies performed by any of the authors with human participants.
Data Availability Statement
No new data were created or analyzed in this study.
Acknowledgments
We thank the staff of the National Swine Research and Resource Center, the University of Missouri, funded by National Institutes of Health (NIH) grant U42 OD011140, as well as Randall Prather and his associates for sow ovary and boar semen collections for experiments reviewed in this article. We thank Mayra Ortiz D’Avila Assumpcao for her comments on the manuscript. Clerical assistance provided by Miriam Sutovsky is appreciated.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
References
- Kim, A.; Lalonde, K.; Truesdell, A.; Gomes Welter, P.; Brocardo, P.S.; Rosenstock, T.R.; Gil-Mohapel, J. New Avenues for the Treatment of Huntington’s Disease. Int. J. Mol. Sci. 2021, 22, 8363. [Google Scholar] [CrossRef] [PubMed]
- Evans, S.J.; Douglas, I.; Rawlins, M.D.; Wexler, N.S.; Tabrizi, S.J.; Smeeth, L. Prevalence of adult Huntington’s disease in the UK based on diagnoses recorded in general practice records. J. Neurol. Neurosurg. Psychiatry 2013, 84, 1156–1160. [Google Scholar] [CrossRef] [PubMed]
- Kay, C.; Fisher, E.; Hyden, M.R. Epidemiology. In Huntington’s Disease, 4th ed.; Bates, G.P., Tabrizi, S.J., Jones, L., Eds.; Oxford University Press: New York, NY, USA, 2014. [Google Scholar] [CrossRef]
- Langbehn, D.R.; Hayden, M.R.; Paulsen, J.S. CAG-repeat length and the age of onset in Huntington disease (HD): A review and validation study of statistical approaches. Am. J. Med. Genetics Part B Neuropsychiatr. Genet. Off. Publ. Int. Soc. Psychiatr. Genet. 2010, 153, 397–408. [Google Scholar] [CrossRef] [PubMed]
- Gatto, E.M.; Rojas, N.G.; Persi, G.; Etcheverry, J.L.; Cesarini, M.E.; Perandones, C. Huntington disease: Advances in the understanding of its mechanisms. Clin. Parkinsonism Relat. Disord. 2020, 3, 100056. [Google Scholar] [CrossRef]
- Tabrizi, S.J.; Flower, M.D.; Ross, C.A.; Wild, E.J. Huntington disease: New insights into molecular pathogenesis and therapeutic opportunities. Nat. Rev. Neurol. 2020, 16, 529–546. [Google Scholar] [CrossRef]
- Caterino, M.; Squillaro, T.; Montesarchio, D.; Giordano, A.; Giancola, C.; Melone, M.A.B. Huntingtin protein: A new option for fixing the Huntington’s disease countdown clock. Neuropharmacology 2018, 135, 126–138. [Google Scholar] [CrossRef]
- Desmond, C.R.; Atwal, R.S.; Xia, J.; Truant, R. Identification of a karyopherin β1/β2 proline-tyrosine nuclear localization signal in huntingtin protein. J. Biol. Chem. 2012, 287, 39626–39633. [Google Scholar] [CrossRef]
- Xia, J.; Lee, D.H.; Taylor, J.; Vandelft, M.; Truant, R. Huntingtin contains a highly conserved nuclear export signal. Hum. Mol. Genet. 2003, 12, 1393–1403. [Google Scholar] [CrossRef]
- Zheng, Z.; Li, A.; Holmes, B.B.; Marasa, J.C.; Diamond, M.I. An N-terminal nuclear export signal regulates trafficking and aggregation of Huntingtin (Htt) protein exon 1. J. Biol. Chem. 2013, 288, 6063–6071. [Google Scholar] [CrossRef]
- Nasir, J.; Floresco, S.B.; O’Kusky, J.R.; Diewert, V.M.; Richman, J.M.; Zeisler, J.; Borowski, A.; Marth, J.D.; Phillips, A.G.; Hayden, M.R. Targeted disruption of the Huntington’s disease gene results in embryonic lethality and behavioral and morphological changes in heterozygotes. Cell 1995, 81, 811–823. [Google Scholar] [CrossRef]
- Zeitlin, S.; Liu, J.P.; Chapman, D.L.; Papaioannou, V.E.; Efstratiadis, A. Increased apoptosis and early embryonic lethality in mice nullizygous for the Huntington’s disease gene homologue. Nat. Genet. 1995, 11, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, G.D.; Gokhan, S.; Molero, A.E.; Mehler, M.F. Selective roles of normal and mutant huntingtin in neural induction and early neurogenesis. PLoS ONE 2013, 8, e64368. [Google Scholar] [CrossRef]
- Hoffner, G.; Kahlem, P.; Djian, P. Perinuclear localization of huntingtin as a consequence of its binding to microtubules through an interaction with beta-tubulin: Relevance to Huntington’s disease. J. Cell Sci. 2002, 115, 941–948. [Google Scholar] [CrossRef] [PubMed]
- Caviston, J.P.; Ross, J.L.; Antony, S.M.; Tokito, M.; Holzbaur, E.L. Huntingtin facilitates dynein/dynactin-mediated vesicle transport. Proc. Natl. Acad. Sci. USA 2007, 104, 10045–10050. [Google Scholar] [CrossRef] [PubMed]
- Godin, J.D.; Colombo, K.; Molina-Calavita, M.; Keryer, G.; Zala, D.; Charrin, B.C.; Dietrich, P.; Volvert, M.L.; Guillemot, F.; Dragatsis, I.; et al. Huntingtin is required for mitotic spindle orientation and mammalian neurogenesis. Neuron 2010, 67, 392–406. [Google Scholar] [CrossRef]
- Zuccato, C.; Tartari, M.; Crotti, A.; Goffredo, D.; Valenza, M.; Conti, L.; Cataudella, T.; Leavitt, B.R.; Hayden, M.R.; Timmusk, T.; et al. Huntingtin interacts with REST/NRSF to modulate the transcription of NRSE-controlled neuronal genes. Nat. Genet. 2003, 35, 76–83. [Google Scholar] [CrossRef]
- McFarland, K.N.; Huizenga, M.N.; Darnell, S.B.; Sangrey, G.R.; Berezovska, O.; Cha, J.H.; Outeiro, T.F.; Sadri-Vakili, G. MeCP2: A novel Huntingtin interactor. Hum. Mol. Genet. 2014, 23, 1036–1044. [Google Scholar] [CrossRef]
- McKinstry, S.U.; Karadeniz, Y.B.; Worthington, A.K.; Hayrapetyan, V.Y.; Ozlu, M.I.; Serafin-Molina, K.; Risher, W.C.; Ustunkaya, T.; Dragatsis, I.; Zeitlin, S.; et al. Huntingtin is required for normal excitatory synapse development in cortical and striatal circuits. J. Neurosci. Off. J. Soc. Neurosci. 2014, 34, 9455–9472. [Google Scholar] [CrossRef]
- Martin, D.D.; Heit, R.J.; Yap, M.C.; Davidson, M.W.; Hayden, M.R.; Berthiaume, L.G. Identification of a post-translationally myristoylated autophagy-inducing domain released by caspase cleavage of huntingtin. Hum. Mol. Genet. 2014, 23, 3166–3179. [Google Scholar] [CrossRef]
- Saudou, F.; Humbert, S. The Biology of Huntingtin. Neuron 2016, 89, 910–926. [Google Scholar] [CrossRef]
- Deng, Y.P.; Albin, R.L.; Penney, J.B.; Young, A.B.; Anderson, K.D.; Reiner, A. Differential loss of striatal projection systems in Huntington’s disease: A quantitative immunohistochemical study. J. Chem. Neuroanat. 2004, 27, 143–164. [Google Scholar] [CrossRef] [PubMed]
- Ehrnhoefer, D.E.; Sutton, L.; Hayden, M.R. Small changes, big impact: Posttranslational modifications and function of huntingtin in Huntington disease. Neurosci. A Rev. J. Bringing Neurobiol. Neurol. Psychiatry 2011, 17, 475–492. [Google Scholar] [CrossRef] [PubMed]
- Hensman Moss, D.J.; Flower, M.D.; Lo, K.K.; Miller, J.R.; van Ommen, G.B.; t Hoen, P.A.; Stone, T.C.; Guinee, A.; Langbehn, D.R.; Jones, L.; et al. Huntington’s disease blood and brain show a common gene expression pattern and share an immune signature with Alzheimer’s disease. Sci. Rep. 2017, 7, 44849. [Google Scholar] [CrossRef] [PubMed]
- Golas, M.M. Human cellular models of medium spiny neuron development and Huntington disease. Life Sci. 2018, 209, 179–196. [Google Scholar] [CrossRef]
- Liang, J.J.H.; McKinnon, I.A.; Rankin, C.H. The contribution of C. elegans neurogenetics to understanding neurodegenerative diseases. J. Neurogenet. 2020, 34, 527–548. [Google Scholar] [CrossRef]
- Lewis, E.A.; Smith, G.A. Using Drosophila models of Huntington’s disease as a translatable tool. J. Neurosci. Methods 2016, 265, 89–98. [Google Scholar] [CrossRef]
- Farshim, P.P.; Bates, G.P. Mouse Models of Huntington’s Disease. Methods Mol. Biol. (Clifton N.J.) 2018, 1780, 97–120. [Google Scholar] [CrossRef]
- Jacobsen, J.C.; Bawden, C.S.; Rudiger, S.R.; McLaughlan, C.J.; Reid, S.J.; Waldvogel, H.J.; MacDonald, M.E.; Gusella, J.F.; Walker, S.K.; Kelly, J.M.; et al. An ovine transgenic Huntington’s disease model. Hum. Mol. Genet. 2010, 19, 1873–1882. [Google Scholar] [CrossRef]
- Baxa, M.; Hruska-Plochan, M.; Juhas, S.; Vodicka, P.; Pavlok, A.; Juhasova, J.; Miyanohara, A.; Nejime, T.; Klima, J.; Macakova, M.; et al. A transgenic minipig model of Huntington’s Disease. J. Huntingt. Dis. 2013, 2, 47–68. [Google Scholar] [CrossRef]
- Yan, S.; Tu, Z.; Liu, Z.; Fan, N.; Yang, H.; Yang, S.; Yang, W.; Zhao, Y.; Ouyang, Z.; Lai, C.; et al. A Huntingtin Knockin Pig Model Recapitulates Features of Selective Neurodegeneration in Huntington’s Disease. Cell 2018, 173, 989–1002.e1013. [Google Scholar] [CrossRef]
- Yang, S.H.; Cheng, P.H.; Banta, H.; Piotrowska-Nitsche, K.; Yang, J.J.; Cheng, E.C.; Snyder, B.; Larkin, K.; Liu, J.; Orkin, J.; et al. Towards a transgenic model of Huntington’s disease in a non-human primate. Nature 2008, 453, 921–924. [Google Scholar] [CrossRef] [PubMed]
- Chan, A.W.; Jiang, J.; Chen, Y.; Li, C.; Prucha, M.S.; Hu, Y.; Chi, T.; Moran, S.; Rahim, T.; Li, S.; et al. Progressive cognitive deficit, motor impairment and striatal pathology in a transgenic Huntington disease monkey model from infancy to adulthood. PLoS ONE 2015, 10, e0122335. [Google Scholar] [CrossRef] [PubMed]
- Howland, D.; Ellederova, Z.; Aronin, N.; Fernau, D.; Gallagher, J.; Taylor, A.; Hennebold, J.; Weiss, A.R.; Gray-Edwards, H.; McBride, J. Large Animal Models of Huntington’s Disease: What We Have Learned and Where We Need to Go Next. J. Huntingt. Dis. 2020, 9, 201–216. [Google Scholar] [CrossRef] [PubMed]
- Zigo, M.; Kerns, K.; Sen, S.; Essien, C.; Oko, R.; Xu, D.; Sutovsky, P. Zinc is a master-regulator of sperm function associated with binding, motility, and metabolic modulation during porcine sperm capacitation. Commun. Biol. 2022, 5, 538. [Google Scholar] [CrossRef] [PubMed]
- Illarioshkin, S.N.; Klyushnikov, S.A.; Vigont, V.A.; Seliverstov, Y.A.; Kaznacheyeva, E.V. Molecular Pathogenesis in Huntington’s Disease. Biochemistry 2018, 83, 1030–1039. [Google Scholar] [CrossRef] [PubMed]
- Jimenez-Sanchez, M.; Licitra, F.; Underwood, B.R.; Rubinsztein, D.C. Huntington’s Disease: Mechanisms of Pathogenesis and Therapeutic Strategies. Cold Spring Harb. Perspect. Med. 2017, 7, a024240. [Google Scholar] [CrossRef]
- Wright, D.J.; Renoir, T.; Gray, L.J.; Hannan, A.J. Huntington’s Disease: Pathogenic Mechanisms and Therapeutic Targets. Adv. Neurobiol. 2017, 15, 93–128. [Google Scholar] [CrossRef]
- Arrasate, M.; Mitra, S.; Schweitzer, E.S.; Segal, M.R.; Finkbeiner, S. Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature 2004, 431, 805–810. [Google Scholar] [CrossRef]
- Nucifora, L.G.; Burke, K.A.; Feng, X.; Arbez, N.; Zhu, S.; Miller, J.; Yang, G.; Ratovitski, T.; Delannoy, M.; Muchowski, P.J.; et al. Identification of novel potentially toxic oligomers formed in vitro from mammalian-derived expanded huntingtin exon-1 protein. J. Biol. Chem. 2012, 287, 16017–16028. [Google Scholar] [CrossRef]
- Liu, K.Y.; Shyu, Y.C.; Barbaro, B.A.; Lin, Y.T.; Chern, Y.; Thompson, L.M.; James Shen, C.K.; Marsh, J.L. Disruption of the nuclear membrane by perinuclear inclusions of mutant huntingtin causes cell-cycle re-entry and striatal cell death in mouse and cell models of Huntington’s disease. Hum. Mol. Genet. 2015, 24, 1602–1616. [Google Scholar] [CrossRef]
- Zuccato, C.; Cattaneo, E. Role of brain-derived neurotrophic factor in Huntington’s disease. Prog. Neurobiol. 2007, 81, 294–330. [Google Scholar] [CrossRef] [PubMed]
- Bassi, S.; Tripathi, T.; Monziani, A.; Di Leva, F.; Biagioli, M. Epigenetics of Huntington’s Disease. Adv. Exp. Med. Biol. 2017, 978, 277–299. [Google Scholar] [CrossRef] [PubMed]
- Zhao, T.; Hong, Y.; Li, X.J.; Li, S.H. Subcellular Clearance and Accumulation of Huntington Disease Protein: A Mini-Review. Front. Mol. Neurosci. 2016, 9, 27. [Google Scholar] [CrossRef]
- Valionyte, E.; Yang, Y.; Roberts, S.L.; Kelly, J.; Lu, B.; Luo, S. Lowering Mutant Huntingtin Levels and Toxicity: Autophagy-Endolysosome Pathways in Huntington’s Disease. J. Mol. Biol. 2020, 432, 2673–2691. [Google Scholar] [CrossRef]
- Harding, R.J.; Tong, Y.F. Proteostasis in Huntington’s disease: Disease mechanisms and therapeutic opportunities. Acta Pharmacol. Sin. 2018, 39, 754–769. [Google Scholar] [CrossRef]
- Jodeiri Farshbaf, M.; Ghaedi, K. Huntington’s Disease and Mitochondria. Neurotox. Res. 2017, 32, 518–529. [Google Scholar] [CrossRef]
- Sharma, A.; Behl, T.; Sharma, L.; Aelya, L.; Bungau, S. Mitochondrial Dysfunction in Huntington’s disease: Pathogenesis and Therapeutic Opportunities. Curr. Drug Targets 2021, 22, 1637–1667. [Google Scholar] [CrossRef]
- Roe, A.J.; Qi, X. Drp1 phosphorylation by MAPK1 causes mitochondrial dysfunction in cell culture model of Huntington’s disease. Biochem. Biophys. Res. Commun. 2018, 496, 706–711. [Google Scholar] [CrossRef]
- Sawant, N.; Morton, H.; Kshirsagar, S.; Reddy, A.P.; Reddy, P.H. Mitochondrial Abnormalities and Synaptic Damage in Huntington’s Disease: A Focus on Defective Mitophagy and Mitochondria-Targeted Therapeutics. Mol. Neurobiol. 2021, 58, 6350–6377. [Google Scholar] [CrossRef]
- Franco-Iborra, S.; Plaza-Zabala, A.; Montpeyo, M.; Sebastian, D.; Vila, M.; Martinez-Vicente, M. Mutant HTT (huntingtin) impairs mitophagy in a cellular model of Huntington disease. Autophagy 2021, 17, 672–689. [Google Scholar] [CrossRef] [PubMed]
- Shirendeb, U.P.; Calkins, M.J.; Manczak, M.; Anekonda, V.; Dufour, B.; McBride, J.L.; Mao, P.; Reddy, P.H. Mutant huntingtin’s interaction with mitochondrial protein Drp1 impairs mitochondrial biogenesis and causes defective axonal transport and synaptic degeneration in Huntington’s disease. Hum. Mol. Genet. 2012, 21, 406–420. [Google Scholar] [CrossRef] [PubMed]
- Sepers, M.D.; Raymond, L.A. Mechanisms of synaptic dysfunction and excitotoxicity in Huntington’s disease. Drug Discov. Today 2014, 19, 990–996. [Google Scholar] [CrossRef]
- Labbadia, J.; Morimoto, R. Huntington’s Disease: Underlying molecular mechanisms and emerging concepts. Trends Biochem. Sci. 2013, 38, 378–385. [Google Scholar] [CrossRef] [PubMed]
- Tai, Y.F.; Pavese, N.; Gerhard, A.; Tabrizi, S.J.; Barker, R.A.; Brooks, D.J.; Piccini, P. Microglial activation in presymptomatic Huntington’s disease gene carriers. Brain J. Neurol. 2007, 130, 1759–1766. [Google Scholar] [CrossRef] [PubMed]
- Khoshnan, A.; Ko, J.; Watkin, E.E.; Paige, L.A.; Reinhart, P.H.; Patterson, P.H. Activation of the IkappaB kinase complex and nuclear factor-kappaB contributes to mutant huntingtin neurotoxicity. J. Neurosci. Off. J. Soc. Neurosci. 2004, 24, 7999–8008. [Google Scholar] [CrossRef]
- Björkqvist, M.; Wild, E.J.; Thiele, J.; Silvestroni, A.; Andre, R.; Lahiri, N.; Raibon, E.; Lee, R.V.; Benn, C.L.; Soulet, D.; et al. A novel pathogenic pathway of immune activation detectable before clinical onset in Huntington’s disease. J. Exp. Med. 2008, 205, 1869–1877. [Google Scholar] [CrossRef]
- Valadão, P.A.C.; Santos, K.B.S.; Ferreira, E.V.T.H.; Macedo, E.C.T.; Teixeira, A.L.; Guatimosim, C.; de Miranda, A.S. Inflammation in Huntington’s disease: A few new twists on an old tale. J. Neuroimmunol. 2020, 348, 577380. [Google Scholar] [CrossRef]
- Grima, J.C.; Daigle, J.G.; Arbez, N.; Cunningham, K.C.; Zhang, K.; Ochaba, J.; Geater, C.; Morozko, E.; Stocksdale, J.; Glatzer, J.C.; et al. Mutant Huntingtin Disrupts the Nuclear Pore Complex. Neuron 2017, 94, 93–107. [Google Scholar] [CrossRef]
- Dash, D.; Mestre, T.A. Therapeutic Update on Huntington’s Disease: Symptomatic Treatments and Emerging Disease-Modifying Therapies. Neurotherapeutics 2020, 17, 1645–1659. [Google Scholar] [CrossRef]
- Estevez-Fraga, C.; Flower, M.D.; Tabrizi, S.J. Therapeutic strategies for Huntington’s disease. Curr. Opin. Neurol. 2020, 33, 508–518. [Google Scholar] [CrossRef]
- Heo, Y.A.; Scott, L.J. Deutetrabenazine: A Review in Chorea Associated with Huntington’s Disease. Drugs 2017, 77, 1857–1864. [Google Scholar] [CrossRef] [PubMed]
- Poon, L.H.; Kang, G.A.; Lee, A.J. Role of tetrabenazine for Huntington’s disease-associated chorea. Ann. Pharm. 2010, 44, 1080–1089. [Google Scholar] [CrossRef] [PubMed]
- Beister, A.; Kraus, P.; Kuhn, W.; Dose, M.; Weindl, A.; Gerlach, M. The N-methyl-D-aspartate antagonist memantine retards progression of Huntington’s disease. J. Neural Transm. Suppl. 2004, 68, 117–122. [Google Scholar] [CrossRef]
- Galts, C.P.C.; Bettio, L.E.B.; Jewett, D.C.; Yang, C.C.; Brocardo, P.S.; Rodrigues, A.L.S.; Thacker, J.S.; Gil-Mohapel, J. Depression in neurodegenerative diseases: Common mechanisms and current treatment options. Neurosci. Biobehav. Rev. 2019, 102, 56–84. [Google Scholar] [CrossRef] [PubMed]
- Wyant, K.J.; Ridder, A.J.; Dayalu, P. Huntington’s Disease-Update on Treatments. Curr. Neurol. Neurosci. Rep. 2017, 17, 33. [Google Scholar] [CrossRef] [PubMed]
- Mullard, A. Pioneering antisense drug heads into pivotal trials for Huntington disease. Nat. Rev. Drug Discov. 2019, 18, 161–163. [Google Scholar] [CrossRef]
- Leavitt, B.R.; Kordasiewicz, H.B.; Schobel, S.A. Huntingtin-Lowering Therapies for Huntington Disease: A Review of the Evidence of Potential Benefits and Risks. JAMA Neurol. 2020, 77, 764–772. [Google Scholar] [CrossRef]
- Valero, T. Mitochondrial biogenesis: Pharmacological approaches. Curr. Pharm. Des. 2014, 20, 5507–5509. [Google Scholar] [CrossRef]
- Adanyeguh, I.M.; Rinaldi, D.; Henry, P.G.; Caillet, S.; Valabregue, R.; Durr, A.; Mochel, F. Triheptanoin improves brain energy metabolism in patients with Huntington disease. Neurology 2015, 84, 490–495. [Google Scholar] [CrossRef]
- Vázquez-Manrique, R.P.; Farina, F.; Cambon, K.; Dolores Sequedo, M.; Parker, A.J.; Millán, J.M.; Weiss, A.; Déglon, N.; Neri, C. AMPK activation protects from neuronal dysfunction and vulnerability across nematode, cellular and mouse models of Huntington’s disease. Hum. Mol. Genet. 2016, 25, 1043–1058. [Google Scholar] [CrossRef]
- Sanchis, A.; García-Gimeno, M.A.; Cañada-Martínez, A.J.; Sequedo, M.D.; Millán, J.M.; Sanz, P.; Vázquez-Manrique, R.P. Metformin treatment reduces motor and neuropsychiatric phenotypes in the zQ175 mouse model of Huntington disease. Exp. Mol. Med. 2019, 51, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Ma, T.C.; Buescher, J.L.; Oatis, B.; Funk, J.A.; Nash, A.J.; Carrier, R.L.; Hoyt, K.R. Metformin therapy in a transgenic mouse model of Huntington’s disease. Neurosci. Lett. 2007, 411, 98–103. [Google Scholar] [CrossRef] [PubMed]
- Hervás, D.; Fornés-Ferrer, V.; Gómez-Escribano, A.P.; Sequedo, M.D.; Peiró, C.; Millán, J.M.; Vázquez-Manrique, R.P. Metformin intake associates with better cognitive function in patients with Huntington’s disease. PLoS ONE 2017, 12, e0179283. [Google Scholar] [CrossRef]
- Eddings, C.R.; Arbez, N.; Akimov, S.; Geva, M.; Hayden, M.R.; Ross, C.A. Pridopidine protects neurons from mutant-huntingtin toxicity via the sigma-1 receptor. Neurobiol. Dis. 2019, 129, 118–129. [Google Scholar] [CrossRef] [PubMed]
- Madkour, M.M.; Anbar, H.S.; El-Gamal, M.I. Current status and future prospects of p38α/MAPK14 kinase and its inhibitors. Eur. J. Med. Chem. 2021, 213, 113216. [Google Scholar] [CrossRef] [PubMed]
- Choi, K.A.; Choi, Y.; Hong, S. Stem cell transplantation for Huntington’s diseases. Methods 2018, 133, 104–112. [Google Scholar] [CrossRef]
- Mu, S.; Han, L.; Zhou, G.; Mo, C.; Duan, J.; He, Z.; Wang, Z.; Ren, L.; Zhang, J. Protein regulation of induced pluripotent stem cells by transplanting in a Huntington’s animal model. Neuropathol. Appl. Neurobiol. 2016, 42, 521–534. [Google Scholar] [CrossRef]
- Mu, S.; Wang, J.; Zhou, G.; Peng, W.; He, Z.; Zhao, Z.; Mo, C.; Qu, J.; Zhang, J. Transplantation of induced pluripotent stem cells improves functional recovery in Huntington’s disease rat model. PLoS ONE 2014, 9, e101185. [Google Scholar] [CrossRef]
- Grochowski, C.; Blicharska, E.; Krukow, P.; Jonak, K.; Maciejewski, M.; Szczepanek, D.; Jonak, K.; Flieger, J.; Maciejewski, R. Analysis of Trace Elements in Human Brain: Its Aim, Methods, and Concentration Levels. Front. Chem. 2019, 7, 115. [Google Scholar] [CrossRef]
- Oldereid, N.B.; Thomassen, Y.; Attramadal, A.; Olaisen, B.; Purvis, K. Concentrations of lead, cadmium and zinc in the tissues of reproductive organs of men. J. Reprod. Fertil. 1993, 99, 421–425. [Google Scholar] [CrossRef]
- Morris, D.R.; Levenson, C.W. Neurotoxicity of Zinc. Adv. Neurobiol. 2017, 18, 303–312. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.T.; Graham, T.C.; Ogden, L.; Ali, S.; Salwa; Thompson, S.J.; Shireen, K.F.; Mahboob, M. A two-generational reproductive toxicity study of zinc in rats. J. Environ. Sci. Health Part B Pestic. Food Contam. Agric. Wastes 2007, 42, 403–415. [Google Scholar] [CrossRef] [PubMed]
- Sandstead, H.H. Subclinical zinc deficiency impairs human brain function. J. Trace Elem. Med. Biol. Organ Soc. Miner. Trace Elem. (GMS) 2012, 26, 70–73. [Google Scholar] [CrossRef]
- Yamaguchi, S.; Miura, C.; Kikuchi, K.; Celino, F.T.; Agusa, T.; Tanabe, S.; Miura, T. Zinc is an essential trace element for spermatogenesis. Proc. Natl. Acad. Sci. USA 2009, 106, 10859–10864. [Google Scholar] [CrossRef] [PubMed]
- Krall, R.F.; Tzounopoulos, T.; Aizenman, E. The Function and Regulation of Zinc in the Brain. Neuroscience 2021, 457, 235–258. [Google Scholar] [CrossRef] [PubMed]
- Kambe, T.; Taylor, K.M.; Fu, D. Zinc transporters and their functional integration in mammalian cells. J. Biol. Chem. 2021, 296, 100320. [Google Scholar] [CrossRef]
- Baltaci, A.K.; Yuce, K.; Mogulkoc, R. Zinc Metabolism and Metallothioneins. Biol. Trace Elem. Res. 2018, 183, 22–31. [Google Scholar] [CrossRef]
- Niu, L.; Li, L.; Yang, S.; Wang, W.; Ye, C.; Li, H. Disruption of zinc transporter ZnT3 transcriptional activity and synaptic vesicular zinc in the brain of Huntington’s disease transgenic mouse. Cell Biosci. 2020, 10, 106. [Google Scholar] [CrossRef]
- Granzotto, A.; Canzoniero, L.M.T.; Sensi, S.L. A Neurotoxic Ménage-à-trois: Glutamate, Calcium, and Zinc in the Excitotoxic Cascade. Front. Mol. Neurosci. 2020, 13, 600089. [Google Scholar] [CrossRef]
- Watt, N.T.; Griffiths, H.H.; Hooper, N.M. Neuronal zinc regulation and the prion protein. Prion 2013, 7, 203–208. [Google Scholar] [CrossRef]
- Choi, S.; Hong, D.K.; Choi, B.Y.; Suh, S.W. Zinc in the Brain: Friend or Foe? Int. J. Mol. Sci. 2020, 21, 8941. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.J.; Park, M.H.; Choi, S.Y.; Koh, J.Y. Activation of the Trk signaling pathway by extracellular zinc. Role of metalloproteinases. J. Biol. Chem. 2005, 280, 11995–12001. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.Z.; Pan, E.; Xiong, Z.Q.; McNamara, J.O. Zinc-mediated transactivation of TrkB potentiates the hippocampal mossy fiber-CA3 pyramid synapse. Neuron 2008, 57, 546–558. [Google Scholar] [CrossRef] [PubMed]
- Portbury, S.D.; Adlard, P.A. Zinc Signal in Brain Diseases. Int. J. Mol. Sci. 2017, 18, 2506. [Google Scholar] [CrossRef]
- Kerns, K.; Zigo, M.; Sutovsky, P. Zinc: A Necessary Ion for Mammalian Sperm Fertilization Competency. Int. J. Mol. Sci. 2018, 19, 4097. [Google Scholar] [CrossRef]
- Foresta, C.; Garolla, A.; Cosci, I.; Menegazzo, M.; Ferigo, M.; Gandin, V.; De Toni, L. Role of zinc trafficking in male fertility: From germ to sperm. Hum. Reprod. 2014, 29, 1134–1145. [Google Scholar] [CrossRef]
- Ellis, R.E.; Stanfield, G.M. The regulation of spermatogenesis and sperm function in nematodes. Semin. Cell Dev. Biol. 2014, 29, 17–30. [Google Scholar] [CrossRef]
- Zhao, Y.; Tan, C.H.; Krauchunas, A.; Scharf, A.; Dietrich, N.; Warnhoff, K.; Yuan, Z.; Druzhinina, M.; Gu, S.G.; Miao, L.; et al. The zinc transporter ZIPT-7.1 regulates sperm activation in nematodes. PLoS Biol. 2018, 16, e2005069. [Google Scholar] [CrossRef]
- Kumari, D.; Nair, N.; Bedwal, R.S. Effect of dietary zinc deficiency on testes of Wistar rats: Morphometric and cell quantification studies. J. Trace Elem. Med. Biol. Organ Soc. Miner. Trace Elem. (GMS) 2011, 25, 47–53. [Google Scholar] [CrossRef]
- Kumari, D.; Nair, N.; Bedwal, R.S. Testicular apoptosis after dietary zinc deficiency: Ultrastructural and TUNEL studies. Syst. Biol. Reprod. Med. 2011, 57, 233–243. [Google Scholar] [CrossRef]
- Tuncer, I.; Sunar, F.; Toy, H.; Baltaci, A.K.; Mogulkoc, R. Histological effects of zinc and melatonin on rat testes. Bratisl. Lek. Listy 2011, 112, 425–427. [Google Scholar] [PubMed]
- Croxford, T.P.; McCormick, N.H.; Kelleher, S.L. Moderate zinc deficiency reduces testicular Zip6 and Zip10 abundance and impairs spermatogenesis in mice. J. Nutr. 2011, 141, 359–365. [Google Scholar] [CrossRef] [PubMed]
- Ghasemzadeh-Hasankolai, M.; Batavani, R.; Eslaminejad, M.B.; Sedighi-Gilani, M. Effect of zinc ions on differentiation of bone marrow-derived mesenchymal stem cells to male germ cells and some germ cell-specific gene expression in rams. Biol. Trace Elem. Res. 2012, 150, 137–146. [Google Scholar] [CrossRef]
- Vickram, S.; Rohini, K.; Srinivasan, S.; Nancy Veenakumari, D.; Archana, K.; Anbarasu, K.; Jeyanthi, P.; Thanigaivel, S.; Gulothungan, G.; Rajendiran, N.; et al. Role of Zinc (Zn) in Human Reproduction: A Journey from Initial Spermatogenesis to Childbirth. Int. J. Mol. Sci. 2021, 22, 2188. [Google Scholar] [CrossRef] [PubMed]
- You, X.; Wei, L.; Fan, S.; Yang, W.; Liu, X.; Wang, G.; Man, Y.; Pan, Z.; Feng, W. Expression pattern of Zinc finger protein 185 in mouse testis and its role in regulation of testosterone secretion. Mol. Med. Rep. 2017, 16, 2101–2106. [Google Scholar] [CrossRef]
- Ishizuka, M.; Ohtsuka, E.; Inoue, A.; Odaka, M.; Ohshima, H.; Tamura, N.; Yoshida, K.; Sako, N.; Baba, T.; Kashiwabara, S.; et al. Abnormal spermatogenesis and male infertility in testicular zinc finger protein Zfp318-knockout mice. Dev. Growth Differ. 2016, 58, 600–608. [Google Scholar] [CrossRef]
- Bjorndahl, L.; Kvist, U. Human sperm chromatin stabilization: A proposed model including zinc bridges. Mol. Hum. Reprod. 2010, 16, 23–29. [Google Scholar] [CrossRef]
- Henkel, R.; Baldauf, C.; Bittner, J.; Weidner, W.; Miska, W. Elimination of zinc from the flagella of spermatozoa during epididymal transit is important for motility. Reprod. Technol. 2001, 10, 280–285. [Google Scholar]
- Clermont, Y.; Oko, R.; Hermo, L. Immunocytochemical localization of proteins utilized in the formation of outer dense fibers and fibrous sheath in rat spermatids: An electron microscope study. Anat. Rec. 1990, 227, 447–457. [Google Scholar] [CrossRef]
- Beigi Harchegani, A.; Dahan, H.; Tahmasbpour, E.; Bakhtiari Kaboutaraki, H.; Shahriary, A. Effects of zinc deficiency on impaired spermatogenesis and male infertility: The role of oxidative stress, inflammation and apoptosis. Hum. Fertil. 2020, 23, 5–16. [Google Scholar] [CrossRef]
- Calvin, H.I. Comparative labelling of rat epididymal spermatozoa by intratesticularly administered 65ZnCl2 and [35S]cysteine. J. Reprod. Fertil. 1981, 61, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Kaminska, B.; Rozewicka, L.; Dominiak, B.; Mielnicka, M.; Mikulska, D. Zinc content in epididymal spermatozoa of metoclopramide-treated rats. Andrologia 1987, 19, 677–683. [Google Scholar] [CrossRef] [PubMed]
- Henkel, R.; Baldauf, C.; Schill, W.B. Resorption of the element zinc from spermatozoa by the epididymal epithelium. Reprod. Domest. Anim. Zuchthyg. 2003, 38, 97–101. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Macias, V.; Martinez-Pastor, F.; Alvarez, M.; Garde, J.J.; Anel, E.; Anel, L.; de Paz, P. Assessment of chromatin status (SCSA) in epididymal and ejaculated sperm in Iberian red deer, ram and domestic dog. Theriogenology 2006, 66, 1921–1930. [Google Scholar] [CrossRef]
- Henkel, R.; Bittner, J.; Weber, R.; Hüther, F.; Miska, W. Relevance of zinc in human sperm flagella and its relation to motility. Fertil. Steril. 1999, 71, 1138–1143. [Google Scholar] [CrossRef]
- Kerns, K.; Zigo, M.; Drobnis, E.Z.; Sutovsky, M.; Sutovsky, P. Zinc ion flux during mammalian sperm capacitation. Nat. Commun. 2018, 9, 2061. [Google Scholar] [CrossRef]
- Allouche-Fitoussi, D.; Breitbart, H. The Role of Zinc in Male Fertility. Int. J. Mol. Sci. 2020, 21, 7796. [Google Scholar] [CrossRef]
- Bjorndahl, L.; Kvist, U. A model for the importance of zinc in the dynamics of human sperm chromatin stabilization after ejaculation in relation to sperm DNA vulnerability. Syst. Biol. Reprod. Med. 2011, 57, 86–92. [Google Scholar] [CrossRef]
- Kvist, U. Importance of spermatozoal zinc as temporary inhibitor of sperm nuclear chromatin decondensation ability in man. Acta Physiol. Scand. 1980, 109, 79–84. [Google Scholar] [CrossRef]
- Kvist, U. Sperm nuclear chromatin decondensation ability. An in vitro study on ejaculated human spermatozoa. Acta Physiol. Scand. Suppl. 1980, 486, 1–24. [Google Scholar]
- Riffo, M.; Leiva, S.; Astudillo, J. Effect of zinc on human sperm motility and the acrosome reaction. Int. J. Androl. 1992, 15, 229–237. [Google Scholar] [CrossRef] [PubMed]
- Bettger, W.J.; O’Dell, B.L. A critical physiological role of zinc in the structure and function of biomembranes. Life Sci. 1981, 28, 1425–1438. [Google Scholar] [CrossRef]
- O’Dell, B.L. Role of zinc in plasma membrane function. J. Nutr. 2000, 130, 1432s–1436s. [Google Scholar] [CrossRef] [PubMed]
- Narasimhaiah, M.; Arunachalam, A.; Sellappan, S.; Mayasula, V.K.; Guvvala, P.R.; Ghosh, S.K.; Chandra, V.; Ghosh, J.; Kumar, H. Organic zinc and copper supplementation on antioxidant protective mechanism and their correlation with sperm functional characteristics in goats. Reprod. Domest. Anim. Zuchthyg. 2018, 53, 644–654. [Google Scholar] [CrossRef] [PubMed]
- Nazari, A.; Sabeti, P.; Pourmasumi, S. Comparison between sperm parameters and chromatin in recurrent pregnancy loss couples after antioxidant therapy. J. Fam. Med. Prim. Care 2020, 9, 597–601. [Google Scholar] [CrossRef]
- Suarez, S.S. Mammalian sperm interactions with the female reproductive tract. Cell Tissue Res. 2016, 363, 185–194. [Google Scholar] [CrossRef]
- Kerns, K.; Sharif, M.; Zigo, M.; Xu, W.; Hamilton, L.E.; Sutovsky, M.; Ellersieck, M.; Drobnis, E.Z.; Bovin, N.; Oko, R.; et al. Sperm Cohort-Specific Zinc Signature Acquisition and Capacitation-Induced Zinc Flux Regulate Sperm-Oviduct and Sperm-Zona Pellucida Interactions. Int. J. Mol. Sci. 2020, 21, 2121. [Google Scholar] [CrossRef]
- Sanchez, R.; Deppe, M.; Schulz, M.; Bravo, P.; Villegas, J.; Morales, P.; Risopatron, J. Participation of the sperm proteasome during in vitro fertilisation and the acrosome reaction in cattle. Andrologia 2011, 43, 114–120. [Google Scholar] [CrossRef]
- Ferrer, M.; Rodriguez, H.; Zara, L.; Yu, Y.; Xu, W.; Oko, R. MMP2 and acrosin are major proteinases associated with the inner acrosomal membrane and may cooperate in sperm penetration of the zona pellucida during fertilization. Cell Tissue Res. 2012, 349, 881–895. [Google Scholar] [CrossRef]
- Seeler, J.F.; Sharma, A.; Zaluzec, N.J.; Bleher, R.; Lai, B.; Schultz, E.G.; Hoffman, B.M.; LaBonne, C.; Woodruff, T.K.; O’Halloran, T.V. Metal ion fluxes controlling amphibian fertilization. Nat. Chem. 2021, 13, 683–691. [Google Scholar] [CrossRef]
- Kim, A.M.; Bernhardt, M.L.; Kong, B.Y.; Ahn, R.W.; Vogt, S.; Woodruff, T.K.; O’Halloran, T.V. Zinc sparks are triggered by fertilization and facilitate cell cycle resumption in mammalian eggs. ACS Chem. Biol. 2011, 6, 716–723. [Google Scholar] [CrossRef] [PubMed]
- Duncan, F.E.; Que, E.L.; Zhang, N.; Feinberg, E.C.; O’Halloran, T.V.; Woodruff, T.K. The zinc spark is an inorganic signature of human egg activation. Sci. Rep. 2016, 6, 24737. [Google Scholar] [CrossRef] [PubMed]
- Que, E.L.; Duncan, F.E.; Lee, H.C.; Hornick, J.E.; Vogt, S.; Fissore, R.A.; O’Halloran, T.V.; Woodruff, T.K. Bovine eggs release zinc in response to parthenogenetic and sperm-induced egg activation. Theriogenology 2019, 127, 41–48. [Google Scholar] [CrossRef]
- Meizel, S. The sperm, a neuron with a tail: ‘Neuronal’ receptors in mammalian sperm. Biol. Rev. Camb. Philos. Soc. 2004, 79, 713–732. [Google Scholar] [CrossRef]
- Meizel, S.; Son, J.H. Studies of sperm from mutant mice suggesting that two neurotransmitter receptors are important to the zona pellucida-initiated acrosome reaction. Mol. Reprod. Dev. 2005, 72, 250–258. [Google Scholar] [CrossRef] [PubMed]
- Urra, J.A.; Villaroel-Espíndola, F.; Covarrubias, A.A.; Rodríguez-Gil, J.E.; Ramírez-Reveco, A.; Concha, I.I. Presence and function of dopamine transporter (DAT) in stallion sperm: Dopamine modulates sperm motility and acrosomal integrity. PLoS ONE 2014, 9, e112834. [Google Scholar] [CrossRef]
- Gorodeski, G.I. Purinergic Signalling in the Reproductive System. Auton. Neurosci. Basic. Clin. 2015, 191, 82–101. [Google Scholar] [CrossRef]
- Kumar, P.; Meizel, S. Nicotinic acetylcholine receptor subunits and associated proteins in human sperm. J. Biol. Chem. 2005, 280, 25928–25935. [Google Scholar] [CrossRef]
- Gianzo, M.; Muñoa-Hoyos, I.; Urizar-Arenaza, I.; Larreategui, Z.; Quintana, F.; Garrido, N.; Subirán, N.; Irazusta, J. Angiotensin II type 2 receptor is expressed in human sperm cells and is involved in sperm motility. Fertil. Steril. 2016, 105, 608–616. [Google Scholar] [CrossRef]
- Amoako, A.A.; Marczylo, T.H.; Marczylo, E.L.; Elson, J.; Willets, J.M.; Taylor, A.H.; Konje, J.C. Anandamide modulates human sperm motility: Implications for men with asthenozoospermia and oligoasthenoteratozoospermia. Hum. Reprod. 2013, 28, 2058–2066. [Google Scholar] [CrossRef]
- Flegel, C.; Vogel, F.; Hofreuter, A.; Schreiner, B.S.; Osthold, S.; Veitinger, S.; Becker, C.; Brockmeyer, N.H.; Muschol, M.; Wennemuth, G.; et al. Characterization of the Olfactory Receptors Expressed in Human Spermatozoa. Front. Mol. Biosci. 2015, 2, 73. [Google Scholar] [CrossRef] [PubMed]
- Cortés-Rodriguez, M.; Royo, J.L.; Reyes-Palomares, A.; Lendínez, A.M.; Ruiz-Galdón, M.; Reyes-Engel, A. Sperm count and motility are quantitatively affected by functional polymorphisms of HTR2A, MAOA and SLC18A. Reprod. Biomed. Online 2018, 36, 560–567. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Zhu, P.; Wu, C.; Yu, L.; Zhao, S.; Gu, X. In silico analysis indicates a similar gene expression pattern between human brain and testis. Cytogenet. Genome Res. 2003, 103, 58–62. [Google Scholar] [CrossRef] [PubMed]
- Sharp, A.H.; Ross, C.A. Neurobiology of Huntington’s disease. Neurobiol. Dis. 1996, 3, 3–15. [Google Scholar] [CrossRef]
- Clever, F.; Cho, I.K.; Yang, J.; Chan, A.W.S. Progressive Polyglutamine Repeat Expansion in Peripheral Blood Cells and Sperm of Transgenic Huntington’s Disease Monkeys. J. Huntingt. Dis. 2019, 8, 443–448. [Google Scholar] [CrossRef] [PubMed]
- Telenius, H.; Almqvist, E.; Kremer, B.; Spence, N.; Squitieri, F.; Nichol, K.; Grandell, U.; Starr, E.; Benjamin, C.; Castaldo, I.; et al. Somatic mosaicism in sperm is associated with intergenerational (CAG)n changes in Huntington disease. Hum. Mol. Genet. 1995, 4, 189–195. [Google Scholar] [CrossRef]
- Mangiarini, L.; Sathasivam, K.; Mahal, A.; Mott, R.; Seller, M.; Bates, G.P. Instability of highly expanded CAG repeats in mice transgenic for the Huntington’s disease mutation. Nat Genet. 1997, 15, 197–200. [Google Scholar] [CrossRef]
- Dragileva, E.; Hendricks, A.; Teed, A.; Gillis, T.; Lopez, E.T.; Friedberg, E.C.; Kucherlapati, R.; Edelmann, W.; Lunetta, K.L.; MacDonald, M.E.; et al. Intergenerational and striatal CAG repeat instability in Huntington’s disease knock-in mice involve different DNA repair genes. Neurobiol. Dis. 2009, 33, 37–47. [Google Scholar] [CrossRef]
- Mollersen, L.; Rowe, A.D.; Larsen, E.; Rognes, T.; Klungland, A. Continuous and periodic expansion of CAG repeats in Huntington’s disease R6/1 mice. PLoS Genet. 2010, 6, e1001242. [Google Scholar] [CrossRef]
- Im, W.; Chung, J.; Lee, S.T.; Chu, K.; Kim, M.W.; Kim, M. Nuclear localization of huntingtin during spermatogenesis. Neurol. Sci. Off. J. Ital. Neurol. Soc. Ital. Soc. Clin. Neurophysiol. 2014, 35, 459–462. [Google Scholar] [CrossRef]
- Leavitt, B.R.; Guttman, J.A.; Hodgson, J.G.; Kimel, G.H.; Singaraja, R.; Vogl, A.W.; Hayden, M.R. Wild-type huntingtin reduces the cellular toxicity of mutant huntingtin in vivo. Am. J. Hum. Genet. 2001, 68, 313–324. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Zhang, H.; Liu, Y.; Zhao, F.; Zhu, S.; Xie, C.; Tang, T.S.; Guo, C. Germline deletion of huntingtin causes male infertility and arrested spermiogenesis in mice. J. Cell Sci. 2016, 129, 492–501. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Van Raamsdonk, J.M.; Murphy, Z.; Selva, D.M.; Hamidizadeh, R.; Pearson, J.; Petersén, A.; Björkqvist, M.; Muir, C.; Mackenzie, I.R.; Hammond, G.L.; et al. Testicular degeneration in Huntington disease. Neurobiol. Dis. 2007, 26, 512–520. [Google Scholar] [CrossRef] [PubMed]
- Vidinská, D.; Vochozková, P.; Šmatlíková, P.; Ardan, T.; Klíma, J.; Juhás, Š.; Juhásová, J.; Bohuslavová, B.; Baxa, M.; Valeková, I.; et al. Gradual Phenotype Development in Huntington Disease Transgenic Minipig Model at 24 Months of Age. Neuro-Degener. Dis. 2018, 18, 107–119. [Google Scholar] [CrossRef]
- Macakova, M.; Bohuslavova, B.; Vochozkova, P.; Pavlok, A.; Sedlackova, M.; Vidinska, D.; Vochyanova, K.; Liskova, I.; Valekova, I.; Baxa, M.; et al. Mutated Huntingtin Causes Testicular Pathology in Transgenic Minipig Boars. Neuro. Degener. Dis. 2016, 16, 245–259. [Google Scholar] [CrossRef]
- Krizova, J.; Stufkova, H.; Rodinova, M.; Macakova, M.; Bohuslavova, B.; Vidinska, D.; Klima, J.; Ellederova, Z.; Pavlok, A.; Howland, D.S.; et al. Mitochondrial Metabolism in a Large-Animal Model of Huntington Disease: The Hunt for Biomarkers in the Spermatozoa of Presymptomatic Minipigs. Neuro-Degener. Dis. 2017, 17, 213–226. [Google Scholar] [CrossRef]
- Paul, N.; Kumaresan, A.; Das Gupta, M.; Nag, P.; Guvvala, P.R.; Kuntareddi, C.; Sharma, A.; Selvaraju, S.; Datta, T.K. Transcriptomic Profiling of Buffalo Spermatozoa Reveals Dysregulation of Functionally Relevant mRNAs in Low-Fertile Bulls. Front. Vet. Sci. 2020, 7, 609518. [Google Scholar] [CrossRef]
- Prakash, M.A.; Kumaresan, A.; Ebenezer Samuel King, J.P.; Nag, P.; Sharma, A.; Sinha, M.K.; Kamaraj, E.; Datta, T.K. Comparative Transcriptomic Analysis of Spermatozoa From High- and Low-Fertile Crossbred Bulls: Implications for Fertility Prediction. Front. Cell Dev. Biol. 2021, 9, 647717. [Google Scholar] [CrossRef]
- Walters, E.M.; Wells, K.D.; Bryda, E.C.; Schommer, S.; Prather, R.S. Swine models, genomic tools and services to enhance our understanding of human health and diseases. Lab. Anim. 2017, 46, 167–172. [Google Scholar] [CrossRef]
- Archibald, A.L.; Bolund, L.; Churcher, C.; Fredholm, M.; Groenen, M.A.; Harlizius, B.; Lee, K.T.; Milan, D.; Rogers, J.; Rothschild, M.F.; et al. Pig genome sequence--analysis and publication strategy. BMC Genom. 2010, 11, 438. [Google Scholar] [CrossRef]
- Marsh, J.L.; Pallos, J.; Thompson, L.M. Fly models of Huntington’s disease. Hum. Mol. Genet. 2003, 12, R187–R193. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Wang, M.; Yuan, Y.; Wang, X.; Fu, R.; Wan, H.; Xie, M.; Liu, M.; Guo, X.; Zheng, Y.; et al. Complete Meiosis from Embryonic Stem Cell-Derived Germ Cells In Vitro. Cell Stem Cell 2016, 18, 330–340. [Google Scholar] [CrossRef] [PubMed]
- Easley, C.A.t.; Phillips, B.T.; McGuire, M.M.; Barringer, J.M.; Valli, H.; Hermann, B.P.; Simerly, C.R.; Rajkovic, A.; Miki, T.; Orwig, K.E.; et al. Direct differentiation of human pluripotent stem cells into haploid spermatogenic cells. Cell Rep. 2012, 2, 440–446. [Google Scholar] [CrossRef] [PubMed]
- Khampang, S.; Cho, I.K.; Punyawai, K.; Gill, B.; Langmo, J.N.; Nath, S.; Greeson, K.W.; Symosko, K.M.; Fowler, K.L.; Tian, S.; et al. Blastocyst Development after Fertilization with in vitro Spermatids Derived from Non-Human Primate Embryonic Stem Cells. F S Sci. 2021, 2, 365–375. [Google Scholar] [CrossRef] [PubMed]
- Saitou, M.; Hayashi, K. Mammalian in vitro gametogenesis. Science 2021, 374, eaaz6830. [Google Scholar] [CrossRef]
- Symosko, K.M.; Schatten, G.; Easley, C.A. Gamete Production from Stem Cells. In Female and Male Fertility Preservation; Grynberg, M., Patrizio, P., Eds.; Springer Nature: Cham, Switzerland, 2022; pp. 395–407. [Google Scholar] [CrossRef]
- Steinberger, A. Factors affecting spermatogenesis in organ cultures of mammalian testes. J. Reprod. Fertil. Suppl. 1967, 2, 117–124. [Google Scholar]
- Ishikura, Y.; Yabuta, Y.; Ohta, H.; Hayashi, K.; Nakamura, T.; Okamoto, I.; Yamamoto, T.; Kurimoto, K.; Shirane, K.; Sasaki, H.; et al. In Vitro Derivation and Propagation of Spermatogonial Stem Cell Activity from Mouse Pluripotent Stem Cells. Cell Rep. 2016, 17, 2789–2804. [Google Scholar] [CrossRef]
- Irie, N.; Weinberger, L.; Tang, W.W.; Kobayashi, T.; Viukov, S.; Manor, Y.S.; Dietmann, S.; Hanna, J.H.; Surani, M.A. SOX17 is a critical specifier of human primordial germ cell fate. Cell 2015, 160, 253–268. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, K.; Yokobayashi, S.; Nakamura, T.; Okamoto, I.; Yabuta, Y.; Kurimoto, K.; Ohta, H.; Moritoki, Y.; Iwatani, C.; Tsuchiya, H.; et al. Robust In Vitro Induction of Human Germ Cell Fate from Pluripotent Stem Cells. Cell Stem Cell 2015, 17, 178–194. [Google Scholar] [CrossRef]
- Bucay, N.; Yebra, M.; Cirulli, V.; Afrikanova, I.; Kaido, T.; Hayek, A.; Montgomery, A.M. A novel approach for the derivation of putative primordial germ cells and sertoli cells from human embryonic stem cells. Stem. Cells 2009, 27, 68–77. [Google Scholar] [CrossRef]
- Fukunaga, N.; Teramura, T.; Onodera, Y.; Takehara, T.; Fukuda, K.; Hosoi, Y. Leukemia inhibitory factor (LIF) enhances germ cell differentiation from primate embryonic stem cells. Cell Reprogram 2010, 12, 369–376. [Google Scholar] [CrossRef] [PubMed]
- Kee, K.; Angeles, V.T.; Flores, M.; Nguyen, H.N.; Reijo Pera, R.A. Human DAZL, DAZ and BOULE genes modulate primordial germ-cell and haploid gamete formation. Nature 2009, 462, 222–225. [Google Scholar] [CrossRef] [PubMed]
- Panula, S.; Medrano, J.V.; Kee, K.; Bergstrom, R.; Nguyen, H.N.; Byers, B.; Wilson, K.D.; Wu, J.C.; Simon, C.; Hovatta, O.; et al. Human germ cell differentiation from fetal- and adult-derived induced pluripotent stem cells. Hum. Mol. Genet. 2011, 20, 752–762. [Google Scholar] [CrossRef] [PubMed]
- Park, T.S.; Galic, Z.; Conway, A.E.; Lindgren, A.; van Handel, B.J.; Magnusson, M.; Richter, L.; Teitell, M.A.; Mikkola, H.K.; Lowry, W.E.; et al. Derivation of primordial germ cells from human embryonic and induced pluripotent stem cells is significantly improved by coculture with human fetal gonadal cells. Stem Cells 2009, 27, 783–795. [Google Scholar] [CrossRef]
- Teramura, T.; Takehara, T.; Kawata, N.; Fujinami, N.; Mitani, T.; Takenoshita, M.; Matsumoto, K.; Saeki, K.; Iritani, A.; Sagawa, N.; et al. Primate embryonic stem cells proceed to early gametogenesis in vitro. Cloning Stem Cells 2007, 9, 144–156. [Google Scholar] [CrossRef] [PubMed]
- Tilgner, K.; Atkinson, S.P.; Golebiewska, A.; Stojkovic, M.; Lako, M.; Armstrong, L. Isolation of primordial germ cells from differentiating human embryonic stem cells. Stem Cells 2008, 26, 3075–3085. [Google Scholar] [CrossRef]
- Yamauchi, K.; Hasegawa, K.; Chuma, S.; Nakatsuji, N.; Suemori, H. In vitro germ cell differentiation from cynomolgus monkey embryonic stem cells. PLoS ONE 2009, 4, e5338. [Google Scholar] [CrossRef]
- Eguizabal, C.; Montserrat, N.; Vassena, R.; Barragan, M.; Garreta, E.; Garcia-Quevedo, L.; Vidal, F.; Giorgetti, A.; Veiga, A.; Izpisua Belmonte, J.C. Complete meiosis from human induced pluripotent stem cells. Stem Cells 2011, 29, 1186–1195. [Google Scholar] [CrossRef]
- Alves-Lopes, J.P.; Stukenborg, J.B. Testicular organoids: A new model to study the testicular microenvironment in vitro? Hum. Reprod. Update 2018, 24, 176–191. [Google Scholar] [CrossRef]
- Huleihel, M.; Nourashrafeddin, S.; Plant, T.M. Application of three-dimensional culture systems to study mammalian spermatogenesis, with an emphasis on the rhesus monkey (Macaca mulatta). Asian J. Androl. 2015, 17, 972–980. [Google Scholar] [CrossRef]
- Pendergraft, S.S.; Sadri-Ardekani, H.; Atala, A.; Bishop, C.E. Three-dimensional testicular organoid: A novel tool for the study of human spermatogenesis and gonadotoxicity in vitro. Biol. Reprod. 2017, 96, 720–732. [Google Scholar] [CrossRef] [PubMed]
- Simard, O.; Gregoire, M.C.; Arguin, M.; Brazeau, M.A.; Leduc, F.; Marois, I.; Richter, M.V.; Boissonneault, G. Instability of trinucleotidic repeats during chromatin remodeling in spermatids. Hum. Mutat. 2014, 35, 1280–1284. [Google Scholar] [CrossRef] [PubMed]
- Usdin, K.; House, N.C.; Freudenreich, C.H. Repeat instability during DNA repair: Insights from model systems. Crit. Rev. Biochem. Mol. Biol. 2015, 50, 142–167. [Google Scholar] [CrossRef]
- Yoon, S.R.; Dubeau, L.; de Young, M.; Wexler, N.S.; Arnheim, N. Huntington disease expansion mutations in humans can occur before meiosis is completed. Proc. Natl. Acad. Sci. USA 2003, 100, 8834–8838. [Google Scholar] [CrossRef]
- Ehmcke, J.; Wistuba, J.; Schlatt, S. Spermatogonial stem cells: Questions, models and perspectives. Hum. Reprod. Update 2006, 12, 275–282. [Google Scholar] [CrossRef]
- Fayomi, A.P.; Orwig, K.E. Spermatogonial stem cells and spermatogenesis in mice, monkeys and men. Stem. Cell Res. 2018, 29, 207–214. [Google Scholar] [CrossRef]
- Khampang, S.; Parnpai, R.; Mahikul, W.; Easley, C.A.t.; Cho, I.K.; Chan, A.W.S. CAG repeat instability in embryonic stem cells and derivative spermatogenic cells of transgenic Huntington’s disease monkey. J. Assist. Reprod. Genet. 2021, 38, 1215–1229. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).