Abstract
Differential evolution of apoptosis, programmed necrosis, and autophagy, parthanatos is a form of cell death mediated by poly(ADP-ribose) polymerase 1 (PARP1), which is caused by DNA damage. PARP1 hyper-activation stimulates apoptosis-inducing factor (AIF) nucleus translocation, and accelerates nicotinamide adenine dinucleotide (NAD+) and adenosine triphosphate (ATP) depletion, leading to DNA fragmentation. The mechanisms of parthanatos mainly include DNA damage, PARP1 hyper-activation, PAR accumulation, NAD+ and ATP depletion, and AIF nucleus translocation. Now, it is reported that parthanatos widely exists in different diseases (tumors, retinal diseases, neurological diseases, diabetes, renal diseases, cardiovascular diseases, ischemia-reperfusion injury...). Excessive or defective parthanatos contributes to pathological cell damage; therefore, parthanatos is critical in the therapy and prevention of many diseases. In this work, the hallmarks and molecular mechanisms of parthanatos and its related disorders are summarized. The questions raised by the recent findings are also presented. Further understanding of parthanatos will provide a new treatment option for associated conditions.
1. Introduction
Parthanatos, a kind of new programmed death mode, has been put forward by Professors Ted and Valina Dawson to indicate a caspase-independent cell death subroutine that critically relies on the hyper-activation of poly(ADP-ribose) polymerase 1 (PARP1) [1,2]. Traumatic injury, excitotoxicity, ischemia, and many neurodegenerative disorders will cause toxic stimuli accumulation, which leads to DNA damage, PARP1 hyper-activation, PAR accumulation, and mitochondrial apoptosis-inducing factor (AIF) nucleus translocation. Ultimately, large-scale DNA fragmentation and later caspase activation lead to mitochondria dysfunction, failure to generate NAD+/ATP, and end up with cell collapse.
There are many differences between some common forms of cell death and parthanatos in morphological features, biochemical features, regulatory pathways, key genetic inhibitors, or inhibition by protein over-expression (Table 1). Parthanatos is characterized by PARP1 hyper-activation, PAR polymers accumulation, mitochondria depolarization, and AIF nucleus translocation [3,4,5]. It is reported that parthanatos is involved in many common important diseases, such as Parkinson’s disease, stroke, cancer, heart attack, retinal disease, and diabetes [6,7,8,9,10], and can be applied to the clinical treatment of related diseases (Figure 1).

Table 1.
Similarities and differences between apoptosis, necrosis, autophagy, and parthanatos.
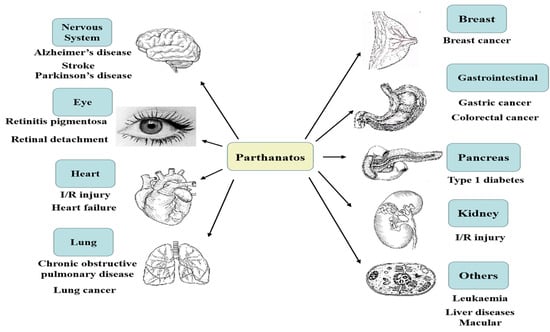
Figure 1.
Parthanatos is associated with multiple system diseases; for example, nervous system diseases, heart diseases, retinal diseases, gastrointestinal diseases, lung diseases, kidney diseases, pancreatic diseases, breast diseases, and so on.
In this work, we review the hallmarks and molecular mechanisms of parthanatos and its related diseases to further learn about its pathogenesis and put forward new treatments for related conditions.
2. Hallmarks of Parthanatos
2.1. Morphological Features
Cells undergoing parthanatos usually show necrosis-like and apoptosis-like morphological changes [2]. These features include the loss of cell membrane integrity, cellular propidium iodide (PI) positive staining, and DNA fragmentations (15 kb to 50 kb) [1,11]. At the ultra-structural level, parthanatic cells usually exhibit mitochondrial abnormalities, such as the dissipation of inner transmembrane potential, nuclear shrinkage, and chromatin condensation [1].
2.2. Biochemical Features
2.2.1. DNA Injury
DNA injury signaling is essential in maintaining genome integrity and cell fate. The main reasons for DNA injury are roughly divided into environmental effects (exogenous damage) and spontaneous injury (endogenous injury). Environmental factors often cover ultraviolet radiation (UVR), ionizing radiation (IR), alkylating agents, and metabolically activated compounds. Mistakes in DNA replication, base tautomerism, base deamination, and loss belong to spontaneous DNA damage. Sometimes chronic UVR is helpful to DNA repair response; most of the time, it results in unrepairable DNA damage [12,13,14]. One publication emphasizes mitochondrial changes and DNA damage mediated by UVR in HaCaT cells, and the significance of PARP in these alterations [15].
IR (α-, β-, γ-, X-ray), a valid and widely used measurement for cancer treatment, is able to control tumors affecting DNA damage response and repair (DRR) processes, which determine the fate of the tumor cells. IR-induced DNA double-strand breaks (DSBs) are the most lethal form of damage [16].
Regarding alkylating agents, the primary mode of their action causes cytotoxic DNA damage. To resist alkylation-induced cell death or mutation, direct DNA damage reversal, base excision repair (BER), and mismatch repair (MMR) come into work. It is essential for a favorable response of the organism to alkylating agents to keep an appropriate balance of activity both within and between these pathways [17]. Widespread as an environmental mutagen, N-methyl-N′-nitro-N-nitrosoguanidine (MNNG) is a commonly used DNA-alkylating agent that potently initiates parthanatic cell death in cell-death research [18].
ROS play a multifaceted and pleomorphic role in DNA damage response (DDR), such as mediating genotoxin-induced damage, DNA damage by oncogenic replication stress, sensing of DSBs, signal transduction within DDR, cell cycle progression, apoptosis, and DNA repair. It is necessary to distinguish the role between oxidative stress and ROS in DDR [19,20,21].
PARP1 is crucial in maintaining genomic stability by facilitating DNA repair to ensure cell survival. It can regulate BER, single-stranded break (SSB), and DSB repair pathways. Meanwhile, PARP1 mediates parthanatos in response to severe DNA damage [22].
2.2.2. NAD+ Depletion
Cellular energy depletion is caused by PARP1 over-activation through NAD+ consumption [23,24]. NAD+ is a co-factor in cellular metabolism, which is needed for generating ATP. Besides, NAD+ resynthesis calls for about 2–4 molecules of ATP [25]. Studies suggest that ATP decreases with the consumption of NAD+, along with a decrease in cellular energy, resulting in cell death [26,27]. NAD+ and energy are preserved when PARP1 inhibitors and the model of PARP1 deficiency are used [28,29]. However, the decrease in NAD+ is not always associated with ATP decline in cerebral ischemia-reperfusion injury (CI/RI) models. A study points out that PARP1 KO mice reduce the infarct size in the CI/RI group, compared with the normal group, but the energy status does not change [30].
2.2.3. Poly(ADP-Ribose) (PAR) Accumulation
The synthesis of PAR polymers depends on certain enzymes called PARPs [31]. PARP1, a member of PARPs, is the most studied and makes the greatest contribution to synthesizing PAR polymers [32]. When there is a toxic stimulus, DNA is damaged, PARP1 becomes hyper-activated and generates superfluous PAR polymer, followed by AIF nucleus translocation [33]. PAR polymers activate a signal to modulate the downstream transcriptional process and the mechanism of DNA repair. PAR interacts with NONO, a novel PAR-binding protein, the binding modulates the physiological functions of protein [34]. Subsequently, PAR has been studied to be directly toxic to neurons. AIF and PAR polymer-binding protein [35,36], a physical interaction between PAR and AIF, induces AIF release from the mitochondria and leads to the occurrence of parthanatos [37,38].
2.3. Genetic Features
A few genes/proteins are considered biomarkers of parthanatos; for example, PARP1 is a therapeutic target in the stroke, trauma, I/R injury, and diabetes model [23]. Cell parthanatos is suppressed in fibroblasts that are isolated from PARP1 KO mice [39]. AIF is identified as a candidate, PAR-binding protein [36,37]. Genetic ablation of NAMPT or FK866 treatment sensitizes lymphocytes to MNNG-induced parthanatos, while over-expression of a catalytically active recombinant NAMPT protects NIH-3T3 cells from the toxicity of the same DNA alkylating agent [40]. The result of AIF KO shows AIF as a cell death effector in PARP1 toxicity and parthanatos [41]. KO PARG dysfunction sensitizes various cancer cells to chemotherapeutic agents and radiation [42,43]. Human osteosarcoma U2OS is exposed to hydrogen peroxide (H2O2) and is compared with the ADP-ribosylation (ADPr) pattern of control, ADP-ribosyl hydrolase 3 (ARH3) KO, HPF1 KO, and PARP1 KO cells. The data indicate that global ADPr in response to DNA damage requires ARH3, HPF1, and PARP1 [44].
2.4. Immune Features
The activation of PARP1 promotes the transcription of pro-inflammatory genes and down-regulates multiple pathways of inflammation and tissue injury [45]. The cells, which infect oligodendrocytes in human natural killer (NK) cells, are killed by virus-induced PARP1 and AIF translocation, rather than the immune system [46]. For instance, the novel mechanism of immune escape in acute myeloid leukemia (AML) is uncovered: mature malignant cells in monocytic forms of leukemia have the capacity to produce ROS via NADPH oxidase and thus trigger parthanatos in adjacent antileukemic lymphocytes [47]. In addition, the lymphocyte function is impaired because those malignant ROS-producing myeloid cells induce parthanatos in NK cells [47,48,49]. NAD+ metabolism makes a contribution to inflammation and immune responses [50]. Human cytomegalovirus (HCMV) also restrains local immune responses through ROS-induced parthanatos [14]. PARG is vital to tumor growth and metastasis of colon carcinoma and may be applied as a new target for treating colon carcinoma [51].
3. Molecular Mechanisms of Parthanatos
The molecular mechanisms of parthanatos include DNA damage, PARP1 hyper-activation, NAD+ and ATP depletion, and AIF translocation from the mitochondrial to the nucleus (Figure 2).
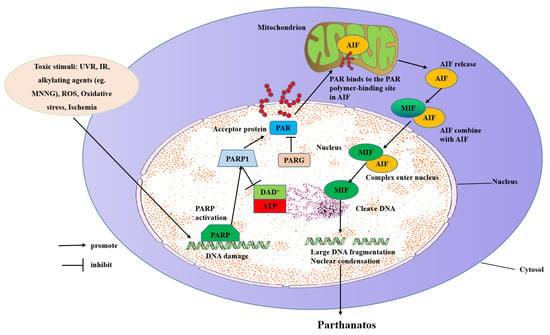
Figure 2.
Molecular mechanisms of parthanatos. Toxic stimuli such as ROS, ischemia, alkylating agents (e.g., MNNG), IR, and UVR activate PARP-1 directly or indirectly through activation of NOS, which makes NO to induce ROS and the subsequent DNA damage. PARP-1 overactivation produces free PAR by PARG-mediated hydrolyzation, which serves as a death signal from the nucleus to mitochondria, inducing the release of AIF. AIF then translocates with MIF to the nucleus where it induces extensive fragmentation of DNA. This form of cell death is called “parthanatos”.
3.1. Inducing Parthanatos by Injuring DNA
DNA damage is caused by toxic stimuli, such as H2O2 or hydroxyl radical, nitrosative stress from NO or peroxynitrite (ONOO−), inflammation, ischemia or I/R, hypoxia, hypoglycemia, and DNA-alkylating agents [25]. There are many DNA repair pathways, including BER, nucleotide excision repair (NER), mismatch repair (MMR), homologous recombination (HR), and non-homologous end-joining (NHEJ). These pathways help to repair DNA damage throughout different stages of the cell cycle [52].
PARP1 is a kind of enzyme that takes part in the process of DNA repair. It may not catalyze the process of DNA repair directly; however, the process of activating multiple DNA repair enzymes such as DNA topoisomerase, DNA helicase, and DNA ligase needs PARP1′s participation [45]. PARP1 composes a DNA base-excision repair system through testing the crack and fracture of DNA strands and promoting its repair via PAR polymer synthesis. When DNA is mildly damaged, PARP1 is hundreds of times more active, and the enzyme uses NAD+ to synthesize PAR polymer by consuming ATP. However, when there is serious DNA damage, PARP1 is over-activated and PAR polymers are generated and accumulated, which leads to phosphatidylserine externalization, mitochondrial membrane potential (MMP) dissipation, AIF nucleus translocation, massive DNA fragmentation, and chromatin condensation, and then parthanatos occurrence [1,2]. PAR also participates in DNA replication and repair; the cell, which is inhibited by PARG, shows an increased sensitivity to DNA-damaging agents [10]. PARG is conducive to DSB and SSB repair, recovery from prolonged replication stress, and unusual replication structures in the S phase [53].
3.2. Inducing Parthanatos by Hyper-Activating PARP1
PARP1, a highly expressed 116 kD eukaryotic nuclear protein, includes an N-terminal, a self-modifying domain, and a C-terminal catalytic domain. In response to DNA damage, histone PARylation factor 1 (HPF1), a required co-factor in PARP1/PARP2-mediated ADP-ribosylation of serine, directly binds to the catalytic domain of PARP1 via its C-terminal domain. The active site, which is formed by HPF1 and PARP1, activates the PARP1 enzyme [54]. PARP1 combines with DNA primarily by the second zinc-finger domain [45,55,56], it catalyzes NAD+ and synthesizes PAR polymers adhered to various nuclear proteins, which significantly influences their function [12].
PARP1 is vital in repairing DNA, leading to stability and transcription of genomics with normal physiological conditions. When DNA damage is slight, PARP1 is more reactive, resulting in ADP-ribosylation of PARP1 and its substrates, which then helps to recruit DNA repair effector proteins to repair DNA [57]. Nevertheless, under immoderate genotoxic stress, PARP1 is hyper-activated and generates excessive PAR and stimulates AIF nucleus translocation, causing a sharp drop in NAD+ storage after DNA breaks, which results in parthanatos. PARP1 is activated by DNA damage, which is caused by external stimuli. Furthermore, PARP1 is stimulated by the calcium (Ca2+) signaling pathway [9].
PARP1 hyper-activation is the first and critical step in the parthanatic cascade. The inhibition or deficiency of PARP1 is protective in models of the cell and rat/mouse injury paradigms, including retinal degeneration diseases, stroke, diabetes, I/R injury, neurodegenerative disease, and heart failure [58]. PARP1 KO mouse models are hypersensitive to alkylating agents and IR, which is related to defective DNA repair [59]. On the contrary, PARP1 KO mouse models are protected from LPS-induced shock, ischemic injury, and 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) excitotoxicity. Therefore, these genetic models emphasize a double function of PARP1 in the cell depending on the stimulation: promotion of DNA repair or mediation of pathological cascades of inflammation/excitotoxicity [9]. These studies contribute to understanding the role of PARP1 in pathological contexts and related diseases. In preclinical zebrafish and human organotypic 3D skin models of psoriasis, PARP1 hyper-activation reacts to ROS-induced DNA damage: it is fueled by nicotinamide phosphoribosyltransferase-derived NAD+ and mediates inflammation through parthanatos [60]. In short, as a cell fate determinant, PARP1 has a significant influence in promoting DNA repair or restraining parthanatos. Now, many researchers have presented a number of potential treatments, of which the PARP1 enzyme has been regarded as a potential target intended to deal with related diseases. Targeting various diseases with chemical inhibitors of PARP1 provides therapeutic outcomes by reducing PARP1-mediated neuronal death. Many PARP1 inhibitors (oxaliplatin, PJ-34, 3-aminobenzamide, olaparib) have been studied in stroke, Parkinson’s disease (PD), Alzheimer’s disease (AD), Huntington’s disease (HD), and amyotrophic lateral sclerosis (ALS), but have not been clinically evaluated [61].
3.3. Inducing Parthanatos by Binding of PAR
Nuclear DNA damage stimulation triggers the synthesis of poly ribose (ADP-ribose), next to the distribution of the PAR polymer to the cytoplasm and mitochondria, and then the release of AIF [13]. PAR inhibits the glycolysis process. ARH3 hydrolyzes protein-free PAR in the nucleus and cytoplasm, inhibits PAR transfer to the cytoplasm, and thus prevents parthanatos [62]. ARH3 has been shown to catalyze PAR-degradation in vitro [63,64]. PARG, the most well-characterized enzyme in humans for PAR hydrolysis, utilizes a macrodomain fold to bind ADP-ribosylation and specifically cleaves the ribose-ribose bonds between the subunits of the PAR chains. PARG needs the cooperation of PARP to repair DNA. Once the strand of PARP and DNA breaks, an enzyme is activated, causing PARP to shuttle and the chromatin to open. PARG enters the nucleus and moves to the PARP substrate to repair the broken DNA strand. With the increase in PARG and the decrease in PAR, chromatin recovers its original structure. PARG has been shown to play a vital role in various diseases [65].
3.4. Inducing Parthanatos by Depleting ATP and NAD+
NAD+ is a factor of ATP generation and its resynthesis needs many molecules of ATP, which its consumption results in ATP depletion and cellular energy downturn, which leads to parthanatos [9,28]. The inhibition of PARP1 by pharmacological inhibitors or genetic deletion could recover NAD+ levels [28]. Neurons that are stimulated with toxic show protection and accompanying energy conservation with PARP inhibitors. Besides, PARP1 KO mice of transient cerebral ischemia-induced damage present preserved NAD+ levels [29,66], supporting the suicide hypothesis of PARP1 activation causing a block in glycolysis. Besides, adding NAD+ to cells or over-expression of NAD+ avoids PARP1-dependent parthanatos, which suggests that the decreased NAD+ relevant to PARP1 hyper-activation causes cell demise [28].
A recent finding suggests that the mitochondrial pool of NAD+ is associated with parthanatos because that parthanatos is saved by the preservation of NAD+ in the mitochondria by adding NAD+ biosynthetic enzyme Nampt or MPT inhibitor cyclosporine-A (CsA), and replenishing NAD+ [67,68]. The decrease in NAD+ after PARP1 hyper-activation reflects the level of NAD+ in the whole cell. Therefore, the conclusions of these studies need to be re-examined as the research shows that mitochondrial NAD+ is still at physiological levels after genotoxic stress, and even the nucleus and cytoplasmic NAD+ are depleted [68]. There are no reports yet to confirm that PARP1-dependent parthanatos entirely depends on mitochondrial NAD+ depletion [69]. Cyclosporine A may reduce parthanatos because it preserves mitochondrial NAD+ and inhibits mitochondrial permeability transitions, which are themselves an essential participant in parthanatos [67].
Researchers have questioned whether PARP1 hyper-activation was killed mainly by NAD+ depletion. NAD+ descent is not always associated with ATP decline in the I/R model [70]. Moreover, the energy status does not change as the infarct size is reduced in PARP1 KO mice after MCAO compared with normal mice [71]. In addition, Bax and calpain KO cells are protected from MNNG to the same degree as wild-type controls treated with the PARP1 inhibitor DPQ [72]. Although the NAD+ level is decreased in Bax and calpain KO cells, it is not in DPQ-treated cells. These results imply that PARP1 activation, which is caused by NAD+ depletion, is insufficient to account for parthanatos. NAD+ is expended by ADP-ribose transferases (ARTs) and many PARPs to generate an ADP-ribose protein modification and form PARP1 [73]. cADP-ribose synthases generate and hydrolyze the Ca2+-mobilizing second-messenger cADP-ribose from NAD+ [25,69,74]. There are two ways to promote the increase in NAD+ levels: stimulating the synthesis of NAD+ or inhibiting its excessive consumption [25,75,76].
3.5. Inducing Parthanatos by Releasing and Translocating AIF from Mitochondrial to the Nucleus
Mitochondria is a crucial element in the regulation of parthanatos. Its inter-membrane gap contains a number of proteins released through the outer membrane for taking part in parthanatos degradation. Mitochondria maintain the physiological integrity of cells by energy generation. However, once a cell is stimulated, the mitochondria become permeabilized, and AIF is released to irritate parthanatos. The change of outer membrane permeabilization (OMP) and permeability transition pore (PTP) will lead to the release of pro-apoptotic factors [77]. Bax, one of the pro-apoptotic members, is downstream of PARP1 and induces OMP needed for AIF release. Besides OMP, AIF cleavage by calpains requires AIF to be entirely released from the mitochondria [2].
According to the function, AIF is divided into three parts: an N-terminal part (binds to FAD), a central portion (which binds to NAD or NADH, which have oxidoreductase ability), and a C-terminal part (which is related to parthanatos) [26]. The harlequin (Hq) mouse is a helpful model to research AIF-mediated parthanatos [78]. It is noteworthy that AIF translocation is a vital step for parthanatos [33,79], so the understanding of this event is important in translating parthanatos research into therapy. In the nucleus, AIF-binding DNA is considered necessary even for parthanatos precipitation [80]. AIF, a parthanatos effector, takes part in the process of generating mitochondrial energy. It is usually restricted to mitochondria but translocates to the nucleus when parthanatos happens. [81,82]. AIF translocation, a marker of parthanatos, usually occurs in conjunction with the detection of specific mitochondrial markers. AIF contains a PAR-binding motif, facilitating the direct association between PARP1 and AIF [36], and takes part in PARP1 toxicity [82]. When PARP1 is hyper-activated, excessive PAR escapes from the nucleus and binds to specific cytosolic or mitochondrial proteins [35,38]. PAR combines with these proteins and eventually causes AIF to be released from the mitochondria.
AIF’s structure is markedly regulated by binding NAD(P)H and forming a stable, long-lived charge-transfer complex (CTC). In the process of PARP1-initiated parthanatos, once the NAD+ level is critically depleted, structural modulation of NADH regulates AIF release from mitochondria [83,84]. Excessive PAR triggers the mitochondrial release of AIF, which combines with macrophage MIF and carries MIF into the nucleus where the combination cleaves genomic DNA into large fragments. Chromatinolysis could be inhibited by depleting MIF and/or disrupting the AIF-MIF interaction then preventing MIF nuclease activity. The prevention of MIF nuclease activity should be a fascinating target for diseases.
4. Parthanatos and Related Diseases
Parthanatos is found in many diseases. Excessive or defective parthanatos will cause pathological cell changes. In this work, we sum up the relationship between parthanatos and cancer, retinal disease, diabetes, renal disease, heart failure, myocardial infarction, leukemia, lung injury, smoke-related lung diseases, stroke, ischemic tissue injury, brain trauma, and neurodegenerative diseases, and some representative agents of the parthanatos-associated components of targeted therapy (Table 2).
Table 2.
Summary of parthanatos in animal/human models of human pathological conditions of related diseases.
4.1. Parthanatos and Tumors
4.1.1. Breast Cancer
The high expression of PARP1 is linked to the scarcity of apoptotic bodies necrosis, which is the cytomorphological feature of parthanatos [129]. PARP1 inhibitors have been well used in clinical practice. For example, talazoparib has a better effect than standard chemotherapy on progression-free survival in patients with advanced BC [85]. BZL101 [86], an aqueous extract from Scutellaria barbata, inhibits BC by promoting AIF translocation and has been verified to be feasible and safe for advanced BC patients in a phase I clinical trial. Ganetespib [87], GA/17AAG [88], and lapatinib [89] show evidence of activity in metastatic HER2-positive and triple-negative BC in a trial through stopping MIF stabilization.
4.1.2. Colon Cancer
The E26 transformation-specific (ETS) factor inhibitor represents a promising therapeutic option for p53-deleted colon cancer. ETS1 over-expression induced by MAPK hyper-activation is thought to result in p53 loss/deregulation, PARP1 over-expression, and consequent YK-4-279-mediated parthanatos induction [130]. As the first PARP1 inhibitor, AG14361 has the pharmacologic properties of high potency, specificity, and stability, and plays a radiation sensitizer to reinforce radiation-induced parthanatos by inhibiting DNA repair [90]. HMA (5-(N,N-hexamethylene amiloride), a sodium-hydrogen antiporter inhibitor, promotes the induction of the parthanatos pathway (PAR accumulation and the translocation of the mitochondrial protein AIF to the nucleus) in SW613-B3 colon carcinoma cells [91].
4.1.3. Ovarian Cancer
In studies of ovarian cancer, three PARP1 inhibitors, olaparib [92], niraparib [93], and rucaparib [94] exploit synthetic lethality in platinum-sensitive, relapsed serous ovarian cancer. PDD00017273, a quinazolinedione, suppresses PARG, stabilizes cellular PAR chains, and is devoid of activity against PARP1 and the ARH3 glycohydrolase. It uses and intensifies DNA replication deficiencies and then leads to ascended DNA damage in ovarian cancer cells [95,96]. COH34 [131], a PARG inhibitor, extends the arylation of DNA damage and captures DNA repair factors by binding to its catalytic domain. It not only kills ovarian cancer cells with DNA repair defects but also becomes sensitive to the cells to other DNA-damaging agents. Remarkably, COH34 is effective at killing ovarian cancer cells that are resistant to the PARP1 inhibitor.
4.1.4. Esophageal Cancer
Sepantronium bromide (YM155) restrains esophageal squamous-cell carcinoma (ESCC) growth and keeps body weight in the ESCC xenografts mice model. The main reason is that YM155 activates PARP1, synthesizes PAR polymer, and stimulates AIF nucleus translocation. Genetic KO of PARP1 or AIF abolished YM155-induced parthanatos [132].
4.1.5. Head and Neck Cancer (HNC)
Oral squamous cell carcinoma (OSCC), a kind of head and neck squamous cell carcinoma (HNSCC), is widespread in cancer [133,134,135]. Rucaparib, a PARP1 inhibitor, depolarizes the mitochondrial membrane potential, up-regulates PARP1, MIF, and AIF nucleus translocation, then attenuates parthanatos in OSCC cells. Furthermore, oxaliplatin suppresses OSCC cells by stimulating PARP1-mediated parthanatos via ascending ROS production [97].
4.1.6. Glioma
It is reported that PARP1 status is positively correlated with the degree of glioma malignancy [136]. The cell death of the glioma, which is caused by H2O2, is accompanied by PAR polymer cytoplasmic formation, PARP1 over-activation, and AIF nuclear translocation. In addition, JNK activation facilitates glioma cell parthanatos through increasing intracellular ROS [137]. Glioma cell lines and mice model of xenograft glioma are used to investigate the parthanatos mechanism of deoxypodophyllotoxin (DPT) in cancer cell death, and the result reveals that DPT stimulates parthanatos in glioma cells through induction of overburdened ROS [98].
4.1.7. Others
Meanwhile, dexmedetomidine suppresses bupivacaine-induced parthanatos in human SH-SY5Y cells via miR-7-5p/PARP1 axis-mediated ROS [99]. SH-SY5Y cell death and ropivacaine-induced apoptosis are related to parthanatos, ropivacaine induces NAD+ depletion and PARP1 activation, and the result of treatment with the PARP1 inhibitor PJ-34 indicates that NAD+ depletion is caused by PARP1 activation [100]. The hot water extract of Korean ginseng is reported to obviously inhibit MNNG-induced cell death, significantly reducing AIF expression and nucleation, and decreasing ROS levels in SH-SY5Y cells [138]. In the N-butyl-N-(4-hydroxybutyl)-nitrosamine (BBN) model, MIF accelerates bladder cancer by promoting cell proliferation and angiogenesis, so MIF inhibitors may be a helpful treatment in the field of this disease [139]. Hela cells’ death upon polybrominated diphenyl ethers quinone metabolite (PBDEQ) exposure results from increased ROS production, which manifests as PARP1 and AIF-mediated parthanatos [140].
4.2. Parthanatos and Retinal Disease
Hyper-activation of PARP enzymes is reported to participate in photoreceptor degeneration in the retinal detachment mouse model [101]. To explore the accurate association of PARP1 with photoreceptor cell death, researchers examined the phenotype of PARP1 KO retina in a mouse model. Their finding demonstrates that PARP1 is an indispensable part of normal retinal function and has essential significance for photoreceptor degeneration under pathological conditions. Furthermore, the result suggests that parthanatos is indispensable for retinal degeneration and highlights the importance of PARP1 inhibitors in treating retinitis pigmentosa (RP). The hyper-activation of PAR results in AIF release and then affects rod photoreceptor death—a certain genetic form of RP. In the RP mouse model, PAR up-regulation occurs only in the mutant retinas. RP mouse retinal explants, which are treated with a PARP inhibitor, show reduced cell stress and decreased responsiveness to markers of cell death [141]. Interestingly, researchers found proof in animals of some other retinal degeneration sustaining the role of parthanatos, but not other cell death [142]. NMNAT1-associated retinal degeneration (LCA9), a kind of retinal disease, is caused by mutations of the enzyme, which produces nuclear NAD+ [143]. Moreover, the NAD+ level is reduced, but the NAD+ precursor nicotinamide mononucleotide is augmented in the model, compared with littermate controls [144].
NAD+/ATP depletion is deemed to be a potential reason for dry age-related macular degeneration, and this premise is verified using a human-derived retinal pigment epithelium cell line (ARPE-19) that suffers from oxidative stress. PARP1 activity is increased in ARPE-19 cells after stimulation, but the expression of NAD+ and ATP is reduced, compared with the normal group. Mitochondrial dysfunction and organelle depolarization are changed by supplementing NAD+ or inhibiting PARP1 [145,146]. The photoreceptors are protected against visible-light-induced parthanatos by regulating the mTOR/PARP1 axis or the downstream factors of AIF, and mTOR interacts with PARP1 via sirtuin 1 to adjust parthanatos. On the one hand, the mTOR signaling pathway directly affects intracellular NAD+ levels, and on the other hand, it regulates NAD+ by regulating the expression and activity of SIRT1. In addition, the activity of SIRT1 is affected by the almost complete depletion of NAD+ and ATP pools caused by the over-activation of PARP1 [147]. Parthanatos occurs because of PARP1 over-activation and cellular NAD+ depletion, and the subsequent reduction in SIRT1 deacetylase activity, which is related to substrate deprivation and a high level of the reaction product nicotinamide. C2-acetylsphingosine induces photoreceptor death by parthanatos mechanisms, involving the activation of PARP1, the decline of mitochondrial membrane potential, and AIF translocation [148]. In response to oxidative stress, activated PARP1 consumes intracellular NAD+ and depletes ATP, resulting in the production of PAR polymers, the nucleus translocation of AIF, and lastly massive DNA fragmentation. PJ-34 is demonstrated to be a promising therapeutic agent that alleviates photoreceptor parthanatos death in retinal detachment by inhibiting the PARP1/AIF pathway [141].
4.3. Parthanatos and Neurological Diseases
Neurodegenerative diseases are typical of the enhancement of PARP1 expression and the accumulation of PAR. The activation of PARP1 results in the PAR-mediated AIF nucleus translocation. PAR accumulation and AIF nucleus translocation are associated with various neurological diseases. Alzheimer’s disease (AD) is featured with cognitive impairment, amyloid β (Aβ) production, and PARP1 activation. Abnormal Aβ leads to NO production on a large scale, resulting in PARP1 activation and DNA damage to sensitive subcellular structures [149]. PARP1 depletion protects the brain against Aβ-evoked microglia activation, hippocampal synaptic integrity alteration, and cognitive impairment using hAPPJ20 mice crossed with PARP1−/− mice [150]. In an in vivo model, an inhibitor of PARP1 not only protected the brain against pro-oxidative processes [151] but also safeguarded against systemic inflammatory response-induced impairment of cognitive function, and observably improved spatial memory and locomotor activity in lipopolysaccharide-treated animals [152,153].
Parkinson’s disease (PD) is featured by dopaminergic neurons (DN). The latest study about PD proves that parthanatos is responsible for selective DN loss [154]. The neurons in PD patients show a marked nucleus translocation of AIF [155]. MPTP/1-methyl-4-phenylpyridinium (MPP+) involves PARP1 and AIF nucleus translocation [79,156,157]. The pharmacological inhibition of PARP1 reduces α-synuclein cytotoxicity and MPP+-induced cell death in the PD in vitro model [127,158,159]. In the MPTP animal model of PD, AIF implicates in mediating MPP+ toxicity in dopaminergic cells, which is prevented by the knockdown of AIF [160]. MPP+ causes dopaminergic neuronal cell death and increases protein arginine methyltransferases 1 (PRMT1) expression in a dopaminergic cell line. MPP+-induced cell death is preceded by PRMT1 knockdown, the expression and activity of PARP1, and arylation which are elevated by MPP+. Moreover, the expression of PRMT1 in the substantia nigra pars compacta of MPTP-injected mice is increased, and PRMT1 positively regulates nucleus translocation of AIF. In addition, MPTP-induced DN death is decreased in PRMT1 haploinsufficient (prmt1+/−) mice [161]. In human systems, PAR-mediated parthanatos is accountable for the drop of DN in PD [162]. In a word, PAR or PAR-AIF interaction targets will be a new promising measure for preventing DN loss in PD.
Parthanatos is involved in MCAO/R-induced model animals, which used different PARP inhibitors (PJ-34, olaparib, 3-AB, DPQ, INO-1001, FR247304, DR2313, MP-24, HYDAMTIQ) or PARP1-null mice (PARP1−/−) [163,164]. The oxygen-glucose deprivation (OGD)-injured mesenchymal stem cells (MSCs) neuron model is used to reveal that co-culturing with MSCs protects the cortical neurons from OGD-induced parthanatos by alleviating AIF nucleus translocation [7]. Remote limb preconditioning (RPC) has a protective effect on neuronal oxidative DNA damage and DNA fragmentation after ischemic stroke. RPC down-regulates the PAR/AIF pathway and neuronal parthanatos by suppressing PAR accumulation, AIF translocation, and AIF/H2AX interaction [165]. Astragaloside IV (AIV), a natural saponin abundant in Astragalus membranaceus, guards mitochondrial HK-II and reduces the release of AIF, resultantly protecting neurons from parthanatos in the MCAO model mouse [166].
4.4. Parthanatos and Diabetes
PARP1 hyper-activation is associated with many various characteristics of early nephropathy related to type 1 diabetes, which provides a theoretical basis for developing and studying PARP1 inhibitors and PARP1 inhibitor-containing combination therapies [102]. PARP1−/− gene deficiency alleviates diabetic kidney disease by using the PARP1 deficient mouse to evaluate the role of PARP1 [167]. In the diabetic peripheral neuropathy (DPN) model, a hydrogen-rich medium effectively suppresses parthanatos by down-regulating the PAR level and AIF nucleus translocation [168]. Resveratrol (3,4′,5-trihydroxystilbene), an antioxidant and non-flavonoid polyphenolic compound, extracted from many natural sources, ameliorates type 1 diabetes mellitus (T1DM)-induced sperm abnormality and DNA damage [103].
4.5. Parthanatos and Renal Disease
Chemical inhibition of PARP1 activity reduces renal fibrosis [102]. PARP1 deficiency reduces cisplatin-induced kidney dysfunction, oxidative stress, pro-inflammatory gene induction, and parthanatos as well [169,170,171]. Renal I/R-induced injury or necrosis is decreased and renal function is preserved in mice, which are pretreated with PARP1 inhibitor or excised with the PARP1 gene [172,173,174,175]. PARP1 is involved in renal fibrosis by binding to the cellular communication network factor 2 promoter in mouse proximal tubule epithelial cells (formerly known as a connective tissue growth factor). Pretreatment with 3,3,5-triiodothyronine (T3) lowers the clinical and histological signs of renal I/R injury in rats and these signs are contacted with reductions in tubular PARP1 expression [104]. The PARP1 inhibitor 4-hydroxy quinazoline attenuates the disintegration of the tubulointerstitial structures in the acute kidney model [105]. PARP1 inactivation by treatment with PJ-34 contributes to the decrease in interstitial fibrosis in murine kidneys; therefore, PARP1 is different in obstructive and ischemic renal injury [106]. Using lupus and anti-glomerular basement membrane models of nephritis to determine the effect of PARP1 on renal inflammation, researchers found that the activation of PARP1 and subsequent necrotic cell death is involved in the pathogenesis of male glomerulonephritis [176]. PARP1 ablation preserves ATP levels and renal functions and attenuates inflammatory response in the setting of I/R injury in a mouse model [177].
Glyceraldehyde-3-phosphate dehydrogenase (GAPDH)-poly(ADP-ribosyl)ation suppresses glycolysis, exacerbates ATP depletion, and induces parthanatos in a model of renal I/R injury [178]. In addition to tubulotoxicity and I/R injury, acute tubule necrosis is partially prevented by different inhibitors of PARP1 in animals attacked with intravenous and intravascular lipopolysaccharide as a model of septic mediated renal failure [107,108,175].
4.6. Parthanatos and Cardiovascular Diseases
After myocardial I/R, ROS are produced and PARP1 is activated, while PARP1 deficiency improves cardiac contractility and recovers energy metabolites. PARP1 inhibitor alleviates the infarct size of rat heart and ameliorates morphology and function in myocardial I/R rats, for instance, mitigating myocardial necrosis and inflammation response [164].
Besides myocardial I/R, parthanatos promotes the progression of heart failure, PARP inhibitors effectually restore impaired myocardial function. L-2286 prevents cardiac remodeling, enhances systolic function, and delays the development of heart failure in a spontaneously hypertensive rat (SHR) model [109]. 3-aminobenzamide (3-AB) significantly protects against myocardial morphological and functional alterations in a rat model of myocardial infarction. It is worth noting that infarct size and circulating creatine kinase activity are decreased, and myocardial contractility is restored by 3-AB [110]. INO-1001 is confirmed to arrest the pressure overload-induced attenuation in cardiac contractility and reduce the collagen formation and AIF nuclear translocation in the model of transverse aortic constriction (banding)-induced heart failure [111]. Patients with chronic heart failure (CHF) and volunteers with a normal heart function are investigated for examining oxidative stress-related activation of PARP1. Markers of oxidative-nitrative stress, PARP1 activation, and AIF translocation in blood components are associated with reduced cardiac function and the clinical manifestations of CHF [179].
4.7. Parthanatos and Other Diseases
Many other diseases have a close connection with parthanatos. For instance, zVAD and Nec-1 treatment significantly augment the incidence of cleaved PARP in live shikonin-induced Jurkat cells and lessen the level of DNA damage, in which the characteristics of parthanatos appear in Jurkat cells [112]. APO866 is a small molecule drug that indirectly interferes with NAD+ biosynthesis. APO866 has anti-leukemia activity and induces a large number of ROS/RNS, which lies on PARP1 integrity. This provides a promising way to enhance the anti-tumor activity of APO866 by regulating the parthanatos pathway [113]. Another study points out that hepatocytes are indeed susceptible to parthanatos in vitro. PARP1 inhibitor restores impaired hepatocytes, which is caused by the induction of parthanatos. This provides an experimental basis for searching for potential therapeutic targets for acute and chronic liver diseases [180]. Researchers identified the involvement of parthanatos in remote lung injury due to I/R or acute immune rejection in the renal allografts [114]. Smoke-mediated activation of the parthanatos pathway increases in human bronchial epithelial cells originating from habitual smokers compared to non-smokers, and the use of a specific PARP1 inhibitor, BMN673, abolishes the effect of smoke-induced activation of the parthanatos [115]. In addition, parthanatos is related to age-related macular degeneration [145]. Furthermore, the link between parthanatos and inflammation aroused great attention. PARP1 is able to enhance the transcription of pro-inflammatory genes and down-regulate multiple simultaneous pathways of inflammation and tissue injury [14,45]. Therefore, some ultrapotent novel PARP inhibitors such as rucaparib, niraparib, and olaparib have entered human clinical trials.
5. Discussion and Perspectives
Parthanatos is regulated by PARP1 and activated by oxidative stress-induced DNA damage and chromatolysis. It should be strictly controlled at multiple indexes that include PARP1, NAD+, ATP, AIF, MIF, PAR, PARG, and ARH3. In a range of different disease models, several pharmacological agents or genetic manipulations are used to modulate parthanatic responses, often reducing morbidity and mortality. However, parthanatos is still in its infancy and there are some thorny issues in parthanatos research.
In many cases, investigators concluded that parthanatos participated in pathogenic processes due to PARP1, AIF, PARG, and ROS, resulting in the loss of cell membrane integrity and large DNA fragmentations. Although AIF, PARG, and ROS inhibitors have no toxic effects secure in preclinical animal studies, they are no clinical application till now. Related studies will help to guide better the development and application of AIF, PARG, and ROS inhibitors for different diseases. The activity of the PARP1 enzyme takes charge of PAR degradation and must be tightly controlled for avoiding either PAR accumulation or inappropriate PAR degradation. PARP1 is an integral part of the regulation of parthanatos: PARP1 activation will result in PAR polymer accumulation, AIF nucleus translocation, AIF-mediated chromatin condensation/DNA fragmentation, and parthanatos occurs. Autophagy has a hand in the occurrence of parthanatos. During autophagy, the activity of PARP1 is not affected and a large number of PARs are synthesized by PARP1, thus promoting ferroptosis [181]. Furthermore, PARP1 is involved in necroptosis, tumor necrosis factor (TNF)-α regulates necroptosis by inducing ATP depletion along with PARP1 activation. Ferroptosis and parthanatos are cell death caused by intracellular ROS overload and are associated with many diseases. Nowadays, inhibition of parthanatos is a pathway to avoid oxidative stress-induced cell death and is one of the strategies to intervene in the conditions of cardiovascular degeneration, diabetes, tumors, and so on. JNK, MAPK, and mTOR signaling pathways, which are activated by ROS, are vital to regulating cell parthanatos. Activated JNK, MAPK, and mTOR signaling pathways enhance the expression and the hyper-activation of PARP-1 via the improvement of intracellular ROS levels in cells. Hence, the regulation of these cell death modes has some similarities. However, their relationship remains an open question. Whether they can be integrated into a complete regulatory network is still to be further explored and studied. AIF nuclear translocation, a downstream event of PAR signaling, is also an attractive therapeutic target. They are critical steps of parthanatos and play pivotal roles in the marks therapy of cell death. However, what are the mechanical aspects of the AIF-releasing capacity of PAR? MIF possesses many vital advantages over PARP1 inhibition in some cases because long-term inhibition of PARP1 weakens the detection and restoration of DNA injury. Inhibiting MIF’s nuclease activity and interaction of MIF-AIF can bypass this potential problem and provide treatment opportunities for various diseases. However, how MIF interacts with AIF and what influences the interaction of MIF and AIF is still not clear.
Obviously, an in-depth study of the parthanatos pathway is of great significance to explore the pathogenesis and prevention of diseases. The research of drugs that target parthanatos will become a hot spot in the clinical medicine and pharmaceutical industry. Recently, the progress of parthanatos has been greatly achieved in cancer, retinal disease, diabetes, renal disease, heart failure, myocardial infarction, leukemia, lung injury, smoke-related lung diseases, stroke, ischemic tissue injury, brain trauma, and neurodegenerative diseases. For instance, PARP1 inhibitors (olaparib, rucaparib, niraparib, and talazoparib) are approved by the Food and Drug Administration (FDA) for clinical use in ovarian or breast cancer, and the variety of treated tumors is growing. interestingly, PARP1 agonists such as β-lapachone and deoxypodophyllotoxin are reported to trigger parthanatos in hepatoma carcinoma cells and glioma cells through indirectly activating PARP1 via induction of excessive ROS. These compounds, as well as PDD00017273 (acting on PARG) and 3-AB (acting on PARP1), show great potential and prospect for clinical application. To sum up, the discovery of parthanatos carves a new plat of disease research.
Author Contributions
Conceptualization, writing—original draft preparation, visualization, P.H.; writing—review and editing, G.C. and K.M.; supervision, W.J.; project administration, H.W.; conceptualization, writing—review and editing, funding acquisition, Y.H. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by National Natural Science Foundation of China (No. 81873226) and Zhejiang Provincial Science and Technology Innovation Leading Talent Project of “Ten Thousand Talents Plan” (2019).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Andrabi, S.A.; Dawson, T.M.; Dawson, V.L. Mitochondrial and nuclear cross talk in cell death: Parthanatos. Ann. N. Y. Acad. Sci. 2008, 1147, 233–241. [Google Scholar] [CrossRef]
- David, K.K.; Andrabi, S.A.; Dawson, T.M.; Dawson, V.L. Parthanatos, a messenger of death. Front. Biosci (Landmark Ed.) 2009, 14, 1116–1128. [Google Scholar] [CrossRef]
- Aki, T.; Funakoshi, T.; Uemura, K. Regulated necrosis and its implications in toxicology. Toxicology 2015, 333, 118–126. [Google Scholar] [CrossRef]
- Galluzzi, L.; Kepp, O.; Krautwald, S.; Kroemer, G.; Linkermann, A. Molecular mechanisms of regulated necrosis. Semin. Cell Dev. Biol. 2014, 35, 24–32. [Google Scholar] [CrossRef]
- Tang, D.; Kang, R.; Berghe, T.V.; Vandenabeele, P.; Kroemer, G. The molecular machinery of regulated cell death. Cell Res. 2019, 29, 347–364. [Google Scholar] [CrossRef]
- Liu, S.; Luo, W.; Wang, Y. Emerging role of PARP1 and PARthanatos in ischemic stroke. J. Neurochem. 2021, 160, 74–87. [Google Scholar] [CrossRef]
- Kong, D.; Zhu, J.; Liu, Q.; Jiang, Y.; Xu, L.; Luo, N.; Zhao, Z.; Zhai, Q.; Zhang, H.; Zhu, M.; et al. Mesenchymal stem cells protect neurons against hypoxic-ischemic injury via inhibiting parthanatos, necroptosis, and apoptosis, but not autophagy. Cell. Mol. Neurobiol. 2017, 37, 303–313. [Google Scholar] [CrossRef]
- Power, M.; Das, S.; Schutze, K.; Marigo, V.; Ekstrom, P.; Paquet-Durand, F. Cellular mechanisms of hereditary photoreceptor degeneration—Focus on cGMP. Prog. Retin. Eye Res. 2020, 74, 100772. [Google Scholar] [CrossRef]
- Zhou, Y.; Liu, L.; Tao, S.; Yao, Y.; Wang, Y.; Wei, Q.; Shao, A.; Deng, Y. Parthanatos and its associated components: Promising therapeutic targets for cancer. Pharmacol. Res. 2021, 163, 105299. [Google Scholar] [CrossRef]
- Harrision, D.; Gravells, P.; Thompson, R.; Bryant, H.E. Poly(ADP-Ribose) Glycohydrolase (PARG) vs. Poly(ADP-Ribose) Polymerase (PARP)—Function in Genome Maintenance and Relevance of Inhibitors for Anti-cancer Therapy. Front. Mol. Biosci. 2020, 28, 191. [Google Scholar] [CrossRef]
- Wang, H.; Yu, S.W.; Koh, D.W.; Lew, J.; Coombs, C.; Bowers, W.; Federoff, H.J.; Poirier, G.G.; Dawson, T.M.; Dawson, V.L. Apoptosis-inducing factor substitutes for caspase executioners in NMDA-triggered excitotoxic neuronal death. J. Neurosci. 2004, 24, 10963–10973. [Google Scholar] [CrossRef]
- Aredia, F.; Scovassi, A.I. Involvement of PARPs in cell death. Front. Biosci (Elite Ed.) 2014, 6, 308–317. [Google Scholar] [CrossRef]
- Virag, L.; Robaszkiewicz, A.; Rodriguez-Vargas, J.M.; Oliver, F.J. Poly(ADP-ribose) signaling in cell death. Mol. Aspects Med. 2013, 34, 1153–1167. [Google Scholar] [CrossRef]
- Kim, J.H.; Kim, J.; Roh, J.; Park, C.S.; Seoh, J.Y.; Hwang, E.S. Reactive oxygen species-induced parthanatos of immunocytes by human cytomegalovirus-associated substance. Microbiol. Immunol. 2018, 62, 229–242. [Google Scholar] [CrossRef]
- Wang, Y.; Dawson, V.L.; Dawson, T.M. Poly(ADP-ribose) signals to mitochondrial AIF: A key event in parthanatos. Exp. Neurol. 2009, 218, 193–202. [Google Scholar] [CrossRef]
- Santivasi, W.L.; Xia, F. Ionizing radiation-induced DNA damage, response, and repair. Antioxid. Redox Signal. 2014, 21, 251–259. [Google Scholar] [CrossRef]
- Fu, D.; Calvo, J.A.; Samson, L.D. Balancing repair and tolerance of DNA damage caused by alkylating agents. Nat. Rev. Cancer 2012, 12, 104–120. [Google Scholar] [CrossRef]
- Andrabi, S.A.; Umanah, G.K.; Chang, C.; Stevens, D.A.; Karuppagounder, S.S.; Gagne, J.P.; Poirier, G.G.; Dawson, V.L.; Dawson, T.M. Poly(ADP-ribose) polymerase-dependent energy depletion occurs through inhibition of glycolysis. Proc. Natl. Acad. Sci. USA 2014, 111, 10209–10214. [Google Scholar] [CrossRef]
- Bedard, L.L.; Massey, T.E. Aflatoxin B1-induced DNA damage and its repair. Cancer Lett. 2006, 241, 174–183. [Google Scholar] [CrossRef]
- Srinivas, U.S.; Tan, B.W.Q.; Vellayappan, B.A.; Jeyasekharan, A.D. ROS and the DNA damage response in cancer. Redox Biol. 2019, 25, 101084. [Google Scholar] [CrossRef]
- Salehi, F.; Behboudi, H.; Kavoosi, G.; Ardestani, S.K. Oxidative DNA damage induced by ROS-modulating agents with the ability to target DNA: A comparison of the biological characteristics of citrus pectin and apple pectin. Sci. Rep. 2018, 8, 13902. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Luo, W.; Wang, Y. PARP1 and its associated nucleases in DNA damage response. DNA Repair 2019, 81, 102651. [Google Scholar] [CrossRef] [PubMed]
- Stringari, C.; Edwards, R.A.; Pate, K.T.; Waterman, M.L.; Donovan, P.J.; Gratton, E. Metabolic trajectory of cellular differentiation in small intestine by Phasor Fluorescence Lifetime Microscopy of NADH. Sci. Rep. 2012, 2, 568. [Google Scholar] [CrossRef] [PubMed]
- Belenky, P.; Bogan, K.L.; Brenner, C. NAD+ metabolism in health and disease. Trends Biochem Sci. 2007, 32, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Katsyuba, E.; Romani, M.; Hofer, D.; Auwerx, J. NAD+ homeostasis in health and disease. Nat. Metab. 2020, 2, 9–31. [Google Scholar] [CrossRef] [PubMed]
- Berger, N.A.; Berger, S.J. Metabolic consequences of DNA damage: The role of poly (ADP-ribose) polymerase as mediator of the suicide response. Basic Life Sci. 1986, 38, 357–363. [Google Scholar] [CrossRef] [PubMed]
- Ha, H.C.; Snyder, S.H. Poly(ADP-ribose) polymerase is a mediator of necrotic cell death by ATP depletion. Proc. Natl. Acad. Sci. USA 1999, 96, 13978–13982. [Google Scholar] [CrossRef]
- Chiarugi, A. Intrinsic mechanisms of poly(ADP-ribose) neurotoxicity: Three hypotheses. Neurotoxicology 2005, 26, 847–855. [Google Scholar] [CrossRef]
- Yu, S.W.; Wang, H.; Dawson, T.M.; Dawson, V.L. Poly(ADP-ribose) polymerase-1 and apoptosis inducing factor in neurotoxicity. Neurobiol. Dis. 2003, 14, 303–317. [Google Scholar] [CrossRef]
- Hegedus, C.; Boros, G.; Fidrus, E.; Kis, G.N.; Antal, M.; Juhasz, T.; Janka, E.A.; Janko, L.; Paragh, G.; Emri, G.; et al. PARP1 Inhibition Augments UVB-Mediated Mitochondrial Changes-Implications for UV-Induced DNA Repair and Photocarcinogenesis. Cancers 2019, 12, 5. [Google Scholar] [CrossRef]
- Chambon, P.; Weill, J.D.; Mandel, P. Nicotinamide mononucleotide activation of new DNA-dependent polyadenylic acid synthesizing nuclear enzyme. Biochem. Biophys. Res. Commun. 1963, 11, 39–43. [Google Scholar] [CrossRef]
- Dawson, V.L.; Dawson, T.M. Deadly conversations: Nuclear-mitochondrial cross-talk. J. Bioenerg. Biomembr. 2004, 36, 287–294. [Google Scholar] [CrossRef] [PubMed]
- Fatokun, A.A.; Dawson, V.L.; Dawson, T.M. Parthanatos: Mitochondrial-linked mechanisms and therapeutic opportunities. Br. J. Pharmacol. 2014, 171, 2000–2016. [Google Scholar] [CrossRef] [PubMed]
- Krietsch, J.; Caron, M.C.; Gagné, J.P.; Ethier, C.; Vignard, J.; Vincent, M.; Rouleau, M.; Hendzel, M.J.; Poirier, G.G.; Masson, J.Y. PARP activation regulates the RNA-binding protein NONO in the DNA damage response to DNA double-strand breaks. Nucleic Acids Res. 2012, 40, 10287–10301. [Google Scholar] [CrossRef]
- Andrabi, S.A.; Kim, N.S.; Yu, S.W.; Wang, H.; Koh, D.W.; Sasaki, M.; Klaus, J.A.; Otsuka, T.; Zhang, Z.; Koehler, R.C.; et al. Poly(ADP-ribose) (PAR) polymer is a death signal. Proc. Natl. Acad. Sci. USA 2006, 103, 18308–18313. [Google Scholar] [CrossRef]
- Gagné, J.P.; Isabelle, M.; Lo, K.S.; Bourassa, S.; Hendzel, M.J.; Dawson, V.L.; Dawson, T.M.; Poirier, G.G. Proteome-wide identification of poly(ADP-ribose) binding proteins and poly(ADP-ribose)-associated protein complexes. Nucleic Acids Res. 2008, 36, 6959–6976. [Google Scholar] [CrossRef]
- Wang, Y.; Kim, N.S.; Haince, J.F.; Kang, H.C.; David, K.K.; Andrabi, S.A.; Poirier, G.G.; Dawson, V.L.; Dawson, T.M. Poly(ADP-ribose) (PAR) binding to apoptosis-inducing factor is critical for PAR polymerase-1-dependent cell death (parthanatos). Sci. Signal. 2011, 4, ra20. [Google Scholar] [CrossRef]
- Yu, S.W.; Andrabi, S.A.; Wang, H.; Kim, N.S.; Poirier, G.G.; Dawson, T.M.; Dawson, V.L. Apoptosis-inducing factor mediates poly(ADP-ribose) (PAR) polymer-induced cell death. Proc. Natl. Acad. Sci. USA 2006, 103, 18314–18319. [Google Scholar] [CrossRef]
- Yu, S.W.; Wang, H.; Poitras, M.F.; Coombs, C.; Bowers, W.J.; Federoff, H.J.; Poirier, G.G.; Dawson, T.M.; Dawson, V.L. Mediation of poly(ADP-ribose) polymerase-1-dependent cell death by apoptosis-inducing factor. Science 2002, 297, 259–263. [Google Scholar] [CrossRef]
- Rongvaux, A.; Galli, M.; Denanglaire, S.; Van Gool, F.; Dreze, P.L.; Szpirer, C.; Bureau, F.; Andris, F.; Leo, O. Nicotinamide phosphoribosyl transferase/pre-B cell colony-enhancing factor/visfatin is required for lymphocyte development and cellular resistance to genotoxic stress. J. Immunol. 2008, 181, 4685–4695. [Google Scholar] [CrossRef]
- Culmsee, C.; Zhu, C.; Landshamer, S.; Becattini, B.; Wagner, E.; Pellecchia, M.; Blomgren, K.; Plesnila, N. Apoptosis-inducing factor triggered by poly(ADP-ribose) polymerase and Bid mediates neuronal cell death after oxygen-glucose deprivation and focal cerebral ischemia. J. Neurosci. 2005, 25, 10262–10272. [Google Scholar] [CrossRef] [PubMed]
- Amé, J.C.; Fouquerel, E.; Gauthier, L.R.; Biard, D.; Boussin, F.D.; Dantzer, F.; de Murcia, G.; Schreiber, V. Radiation-induced mitotic catastrophe in PARG-deficient cells. J. Cell Sci. 2009, 122, 1990–2002. [Google Scholar] [CrossRef] [PubMed]
- Shirai, H.; Poetsch, A.R.; Gunji, A.; Maeda, D.; Fujimori, H.; Fujihara, H.; Yoshida, T.; Ogino, H.; Masutani, M. PARG dysfunction enhances DNA double strand break formation in S-phase after alkylation DNA damage and augments different cell death pathways. Cell Death Dis. 2013, 4, e656. [Google Scholar] [CrossRef] [PubMed]
- Palazzo, L.; Leidecker, O.; Prokhorova, E.; Dauben, H.; Matic, I.; Ahel, I. Serine is the major residue for ADP-ribosylation upon DNA damage. eLife 2018, 26, e34334. [Google Scholar] [CrossRef]
- Jagtap, P.; Szabo, C. Poly(ADP-ribose) polymerase and the therapeutic effects of its inhibitors. Nat. Rev. Drug Discov. 2005, 4, 421–440. [Google Scholar] [CrossRef]
- Hansson, M.; Asea, A.; Ersson, U.; Hermodsson, S.; Hellstrand, K. Induction of apoptosis in NK cells by monocyte-derived reactive oxygen metabolites. J. Immunol. 1996, 156, 42–47. [Google Scholar]
- Aurelius, J.; Thoren, F.B.; Akhiani, A.A.; Brune, M.; Palmqvist, L.; Hansson, M.; Hellstrand, K.; Martner, A. Monocytic AML cells inactivate antileukemic lymphocytes: Role of NADPH oxidase/gp91(phox) expression and the PARP1/PAR pathway of apoptosis. Blood 2012, 119, 5832–5837. [Google Scholar] [CrossRef]
- Aurelius, J.; Martner, A.; Brune, M.; Palmqvist, L.; Hansson, M.; Hellstrand, K.; Thoren, F.B. Remission maintenance in acute myeloid leukemia: Impact of functional histamine H2 receptors expressed by leukemic cells. Haematologica 2012, 97, 1904–1908. [Google Scholar] [CrossRef]
- Akhiani, A.A.; Werlenius, O.; Aurelius, J.; Movitz, C.; Martner, A.; Hellstrand, K.; Thoren, F.B. Role of the ERK pathway for oxidant-induced parthanatos in human lymphocytes. PLoS ONE 2014, 9, e89646. [Google Scholar] [CrossRef]
- Martinez-Morcillo, F.J.; Canton-Sandoval, J.; Martinez-Menchon, T.; Corbalan-Velez, R.; Mesa-Del-Castillo, P.; Perez-Oliva, A.B.; Garcia-Moreno, D.; Mulero, V. Non-canonical roles of NAMPT and PARP in inflammation. Dev. Comp. Immunol. 2021, 115, 103881. [Google Scholar] [CrossRef]
- Wang, J.Q.; Tang, Y.; Li, Q.S.; Xiao, M.; Li, M.; Sheng, Y.T.; Yang, Y.; Wang, Y.L. PARG regulates the proliferation and differentiation of DCs and T cells via PARP/NF-κB in tumour metastases of colon carcinoma. Oncol. Rep. 2019, 41, 2657–2666. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, N.; Walker, G.C. Mechanisms of DNA damage, repair, and mutagenesis. Environ. Mol. Mutagenes. 2017, 58, 235–263. [Google Scholar] [CrossRef] [PubMed]
- Gravells, P.; Grant, E.; Smith, K.M.; James, D.I.; Bryant, H.E. Specific killing of DNA damage-response deficient cells with inhibitors of poly(ADP-ribose) glycohydrolase. DNA Repair 2017, 52, 81–91. [Google Scholar] [CrossRef] [PubMed]
- Prokhorova, E.; Zobel, F.; Smith, R.; Zentout, S.; Gibbs-Seymour, I.; Schützenhofer, K.; Peters, A.; Groslambert, J.; Zorzini, V.; Agnew, T.; et al. Serine-linked PARP1 auto-modification controls PARP inhibitor response. Nat. Commun. 2021, 12, 4055. [Google Scholar] [CrossRef] [PubMed]
- Smith, S. The world according to PARP. Trends Biochem. Sci. 2001, 26, 174–179. [Google Scholar] [CrossRef]
- Kim, M.Y.; Zhang, T.; Kraus, W.L. Poly(ADP-ribosyl)ation by PARP1: ‘PAR-laying’ NAD+ into a nuclear signal. Genes Dev. 2005, 19, 1951–1967. [Google Scholar] [CrossRef]
- D’Amours, D.; Desnoyers, S.; D’Silva, I.; Poirier, G.G. Poly(ADP-ribosyl)ation reactions in the regulation of nuclear functions. Biochem. J. 1999, 342, 249–268. [Google Scholar] [CrossRef]
- Keuss, M.J.; Hjerpe, R.; Hsia, O.; Gourlay, R.; Burchmore, R.; Trost, M.; Kurz, T. Unanchored tri-NEDD8 inhibits PARP1 to protect from oxidative stress-induced cell death. EMBO J. 2019, 38, 100024. [Google Scholar] [CrossRef]
- Shall, S.; de Murcia, G. Poly(ADP-ribose) polymerase-1: What have we learned from the deficient mouse model? Mutat. Res. 2000, 460, 1–15. [Google Scholar] [CrossRef]
- Martínez-Morcillo, F.J.; Cantón-Sandoval, J.; Martínez-Navarro, F.J.; Cabas, I.; Martínez-Vicente, I.; Armistead, J.; Hatzold, J.; López-Muñoz, A.; Martínez-Menchón, T.; Corbalán-Vélez, R.; et al. NAMPT-derived NAD+ fuels PARP1 to promote skin inflammation through parthanatos cell death. PLoS Biol. 2021, 19, e3001455. [Google Scholar] [CrossRef]
- Thapa, K.; Khan, H.; Sharma, U.; Grewal, A.K.; Singh, T.G. Poly (ADP-ribose) polymerase-1 as a promising drug target for neurodegenerative diseases. Life Sci. 2021, 267, 118975. [Google Scholar] [CrossRef] [PubMed]
- Mashimo, M.; Moss, J. ADP-Ribosyl-Acceptor Hydrolase Activities Catalyzed by the ARH Family of Proteins. Methods Mol. Biol. 2018, 1813, 187–204. [Google Scholar] [CrossRef] [PubMed]
- Niere, M.; Mashimo, M.; Agledal, L.; Dölle, C.; Kasamatsu, A.; Kato, J.; Moss, J.; Ziegler, M. ADP-ribosylhydrolase 3 (ARH3), not poly(ADP-ribose) glycohydrolase (PARG) isoforms, is responsible for degradation of mitochondrial matrix-associated poly(ADP-ribose). J. Biol. Chem. 2012, 287, 16088–16102. [Google Scholar] [CrossRef] [PubMed]
- Munnur, D.; Ahel, I. Reversible mono-ADP-ribosylation of DNA breaks. FEBS J. 2017, 284, 4002–4016. [Google Scholar] [CrossRef]
- Fauzee, N.J.; Pan, J.; Wang, Y.L. PARP and PARG inhibitors—New therapeutic targets in cancer treatment. Pathol. Oncol. Res. 2010, 16, 469–478. [Google Scholar] [CrossRef]
- Sheline, C.T.; Wei, L. Free radical-mediated neurotoxicity may be caused by inhibition of mitochondrial dehydrogenases in vitro and in vivo. Neuroscience 2006, 140, 235–246. [Google Scholar] [CrossRef]
- Alano, C.C.; Tran, A.; Tao, R.; Ying, W.; Karliner, J.S.; Swanson, R.A. Differences among cell types in NAD(+) compartmentalization: A comparison of neurons, astrocytes, and cardiac myocytes. J. Neurosci. Res. 2007, 85, 3378–3385. [Google Scholar] [CrossRef]
- Yang, H.; Yang, T.; Baur, J.A.; Perez, E.; Matsui, T.; Carmona, J.J.; Lamming, D.W.; Souza-Pinto, N.C.; Bohr, V.A.; Rosenzweig, A.; et al. Nutrient-sensitive mitochondrial NAD+ levels dictate cell survival. Cell 2007, 130, 1095–1107. [Google Scholar] [CrossRef]
- Howard, M.; Grimaldi, J.C.; Bazan, J.F.; Lund, F.E.; Santos-Argumedo, L.; Parkhouse, R.M.; Walseth, T.F.; Lee, H.C. Formation and hydrolysis of cyclic ADP-ribose catalyzed by lymphocyte antigen CD38. Science 1993, 262, 1056–1059. [Google Scholar] [CrossRef]
- Paschen, W.; Olah, L.; Mies, G. Effect of transient focal ischemia of mouse brain on energy state and NAD levels: No evidence that NAD depletion plays a major role in secondary disturbances of energy metabolism. J. Neurochem. 2000, 75, 1675–1680. [Google Scholar] [CrossRef]
- Goto, S.; Xue, R.; Sugo, N.; Sawada, M.; Blizzard, K.K.; Poitras, M.F.; Johns, D.C.; Dawson, T.M.; Dawson, V.L.; Crain, B.J.; et al. Poly(ADP-ribose) polymerase impairs early and long-term experimental stroke recovery. Stroke 2002, 33, 1101–1106. [Google Scholar] [CrossRef] [PubMed]
- Moubarak, R.S.; Yuste, V.J.; Artus, C.; Bouharrour, A.; Greer, P.A.; Menissier-de Murcia, J.; Susin, S.A. Sequential activation of poly(ADP-ribose) polymerase 1, calpains, and Bax is essential in apoptosis-inducing factor-mediated programmed necrosis. Mol. Cell. Biol. 2007, 27, 4844–4862. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, V.; Dantzer, F.; Ame, J.C.; de Murcia, G. Poly(ADP-ribose): Novel functions for an old molecule. Nat. Rev. Mol. Cell Biol. 2006, 7, 517–528. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Jacobson, E.L.; Jacobson, M.K. Synthesis and degradation of cyclic ADP-ribose by NAD glycohydrolases. Science 1993, 261, 1330–1333. [Google Scholar] [CrossRef]
- Houtkooper, R.H.; Canto, C.; Wanders, R.J.; Auwerx, J. The secret life of NAD+: An old metabolite controlling new metabolic signaling pathways. Endocr. Rev. 2010, 31, 194–223. [Google Scholar] [CrossRef] [PubMed]
- Yoshino, J.; Baur, J.A.; Imai, S.I. NAD(+) Intermediates: The Biology and Therapeutic Potential of NMN and NR. Cell Metab. 2018, 27, 513–528. [Google Scholar] [CrossRef] [PubMed]
- Beal, M.F. Mitochondria take center stage in aging and neurodegeneration. Ann. Neurol. 2005, 58, 495–505. [Google Scholar] [CrossRef]
- Klein, J.A.; Longo-Guess, C.M.; Rossmann, M.P.; Seburn, K.L.; Hurd, R.E.; Frankel, W.N.; Bronson, R.T.; Ackerman, S.L. The harlequin mouse mutation downregulates apoptosis-inducing factor. Nature 2002, 419, 367–374. [Google Scholar] [CrossRef]
- Wang, H.; Shimoji, M.; Yu, S.W.; Dawson, T.M.; Dawson, V.L. Apoptosis inducing factor and PARP-mediated injury in the MPTP mouse model of Parkinson’s disease. Ann. N. Y. Acad. Sci. 2003, 991, 132–139. [Google Scholar] [CrossRef]
- Ye, H.; Cande, C.; Stephanou, N.C.; Jiang, S.; Gurbuxani, S.; Larochette, N.; Daugas, E.; Garrido, C.; Kroemer, G.; Wu, H. DNA binding is required for the apoptogenic action of apoptosis inducing factor. Nat. Struct. Biol. 2002, 9, 680–684. [Google Scholar] [CrossRef]
- Mate, M.J.; Ortiz-Lombardia, M.; Boitel, B.; Haouz, A.; Tello, D.; Susin, S.A.; Penninger, J.; Kroemer, G.; Alzari, P.M. The crystal structure of the mouse apoptosis-inducing factor AIF. Nat. Struct. Biol. 2002, 9, 442–446. [Google Scholar] [CrossRef] [PubMed]
- Boehler, C.; Gauthier, L.R.; Mortusewicz, O.; Biard, D.S.; Saliou, J.M.; Bresson, A.; Sanglier-Cianferani, S.; Smith, S.; Schreiber, V.; Boussin, F.; et al. Poly(ADP-ribose) polymerase 3 (PARP3), a newcomer in cellular response to DNA damage and mitotic progression. Proc. Natl. Acad. Sci. USA 2011, 108, 2783–2788. [Google Scholar] [CrossRef] [PubMed]
- Alano, C.C.; Garnier, P.; Ying, W.; Higashi, Y.; Kauppinen, T.M.; Swanson, R.A. NAD+ depletion is necessary and sufficient for poly(ADP-ribose) polymerase-1-mediated neuronal death. J. Neurosci. 2010, 30, 2967–2978. [Google Scholar] [CrossRef] [PubMed]
- Brosey, C.A.; Ho, C.; Long, W.Z.; Singh, S.; Burnett, K.; Hura, G.L.; Nix, J.C.; Bowman, G.R.; Ellenberger, T.; Tainer, J.A. Defining NADH-Driven Allostery Regulating Apoptosis-Inducing Factor. Structure 2016, 24, 2067–2079. [Google Scholar] [CrossRef]
- Litton, J.K.; Rugo, H.S.; Ettl, J.; Hurvitz, S.A.; Goncalves, A.; Lee, K.H.; Fehrenbacher, L.; Yerushalmi, R.; Mina, L.A.; Martin, M.; et al. Talazoparib in Patients with Advanced Breast Cancer and a Germline BRCA Mutation. N. Engl. J. Med. 2018, 379, 753–763. [Google Scholar] [CrossRef]
- Rugo, H.; Shtivelman, E.; Perez, A.; Vogel, C.; Franco, S.; Tan Chiu, E.; Melisko, M.; Tagliaferri, M.; Cohen, I.; Shoemaker, M.; et al. Phase I trial and antitumor effects of BZL101 for patients with advanced breast cancer. Breast Cancer Res. Treat. 2007, 105, 17–28. [Google Scholar] [CrossRef] [PubMed]
- Jhaveri, K.; Chandarlapaty, S.; Lake, D.; Gilewski, T.; Robson, M.; Goldfarb, S.; Drullinsky, P.; Sugarman, S.; Wasserheit-Leiblich, C.; Fasano, J.; et al. A phase II open-label study of ganetespib, a novel heat shock protein 90 inhibitor for patients with metastatic breast cancer. Clin. Breast Cancer 2014, 14, 154–160. [Google Scholar] [CrossRef]
- Schulz, R.; Marchenko, N.D.; Holembowski, L.; Fingerle-Rowson, G.; Pesic, M.; Zender, L.; Dobbelstein, M.; Moll, U.M. Inhibiting the HSP90 chaperone destabilizes macrophage migration inhibitory factor and thereby inhibits breast tumor progression. J. Exp. Med. 2012, 209, 275–289. [Google Scholar] [CrossRef]
- Schulz, R.; Streller, F.; Scheel, A.H.; Ruschoff, J.; Reinert, M.C.; Dobbelstein, M.; Marchenko, N.D.; Moll, U.M. HER2/ErbB2 activates HSF1 and thereby controls HSP90 clients including MIF in HER2-overexpressing breast cancer. Cell Death Dis. 2014, 5, e980. [Google Scholar] [CrossRef]
- Calabrese, C.R.; Almassy, R.; Barton, S.; Batey, M.A.; Calvert, A.H.; Canan-Koch, S.; Durkacz, B.W.; Hostomsky, Z.; Kumpf, R.A.; Kyle, S.; et al. Anticancer chemosensitization and radiosensitization by the novel poly(ADP-ribose) polymerase-1 inhibitor AG14361. J. Natl. Cancer Inst. 2004, 96, 56–67. [Google Scholar] [CrossRef]
- Aredia, F.; Giansanti, V.; Mazzini, G.; Savio, M.; Ortiz, L.M.; Jaadane, I.; Zaffaroni, N.; Forlino, A.; Torriglia, A.; Scovassi, A.I. Multiple effects of the Na(+)/H (+) antiporter inhibitor HMA on cancer cells. Apoptosis 2013, 18, 1586–1598. [Google Scholar] [CrossRef] [PubMed]
- Matulonis, U.A.; Harter, P.; Gourley, C.; Friedlander, M.; Vergote, I.; Rustin, G.; Scott, C.; Meier, W.; Shapira-Frommer, R.; Safra, T.; et al. Olaparib maintenance therapy in patients with platinum-sensitive, relapsed serous ovarian cancer and a BRCA mutation: Overall survival adjusted for postprogression poly(adenosine diphosphate ribose) polymerase inhibitor therapy. Cancer 2016, 122, 1844–1852. [Google Scholar] [CrossRef] [PubMed]
- Mirza, M.R.; Monk, B.J.; Herrstedt, J.; Oza, A.M.; Mahner, S.; Redondo, A.; Fabbro, M.; Ledermann, J.A.; Lorusso, D.; Vergote, I.; et al. Investigators E-ON, Niraparib Maintenance Therapy in Platinum-Sensitive, Recurrent Ovarian Cancer. N. Engl. J. Med. 2016, 375, 2154–2164. [Google Scholar] [CrossRef] [PubMed]
- Swisher, E.M.; Lin, K.K.; Oza, A.M.; Scott, C.L.; Giordano, H.; Sun, J.; Konecny, G.E.; Coleman, R.L.; Tinker, A.V.; O’Malley, D.M.; et al. Rucaparib in relapsed, platinum-sensitive high-grade ovarian carcinoma (ARIEL2 Part 1): An international, multicentre, open-label, phase 2 trial. Lancet Oncol. 2017, 18, 75–87. [Google Scholar] [CrossRef]
- Pillay, N.; Tighe, A.; Nelson, L.; Littler, S.; Coulson-Gilmer, C.; Bah, N.; Golder, A.; Bakker, B.; Spierings, D.C.J.; James, D.I.; et al. DNA Replication Vulnerabilities Render Ovarian Cancer Cells Sensitive to Poly(ADP-Ribose) Glycohydrolase Inhibitors. Cancer Cell 2019, 35, 519–533.e8. [Google Scholar] [CrossRef]
- James, D.I.; Smith, K.M.; Jordan, A.M.; Fairweather, E.E.; Griffiths, L.A.; Hamilton, N.S.; Hitchin, J.R.; Hutton, C.P.; Jones, S.; Kelly, P.; et al. First-in-Class Chemical Probes against Poly(ADP-ribose) Glycohydrolase (PARG) Inhibit DNA Repair with Differential Pharmacology to Olaparib. ACS Chem. Biol. 2016, 11, 3179–3190. [Google Scholar] [CrossRef]
- Li, D.; Kou, Y.; Gao, Y.; Liu, S.; Yang, P.; Hasegawa, T.; Su, R.; Guo, J.; Li, M. Oxaliplatin induces the PARP1-mediated parthanatos in oral squamous cell carcinoma by increasing production of ROS. Aging 2021, 13, 4242–4257. [Google Scholar] [CrossRef]
- Ma, D.; Lu, B.; Feng, C.; Wang, C.; Wang, Y.; Luo, T.; Feng, J.; Jia, H.; Chi, G.; Luo, Y.; et al. Deoxypodophyllotoxin triggers parthanatos in glioma cells via induction of excessive ROS. Cancer Lett. 2016, 371, 194–204. [Google Scholar] [CrossRef]
- Zheng, T.; Zheng, C.; Gao, F.; Huang, F.; Hu, B.; Zheng, X. Dexmedetomidine suppresses bupivacaine-induced parthanatos in human SH-SY5Y cells via the miR-7-5p/PARP1 axis-mediated ROS. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2021, 394, 783–796. [Google Scholar] [CrossRef]
- Zheng, T.; Zheng, C.Y.; Zheng, X.C.; Zhao, R.G.; Chen, Y.Q. Effect of parthanatos on ropivacaine-induced damage in SH-SY5Y cells. Clin. Exp. Pharmacol. Physiol. 2017, 44, 586–594. [Google Scholar] [CrossRef]
- Dong, K.; Yan, Y.; Lu, L.; Wang, Y.; Li, J.; Zhang, M.; Ding, J. PJ34 Protects Photoreceptors from Cell Death by Inhibiting PARP1 Induced Parthanatos after Experimental Retinal Detachment. Curr. Eye Res. 2021, 46, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Drel, V.R.; Xu, W.; Zhang, J.; Pavlov, I.A.; Shevalye, H.; Slusher, B.; Obrosova, I.G. Poly(Adenosine 5′-diphosphate-ribose) polymerase inhibition counteracts multiple manifestations of experimental type 1 diabetic nephropathy. Endocrinology 2009, 150, 5273–5283. [Google Scholar] [CrossRef] [PubMed]
- Abdelali, A.; Al-Bader, M.; Kilarkaje, N. Effects of Trans-Resveratrol on hyperglycemia-induced abnormal spermatogenesis, DNA damage and alterations in poly (ADP-ribose) polymerase signaling in rat testis. Toxicol. Appl. Pharmacol. 2016, 311, 61–73. [Google Scholar] [CrossRef] [PubMed]
- Ferreyra, C.; Vargas, F.; Rodriguez-Gomez, I.; Perez-Abud, R.; O’Valle, F.; Osuna, A. Preconditioning with triiodothyronine improves the clinical signs and acute tubular necrosis induced by ischemia/reperfusion in rats. PLoS ONE 2013, 8, e74960. [Google Scholar] [CrossRef]
- Kalmar-Nagy, K.; Degrell, P.; Szabo, A.; Sumegi, K.; Wittmann, I.; Gallyas, F., Jr.; Sumegi, B. PARP inhibition attenuates acute kidney allograft rejection by suppressing cell death pathways and activating PI-3K-Akt cascade. PLoS ONE 2013, 8, e81928. [Google Scholar] [CrossRef]
- Yoon, S.P.; Kim, J. Poly(ADP-ribose) polymerase 1 activation links ischemic acute kidney injury to interstitial fibrosis. J. Physiol. Sci. 2015, 65, 105–111. [Google Scholar] [CrossRef]
- Kapoor, K.; Singla, E.; Sahu, B.; Naura, A.S. PARP inhibitor, olaparib ameliorates acute lung and kidney injury upon intratracheal administration of LPS in mice. Mol. Cell. Biochem. 2015, 400, 153–162. [Google Scholar] [CrossRef]
- Liu, S.B.; Liu, J.; Liu, D.W.; Wang, X.T.; Yang, R.L. Inhibition of Poly-(ADP-Ribose) Polymerase Protects the Kidney in a Canine Model of Endotoxic Shock. Nephron 2015, 130, 281–292. [Google Scholar] [CrossRef]
- Bartha, E.; Solti, I.; Kereskai, L.; Lantos, J.; Plozer, E.; Magyar, K.; Szabados, E.; Kalai, T.; Hideg, K.; Halmosi, R.; et al. PARP inhibition delays transition of hypertensive cardiopathy to heart failure in spontaneously hypertensive rats. Cardiovasc. Res. 2009, 83, 501–510. [Google Scholar] [CrossRef]
- Liaudet, L.; Szabo, E.; Timashpolsky, L.; Virag, L.; Cziraki, A.; Szabo, C. Suppression of poly (ADP-ribose) polymerase activation by 3-aminobenzamide in a rat model of myocardial infarction: Long-term morphological and functional consequences. Br. J. Pharmacol. 2001, 133, 1424–1430. [Google Scholar] [CrossRef]
- Xiao, C.Y.; Chen, M.; Zsengeller, Z.; Li, H.; Kiss, L.; Kollai, M.; Szabo, C. Poly(ADP-Ribose) polymerase promotes cardiac remodeling, contractile failure, and translocation of apoptosis-inducing factor in a murine experimental model of aortic banding and heart failure. J. Pharmacol. Exp. Ther. 2005, 312, 891–898. [Google Scholar] [CrossRef] [PubMed]
- Warnes, G. Flow cytometric detection of hyper-polarized mitochondria in regulated and accidental cell death processes. Apoptosis 2020, 25, 548–557. [Google Scholar] [CrossRef]
- Cloux, A.J.; Aubry, D.; Heulot, M.; Widmann, C.; ElMokh, O.; Piacente, F.; Cea, M.; Nencioni, A.; Bellotti, A.; Bouzourene, K.; et al. Reactive oxygen/nitrogen species contribute substantially to the antileukemia effect of APO866, a NAD lowering agent. Oncotarget 2019, 10, 6723–6738. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zhao, H.; Ning, J.; Lemaire, A.; Koumpa, F.S.; Sun, J.J.; Fung, A.; Gu, J.; Yi, B.; Lu, K.; Ma, D. Necroptosis and parthanatos are involved in remote lung injury after receiving ischemic renal allografts in rats. Kidney Int. 2015, 87, 738–748. [Google Scholar] [CrossRef]
- Kunzi, L.; Holt, G.E. Cigarette smoke activates the parthanatos pathway of cell death in human bronchial epithelial cells. Cell Death Discov. 2019, 5, 127. [Google Scholar] [CrossRef]
- Komjáti, K.; Mabley, J.G.; Virág, L.; Southan, G.J.; Salzman, A.L.; Szabó, C. Poly(ADP-ribose) polymerase inhibition protect neurons and the white matter and regulates the translocation of apoptosis-inducing factor in stroke. Int. J. Mol. Med. 2004, 13, 373–382. [Google Scholar] [CrossRef][Green Version]
- Abdelkarim, G.E.; Gertz, K.; Harms, C.; Katchanov, J.; Dirnagl, U.; Szabó, C.; Endres, M. Protective effects of PJ34, a novel, potent inhibitor of poly(ADP-ribose) polymerase (PARP) in in vitro and in vivo models of stroke. Int. J. Mol. Med. 2001, 7, 255–260. [Google Scholar] [CrossRef]
- Ding, Y.; Zhou, Y.; Lai, Q.; Li, J.; Gordon, V.; Diaz, F.G. Long-term neuroprotective effect of inhibiting poly(ADP-ribose) polymerase in rats with middle cerebral artery occlusion using a behavioral assessment. Brain Res. 2001, 915, 210–217. [Google Scholar] [CrossRef] [PubMed]
- Lo, E.H.; Bosque-Hamilton, P.; Meng, W. Inhibition of poly(ADP-ribose) polymerase: Reduction of ischemic injury and attenuation of N-methyl-D-aspartate-induced neurotransmitter dysregulation. Stroke 1998, 29, 830–836. [Google Scholar] [CrossRef]
- Hamby, A.M.; Suh, S.W.; Kauppinen, T.M.; Swanson, R.A. Use of a poly(ADP-ribose) polymerase inhibitor to suppress inflammation and neuronal death after cerebral ischemia-reperfusion. Stroke 2007, 38, 632–636. [Google Scholar] [CrossRef]
- Takahashi, K.; Greenberg, J.H.; Jackson, P.; Maclin, K.; Zhang, J. Neuroprotective effects of inhibiting poly(ADP-ribose) synthetase on focal cerebral ischemia in rats. J. Cereb. Blood Flow Metab. 1997, 17, 1137–1142. [Google Scholar] [CrossRef] [PubMed]
- Tokime, T.; Nozaki, K.; Sugino, T.; Kikuchi, H.; Hashimoto, N.; Ueda, K. Enhanced poly(ADP-ribosyl)ation after focal ischemia in rat brain. J. Cereb. Blood Flow Metab. 1998, 18, 991–997. [Google Scholar] [CrossRef] [PubMed]
- Yap, E.; Tan, W.L.; Ng, I.; Ng, Y.K. Combinatorial-approached neuroprotection using pan-caspase inhibitor and poly (ADP-ribose) polymerase (PARP) inhibitor following experimental stroke in rats; is there additional benefit? Brain Res. 2008, 21, 130–138. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Park, S.Y.; Shin, H.K.; Kim, C.D.; Lee, W.S.; Hong, K.W. Poly(ADP-ribose) polymerase inhibition by cilostazol is implicated in the neuroprotective effect against focal cerebral ischemic infarct in rat. Brain Res. 2007, 4, 182–190. [Google Scholar] [CrossRef]
- Couturier, J.Y.; Ding-Zhou, L.; Croci, N.; Plotkine, M.; Margaill, I. 3-Aminobenzamide reduces brain infarction and neutrophil infiltration after transient focal cerebral ischemia in mice. Exp. Neurol. 2003, 184, 973–980. [Google Scholar] [CrossRef]
- Strosznajder, R.P.; Gadamski, R.; Czapski, G.A.; Jesko, H.; Strosznajder, J.B. Poly(ADP-ribose) polymerase during reperfusion after transient forebrain ischemia: Its role in brain edema and cell death. J. Mol. Neurosci. 2003, 20, 61–72. [Google Scholar] [CrossRef]
- Yokoyama, H.; Kuroiwa, H.; Tsukada, T.; Uchida, H.; Kato, H.; Araki, T. Poly(ADP-ribose)polymerase inhibitor can attenuate the neuronal death after 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced neurotoxicity in mice. J. Neurosci. Res. 2010, 88, 1522–1536. [Google Scholar] [CrossRef]
- Mao, J.; Price, D.D.; Zhu, J.; Lu, J.; Mayer, D.J. The inhibition of nitric oxide-activated poly(ADP-ribose) synthetase attenuates transsynaptic alteration of spinal cord dorsal horn neurons and neuropathic pain in the rat. Pain 1997, 72, 355–366. [Google Scholar] [CrossRef]
- Donizy, P.; Halon, A.; Surowiak, P.; Pietrzyk, G.; Kozyra, C.; Matkowski, R. Correlation between PARP1 immunoreactivity and cytomorphological features of parthanatos, a specific cellular death in breast cancer cells. Eur. J. Histochem. 2013, 57, e35. [Google Scholar] [CrossRef][Green Version]
- Dinhof, C.; Pirker, C.; Kroiss, P.; Kirchhofer, D.; Gabler, L.; Gojo, J.; Lotsch-Gojo, D.; Stojanovic, M.; Timelthaler, G.; Ferk, F.; et al. p53 Loss Mediates Hypersensitivity to ETS Transcription Factor Inhibition Based on PARylation-Mediated Cell Death Induction. Cancers 2020, 12, 3205. [Google Scholar] [CrossRef]
- Chen, S.H.; Yu, X. Targeting dePARylation selectively suppresses DNA repair-defective and PARP inhibitor-resistant malignancies. Sci. Adv. 2019, 5, eaav4340. [Google Scholar] [CrossRef]
- Zhao, N.; Mao, Y.; Han, G.; Ju, Q.; Zhou, L.; Liu, F.; Xu, Y.; Zhao, X. YM155, a survivin suppressant, triggers PARP-dependent cell death (parthanatos) and inhibits esophageal squamous-cell carcinoma xenografts in mice. Oncotarget 2015, 6, 18445–18459. [Google Scholar] [CrossRef] [PubMed]
- Chen, W. Cancer statistics: Updated cancer burden in China. Chin. J. Cancer Res. 2015, 27, 1. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef] [PubMed]
- Chi, A.C.; Day, T.A.; Neville, B.W. Oral cavity and oropharyngeal squamous cell carcinoma—An update. CA Cancer J. Clin. 2015, 65, 401–421. [Google Scholar] [CrossRef]
- Wang, X.; Ge, P. Parthanatos in the pathogenesis of nervous system diseases. Neuroscience 2020, 449, 241–250. [Google Scholar] [CrossRef]
- Zheng, L.; Wang, C.; Luo, T.; Lu, B.; Ma, H.; Zhou, Z.; Zhu, D.; Chi, G.; Ge, P.; Luo, Y. JNK Activation Contributes to Oxidative Stress-Induced Parthanatos in Glioma Cells via Increase of Intracellular ROS Production. Mol. Neurobiol. 2017, 54, 3492–3505. [Google Scholar] [CrossRef]
- Guo, Y.; Zhang, D.; Long, R.; Shan, S.; Sun, Q.; Cai, B.; Wang, S. Effect of hot water extract of Korean ginseng on neuroblastoma cell parthanatos. Nan Fang Yi Ke Da Xue Xue Bao 2020, 40, 1313–1318. [Google Scholar] [CrossRef]
- Choudhary, S.; Hegde, P.; Pruitt, J.R.; Sielecki, T.M.; Choudhary, D.; Scarpato, K.; Degraff, D.J.; Pilbeam, C.C.; Taylor, J.A., 3rd. Macrophage migratory inhibitory factor promotes bladder cancer progression via increasing proliferation and angiogenesis. Carcinogenesis 2013, 34, 2891–2899. [Google Scholar] [CrossRef]
- Dong, W.; Yang, B.; Wang, Y.; Yuan, J.; Fan, Y.; Song, E.; Song, Y. Polybrominated Diphenyl Ethers Quinone Induced Parthanatos-like Cell Death through a Reactive Oxygen Species-Associated Poly(ADP-ribose) Polymerase 1 Signaling. Chem. Res. Toxicol. 2018, 31, 1164–1171. [Google Scholar] [CrossRef]
- Paquet-Durand, F.; Silva, J.; Talukdar, T.; Johnson, L.E.; Azadi, S.; van Veen, T.; Ueffing, M.; Hauck, S.M.; Ekstrom, P.A. Excessive activation of poly(ADP-ribose) polymerase contributes to inherited photoreceptor degeneration in the retinal degeneration 1 mouse. J. Neurosci. 2007, 27, 10311–10319. [Google Scholar] [CrossRef] [PubMed]
- Arango-Gonzalez, B.; Trifunovic, D.; Sahaboglu, A.; Kranz, K.; Michalakis, S.; Farinelli, P.; Koch, S.; Koch, F.; Cottet, S.; Janssen-Bienhold, U.; et al. Identification of a common non-apoptotic cell death mechanism in hereditary retinal degeneration. PLoS ONE 2014, 9, e112142. [Google Scholar] [CrossRef] [PubMed]
- Falk, M.J.; Zhang, Q.; Nakamaru-Ogiso, E.; Kannabiran, C.; Fonseca-Kelly, Z.; Chakarova, C.; Audo, I.; Mackay, D.S.; Zeitz, C.; Borman, A.D.; et al. NMNAT1 mutations cause Leber congenital amaurosis. Nat. Genet. 2012, 44, 1040–1045. [Google Scholar] [CrossRef] [PubMed]
- Greenwald, S.H.; Charette, J.R.; Staniszewska, M.; Shi, L.Y.; Brown, S.D.M.; Stone, L.; Liu, Q.; Hicks, W.L.; Collin, G.B.; Bowl, M.R.; et al. Mouse Models of NMNAT1-Leber Congenital Amaurosis (LCA9) Recapitulate Key Features of the Human Disease. Am. J. Pathol. 2016, 186, 1925–1938. [Google Scholar] [CrossRef] [PubMed]
- Jang, K.H.; Do, Y.J.; Son, D.; Son, E.; Choi, J.S.; Kim, E. AIF-independent parthanatos in the pathogenesis of dry age-related macular degeneration. Cell Death Dis. 2017, 8, e2526. [Google Scholar] [CrossRef]
- Greenwald, S.H.; Pierce, E.A. Parthanatos as a Cell Death Pathway Underlying Retinal Disease. Adv. Exp. Med. Biol. 2019, 1185, 323–327. [Google Scholar] [CrossRef]
- Pan, Y.R.; Song, J.Y.; Fan, B.; Wang, Y.; Che, L.; Zhang, S.M.; Chang, Y.X.; He, C.; Li, G.Y. mTOR may interact with PARP1 to regulate visible light-induced parthanatos in photoreceptors. Cell Commun. Signal. 2020, 18, 27. [Google Scholar] [CrossRef]
- Prado Spalm, F.H.; Vera, M.S.; Dibo, M.J.; Simon, M.V.; Politi, L.E.; Rotstein, N.P. Ceramide Induces the Death of Retina Photoreceptors Through Activation of Parthanatos. Mol. Neurobiol. 2019, 56, 4760–4777. [Google Scholar] [CrossRef]
- Strosznajder, J.B.; Jesko, H.; Strosznajder, R.P. Effect of amyloid beta peptide on poly(ADP-ribose) polymerase activity in adult and aged rat hippocampus. Acta Biochim. Pol. 2000, 47, 847–854. [Google Scholar] [CrossRef]
- Kauppinen, T.M.; Suh, S.W.; Higashi, Y.; Berman, A.E.; Escartin, C.; Won, S.J.; Wang, C.; Cho, S.H.; Gan, L.; Swanson, R.A. Poly(ADP-ribose)polymerase-1 modulates microglial responses to amyloid beta. J. Neuroinflamm. 2011, 8, 152. [Google Scholar] [CrossRef]
- Czapski, G.A.; Cakala, M.; Gajkowska, B.; Strosznajder, J.B. Poly(ADP-ribose) polymerase-1 inhibition protects the brain against systemic inflammation. Neurochem. Int. 2006, 49, 751–755. [Google Scholar] [CrossRef] [PubMed]
- Jacewicz, M.; Czapski, G.A.; Katkowska, I.; Strosznajder, R.P. Systemic administration of lipopolysaccharide impairs glutathione redox state and object recognition in male mice. The effect of PARP1 inhibitor. Folia Neuropathol. 2009, 47, 321–328. [Google Scholar]
- Strosznajder, J.B.; Czapski, G.A.; Adamczyk, A.; Strosznajder, R.P. Poly(ADP-ribose) polymerase-1 in amyloid beta toxicity and Alzheimer’s disease. Mol. Neurobiol. 2012, 46, 78–84. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Karuppagounder, S.S.; Shin, J.H.; Lee, Y.I.; Ko, H.S.; Swing, D.; Jiang, H.; Kang, S.U.; Lee, B.D.; Kang, H.C.; et al. Parthanatos mediates AIMP2-activated age-dependent dopaminergic neuronal loss. Nat. Neurosci. 2013, 16, 1392–1400. [Google Scholar] [CrossRef]
- Burguillos, M.A.; Hajji, N.; Englund, E.; Persson, A.; Cenci, A.M.; Machado, A.; Cano, J.; Joseph, B.; Venero, J.L. Apoptosis-inducing factor mediates dopaminergic cell death in response to LPS-induced inflammatory stimulus: Evidence in Parkinson’s disease patients. Neurobiol. Dis. 2011, 41, 177–188. [Google Scholar] [CrossRef] [PubMed]
- Mandir, A.S.; Przedborski, S.; Jackson-Lewis, V.; Wang, Z.Q.; Simbulan-Rosenthal, C.M.; Smulson, M.E.; Hoffman, B.E.; Guastella, D.B.; Dawson, V.L.; Dawson, T.M. Poly(ADP-ribose) polymerase activation mediates 1-methyl-4-phenyl-1, 2,3,6-tetrahydropyridine (MPTP)-induced parkinsonism. Proc. Natl. Acad. Sci. USA 1999, 96, 5774–5779. [Google Scholar] [CrossRef] [PubMed]
- Liou, A.K.; Zhou, Z.; Pei, W.; Lim, T.M.; Yin, X.M.; Chen, J. BimEL up-regulation potentiates AIF translocation and cell death in response to MPTP. FASEB J. 2005, 19, 1350–1352. [Google Scholar] [CrossRef]
- Cosi, C.; Colpaert, F.; Koek, W.; Degryse, A.; Marien, M. Poly(ADP-ribose) polymerase inhibitors protect against MPTP-induced depletions of striatal dopamine and cortical noradrenaline in C57B1/6 mice. Brain Res. 1996, 729, 264–269. [Google Scholar] [CrossRef]
- Venderova, K.; Park, D.S. Programmed cell death in Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2012, 2, 009365. [Google Scholar] [CrossRef]
- Chu, C.T.; Zhu, J.H.; Cao, G.; Signore, A.; Wang, S.; Chen, J. Apoptosis inducing factor mediates caspase-independent 1-methyl-4-phenylpyridinium toxicity in dopaminergic cells. J. Neurochem. 2005, 94, 1685–1695. [Google Scholar] [CrossRef]
- Calabresi, P.; Di Filippo, M.; Gallina, A.; Wang, Y.; Stankowski, J.N.; Picconi, B.; Dawson, V.L.; Dawson, T.M. New synaptic and molecular targets for neuroprotection in Parkinson’s disease. Mov. Disord. 2013, 28, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Kang, H.C.; Lee, B.D.; Lee, Y.I.; Kim, Y.P.; Shin, J.H. Poly (ADP-ribose) in the pathogenesis of Parkinson’s disease. BMB Rep. 2014, 47, 424–432. [Google Scholar] [CrossRef] [PubMed]
- Koehler, R.C.; Dawson, V.L.; Dawson, T.M. Targeting Parthanatos in Ischemic Stroke. Front. Neurol. 2021, 12, 662034. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.Q.; Tian, J.; Luo, X.J.; Peng, J. Targeting the pathways of regulated necrosis: A potential strategy for alleviation of cardio-cerebrovascular injury. Cell. Mol. Life Sci. 2021, 78, 63–78. [Google Scholar] [CrossRef] [PubMed]
- Jin, W.; Xu, W.; Chen, J.; Zhang, X.; Shi, L.; Ren, C. Remote limb preconditioning protects against ischemia-induced neuronal death through ameliorating neuronal oxidative DNA damage and parthanatos. J. Neurol. Sci. 2016, 366, 8–17. [Google Scholar] [CrossRef]
- Li, Y.; Yang, Y.; Zhao, Y.; Zhang, J.; Liu, B.; Jiao, S.; Zhang, X. Astragaloside IV reduces neuronal apoptosis and parthanatos in ischemic injury by preserving mitochondrial hexokinase-II. Free Radic. Biol. Med. 2019, 131, 251–263. [Google Scholar] [CrossRef]
- Shevalye, H.; Maksimchyk, Y.; Watcho, P.; Obrosova, I.G. Poly(ADP-ribose) polymerase-1 (PARP1) gene deficiency alleviates diabetic kidney disease. Biochim. Biophys. Acta 2010, 1802, 1020–1027. [Google Scholar] [CrossRef]
- Li, Q.; Jiao, Y.; Yu, Y.; Wang, G.; Yu, Y. Hydrogenrich medium alleviates high glucoseinduced oxidative stress and parthanatos in rat Schwann cells in vitro. Mol. Med. Rep. 2019, 19, 338–344. [Google Scholar] [CrossRef]
- Mukhopadhyay, P.; Horvath, B.; Kechrid, M.; Tanchian, G.; Rajesh, M.; Naura, A.S.; Boulares, A.H.; Pacher, P. Poly(ADP-ribose) polymerase-1 is a key mediator of cisplatin-induced kidney inflammation and injury. Free Radic. Biol. Med. 2011, 51, 1774–1788. [Google Scholar] [CrossRef]
- Kim, J.; Long, K.E.; Tang, K.; Padanilam, B.J. Poly(ADP-ribose) polymerase 1 activation is required for cisplatin nephrotoxicity. Kidney Int. 2012, 82, 193–203. [Google Scholar] [CrossRef]
- Garg, J.P.; Vucic, D. Targeting Cell Death Pathways for Therapeutic Intervention in Kidney Diseases. Semin. Nephrol. 2016, 36, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, P.K.; Chatterjee, B.E.; Pedersen, H.; Sivarajah, A.; McDonald, M.C.; Mota-Filipe, H.; Brown, P.A.; Stewart, K.N.; Cuzzocrea, S.; Threadgill, M.D.; et al. 5-Aminoisoquinolinone reduces renal injury and dysfunction caused by experimental ischemia/reperfusion. Kidney Int. 2004, 65, 499–509. [Google Scholar] [CrossRef] [PubMed]
- Oztas, E.; Guven, A.; Turk, E.; Uysal, B.; Akgul, E.O.; Cayci, T.; Ersoz, N.; Korkmaz, A. 3-aminobenzamide, a poly ADP ribose polymerase inhibitor, attenuates renal ischemia/reperfusion injury. Ren. Fail. 2009, 31, 393–399. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Del Moral, R.M.; Gomez-Morales, M.; Hernandez-Cortes, P.; Aguilar, D.; Caballero, T.; Aneiros-Fernandez, J.; Caba-Molina, M.; Rodriguez-Martinez, M.D.; Peralta, A.; Galindo-Moreno, P.; et al. PARP inhibition attenuates histopathological lesion in ischemia/reperfusion renal mouse model after cold prolonged ischemia. Sci. World J. 2013, 2013, 486574. [Google Scholar] [CrossRef]
- Kers, J.; Leemans, J.C.; Linkermann, A. An Overview of Pathways of Regulated Necrosis in Acute Kidney Injury. Semin. Nephrol. 2016, 36, 139–152. [Google Scholar] [CrossRef]
- Jog, N.R.; Dinnall, J.A.; Gallucci, S.; Madaio, M.P.; Caricchio, R. Poly(ADP-ribose) polymerase-1 regulates the progression of autoimmune nephritis in males by inducing necrotic cell death and modulating inflammation. J. Immunol. 2009, 182, 7297–7306. [Google Scholar] [CrossRef]
- Zheng, J.; Devalaraja-Narashimha, K.; Singaravelu, K.; Padanilam, B.J. Poly(ADP-ribose) polymerase-1 gene ablation protects mice from ischemic renal injury. Am. J. Physiol. Renal. Physiol. 2005, 288, F387–F398. [Google Scholar] [CrossRef]
- Devalaraja-Narashimha, K.; Padanilam, B.J. PARP1 inhibits glycolysis in ischemic kidneys. J. Am. Soc. Nephrol. 2009, 20, 95–103. [Google Scholar] [CrossRef]
- Barany, T.; Simon, A.; Szabo, G.; Benko, R.; Mezei, Z.; Molnar, L.; Becker, D.; Merkely, B.; Zima, E.; Horvath, E.M. Oxidative Stress-Related Parthanatos of Circulating Mononuclear Leukocytes in Heart Failure. Oxid. Med. Cell. Longev. 2017, 2017, 1249614. [Google Scholar] [CrossRef]
- Aizawa, S.; Brar, G.; Tsukamoto, H. Cell Death and Liver Disease. Gut Liver 2020, 14, 20–29. [Google Scholar] [CrossRef]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).