
Impaired Bestrophin Channel Activity in an iPSC-RPE Model of Best Vitelliform Macular Dystrophy (BVMD) from an Early Onset Patient Carrying the P77S Dominant Mutation


Abstract
:1. Introduction
2. Results
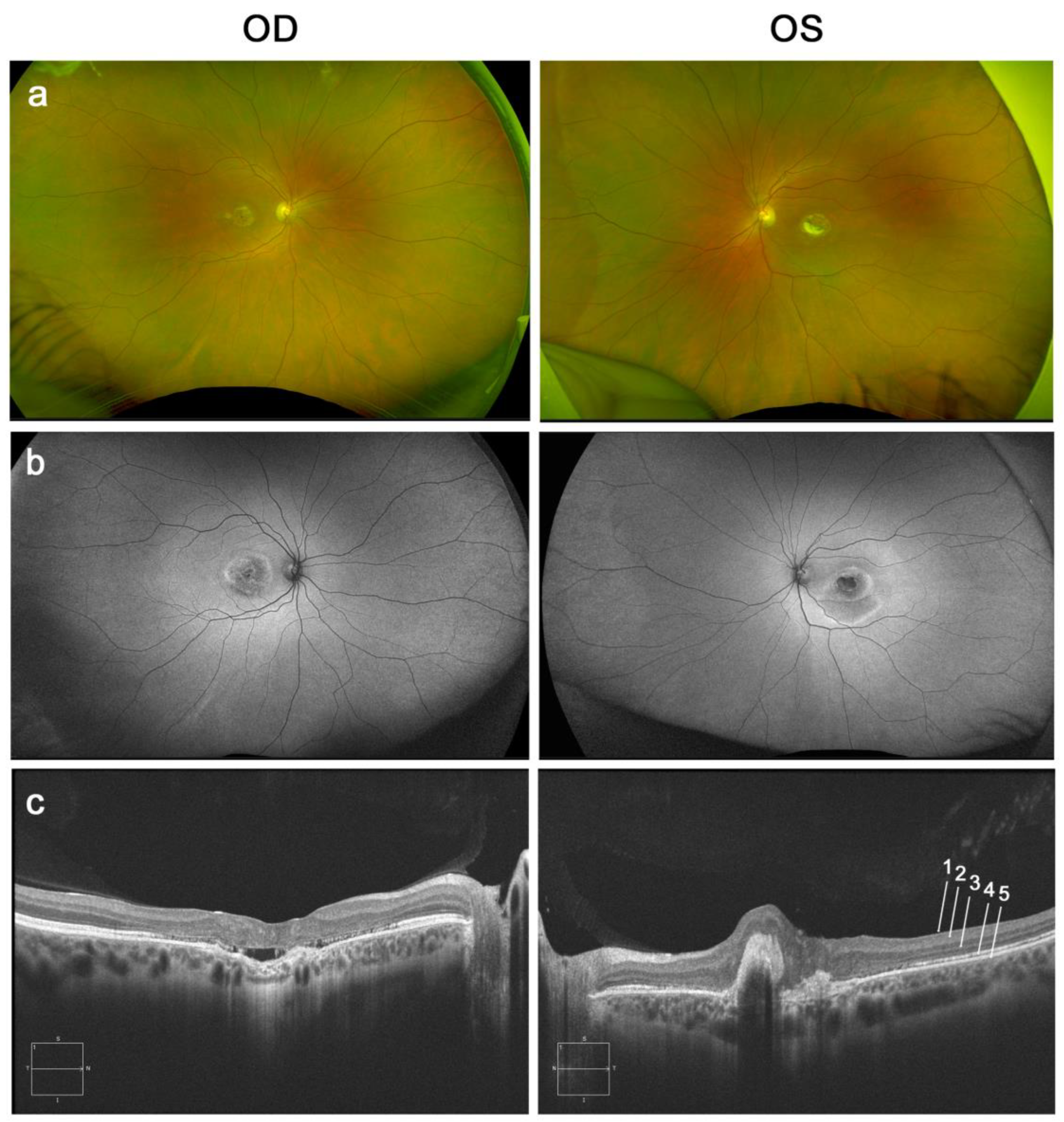
2.1. Patient’s Case Description
2.2. Fi21/01 iPS-RPE Cells Don’t Show Differential Levels of BEST1 mRNA Expression or Bestrophin Expression
2.3. Bestrophin Is Localized Correctly in the Membrane of Mutated iPSC-RPE Cells
2.4. Fi21/01 iPSC-RPE Cells Do Not Show a Significant Apoptotic Profile
2.5. iPSC-RPE Cells with the p.Pro77Ser Mutation Show Increased Halide Entrance
2.6. No Differences in the Phagocytosis Capacity of Fi21/01cells Compared to Control
3. Discussion

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| AminoacidicChange | MODEL | Localization/Expression | Channel Activity | Others | DISEASE | Domain |
|---|---|---|---|---|---|---|
| T6P | pRPE/hiPSC-RPE | Intracellular [65] | BVMD [1] | C-Terminal | ||
| MDCKII | Intracellular [28] | |||||
| MDCKII/HEK293 | Intracellular [27] | ↓* [27] | ||||
| V9M | MDCKII/fhRPE | Intracellular [66] | BVDM [67] | |||
| A10T | HEK293/hiPSC-RPE | ↓ protein [32] | ↓ [32] | BVMD [67] | ||
| HEK293/hiPSC-RPE | ↓ [30] | |||||
| N11K | hiPSC-RPE | ↓ [24] | lower lysosomal pH (POS, 2w) [24] | BVDM [24] | ||
| R19C | HEK293 | ↓ [63] | BVDM [68] | |||
| L21V | MDCKII/HEK293 | Intracellular [27] | ↓* [27] | BVMD [69] | ||
| W24C | MDCKII/HEK293 | Intracellular [27] | ↓* [27] | BVMD [67] | ||
| R25C | HEK293 | ↓ [63] | ND | |||
| K30C | HEK293 | ↓ [63] | ND | TM1 | ||
| L40P | hiPSC-RPE | Decreased fluid flow [70] | ARB [71] | |||
| L41P | MDCKII/HEK293 | Misfolding [72] | ↓ [73] | ARB [73,74] | ||
| MDCKII | ↓ protein/mislocalization [25] | ↓ [25] | ||||
| P77S | hiPSC-RPE | ↑* Ω | BVMD [33] | TM2 | ||
| S79C | MDCKII/HEK293 | ↓* [27] | ND | |||
| F80L | pRPE/hiPSC-RPE | Intracellular [66] | Reduced Ca2+ channel function [66] | BVMD [72] | ||
| MDCKII/HEK293 | ↓* [27] | |||||
| L82V | MDCKII/HEK293 | ↓* [27] | BVMD [75] | |||
| Y85H | MDCKII/HEK293 | ↓ [73] | BVMD [1] | |||
| MDCKII | Apical [29] | |||||
| HEK293 | ↓* [20] | |||||
| V86M | hiPSC-RPE | ↑ [24] | ADVIRC [76] | |||
| R92C | HEK293 | ↓* [20] | BVMD [75] | |||
| R92S | MDCKII/HEK293 | Intracellular [27] | ↓* [27] | BVMD [77] | ||
| W93C | HEK293 | ↓ [78,79] | BVDM [1] | Loop 2 | ||
| HEK293 | ↓* [20] | |||||
| Q96R | MDCKII | Apical [29] | BVMD [80] | |||
| L100R | MDCKII | Apical [29] | BVMD [75] | |||
| L140V | HEK293 | Intracellular [61] | ↓ [61] | RP [61] | ||
| R141H | MDCKII/HEK293/hiPSC-RPE | ↓ [81] | ARB [78,82] | |||
| HEK293 | ↓ [78] | |||||
| MDCKII/HEK293 | Intracellular [72] | |||||
| MDCKII | Intracellular [28] | |||||
| hiPSC-RPE | ↓ protein [24] | ↓ [24] | ||||
| MDCKII | ↓ protein/mislocalization [25] | ↓ [25] | ||||
| R141S | MDCKII | Intracellular [28] | ARB [83] | |||
| S142G | fhRPE | Apoptosis [42] | BVMD [84] | |||
| V143F | fhRPE | Apoptosis [42] | BVMD [85] | |||
| A146K | hiPSC-RPE | Decreased fluid flow, Impaired phagocytosis (POS, 3, 5 months) [86] | BVDM [86] | |||
| hiPSC-RPE | ↓ [26] | |||||
| A146T | fhRPE | Apoptosis [42] | ARB [84] | |||
| P152A | HEK293 | ↓ [78] | ARB [78] | |||
| MDCKII/HEK293 | Intracellular [72] | |||||
| MDCKII | Intracellular [28] | |||||
| L174Qfs*57 | MDCKII | Intracellular [28] | ARB [87] | |||
| L191P | MDCKII | Intracellular [28] | ARB [88] | |||
| A195V | MDCKII/HEK293 | Misfolding [72] | ↓ [73] | ARB [72] | ||
| hiPSC-RPE | ↓ protein [26] | ↓ [26] | ||||
| R200X | MDCKII | Intracellular [28] | ARB [78] | |||
| I201T | hiPSC-RPE | ↓ [89] | BVMD [72] | |||
| R202W | MDCKII/HEK293 | Intracellular [72] | ↓ [73] | ARB [73] | ||
| MDCKII | ↓ protein/mislocalization [25] | ↓ [25] | ||||
| D203A | HEK293 | ↑ [31] | BVMD [31] | |||
| I205T | HEK293 | ↑ [31] | RP [61] | |||
| HEK293 | ↓ [61] | |||||
| HEK293/hiPSC-RPE | ↑ [30] | |||||
| E213G | MDCKII | Intracellular [28] | ARB [28] | |||
| R218C | HEK293 | ↓ [78] | BVDM [67] | |||
| MDCKII/HEK293 | ↓* [27] | |||||
| hiPSC-RPE | ↓ [24] | lower lysosomal pH (POS, 2w) [24] | ||||
| hiPSC-RPE | ↓ [26] | |||||
| R218H | HEK293/hiPSC-RPE | ↓ [32] | BVMD [72] | |||
| hiPSC-RPE | ↓ [44] | |||||
| HEK293/hiPSC-RPE | ↓ [30] | |||||
| L224M | MDCKII/HEK293 | Intracellular [27] | ↓* [27] | BVMD [77] | ||
| Y227E | MDCKII | Apical [29] | BVMD [29] | |||
| MDCKII | Apical [29] | |||||
| MDCKII/HEK293 | Intracellular [27] | ↓* [27] | ||||
| D228N | HEK293 | Intracellular [62] | BVMD [61] | |||
| W229E | HEK293 | ↓ [63] | BVMD [63] | |||
| I230A | HEK293/hiPSC-RPE | ↑ [30] | BVMD [30] | TM3 | ||
| P233A | HEK293 | ↓ [31] | ARB [90] | |||
| L234P | HEK293/hiPSC-RPE | ↓ [32] | BVMD [44] | |||
| hiPSC-RPE | ↓ [44] | |||||
| HEK293/hiPSC-RPE | ↓ [30] | |||||
| L234V | MDCKII | ↓ [91] | BVMD [91] | |||
| V235A | hiPSC-RPE | Apical [92] | ADVIRC [93] | |||
| Y236A | HEK293 | ↓ [31] | ADVIRC [55] | |||
| Y236C | HEK293/hiPSC-RPE | ↑ [30] | ND | |||
| HEK293 | ↑ [31] | |||||
| T237R | MDCKII/HEK293 | ↓ [73] | BVMD [69] | |||
| MDCKII/HEK293 | Intracellular [27] | ↓* [27] | ||||
| Q238R | hiPSC-RPE | Intracellular [24] | ↓ [24] | lower lysosomal pH (POS, 2w) [24] | BVDM [24] | |
| A243T | HEK293/hiPSC-RPE | ↓ [30,32] | BVMD [72] | |||
| MDCKII/HEK293 | ↓* [27] | |||||
| hiPSC-RPE | ↓ [44] | |||||
| A243V | hiPSC-RPE | ↓ [24] | lower lysosomal pH (POS, 2w) [24] | BVMD [69] | ||
| HEK293 | ↓ [94] | |||||
| R255Q | hiPSC-RPE | ↓ [91] | ARB [91] | Loop 3 | ||
| P274R | hiPSC-RPE | Intracellular [91] | ↓ [89] | ARB [85] | TM4 | |
| HEK293/hiPSC-RPE | ↓ [30] | |||||
| W287A | HEK293 | ↓ [31] | ND | |||
| Q293K | HEK293/hiPSC-RPE | ↓ protein [32] | ↓ [32] | BVMD [75] | ||
| HEK293/hiPSC-RPE | ↓ [30] | |||||
| I295del | hiPSC-RPE | Intracellular [24] | ↓ [24] | lower lysosomal pH (POS, 2w) [24] | BVDM [82] | C-terminal (Ca2+ binding) |
| N296H | hiPSC-RPE | ↓ [26] | BVMD [72] | |||
| hiPSC-RPE | Decreased fluid flow, Impaired phagocytosis (POS, 3, 5 months) [88] | BVDM [72] | ||||
| N296S | MDCKII | ↓ [91] | BVMD [72] | |||
| D301N | HEK293 | ↓ [63] | BVMD [82] | |||
| D302A | HEK293/hiPSC-RPE | ↓ [30,32] | BVMD [95] | |||
| hiPSC-RPE | Intracellular [66] | |||||
| F305S | pRPE/hiPSC-RPE | Intracellular [66] | Reduced Ca2+ channel function [66] | BVMD [67] | ||
| MDCKII/HEK293 | Intracellular [27] | ↓* [27] | ||||
| V311G | MDCKII/HEK293 | Intracellular [27] | ↓* [27] | BVMD [69] | ||
| D312N | MDCKII/HEK293 | Intracellular [72] | ↓ [73] | ARB [73,77] | C-terminal | |
| MDCKII | Intracellular [28] | |||||
| HEK293 | ↓ [62] | |||||
| V317M | MDCKII/HEK293 | Intracellular [72] | ↓ [73] | ARB [78] | ||
| MDCKII | Intracellular [28] | |||||
| M325T | MDCKII/HEK293 | Intracellular [72] | ↓ [73] | ARB [78] | ||
| MDCKII | Intracellular [28] | |||||
| MDCKII | ↓ protein/mislocalization [25] | ↓ [25] | ||||
| I366fs*18 | MDCKII/HEK293/hiPSC-RPE | ↑ [81] | ARB [81] | |||
| L40P + A195V | hiPSC-RPE | Decreased fluid flow [73] | ARB [71,72] | |||
| N99K + R141H | hiPSC-RPE | ↓ protein [24] | ↓ [24] | ARB [75,82] | ||
| A195V + L197Pfs*26 | hiPSC-RPE | ↓ mRNA/↓ protein [24] | ↓ [24] | ARB [72,96] | ||
| R141H + I366fs*18 | hiPSC-RPE | ↓ mRNA [23] | Impaired phagocytosis (POS, 5 h) [23] | ARB [77,82] |
4. Materials and Methods
4.1. Generation of Human iPSC
4.2. Differentiation of iPSC into RPE
4.3. RT-PCR and qPCR
4.4. Immunofluorescence
4.5. In Situ Apoptosis TUNEL Assay
4.6. Western Blotting
4.7. Phagocytosis Assay
4.8. Anion Channel Activity Determination
4.9. BEST1 3D Structure
4.10. Statistics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Petrukhin, K.; Koisti, M.J.; Bakall, B.; Li, W.; Xie, G.; Marknell, T.; Sandgren, O.; Forsman, K.; Holmgren, G.; Andreasson, S.; et al. Identification of the gene responsible for best macular dystrophy. Nat. Genet. 1998, 19. [Google Scholar] [CrossRef] [PubMed]
- Stone, E.M.; Nichols, B.E.; Streb, L.M.; Kimura, A.E.; Sheffield, V.C. Genetic linkage of vitelliform macular degeneration (Best’s disease) to chromosome 11q13. Nat. Genet. 1992, 1, 246–250. [Google Scholar] [CrossRef] [PubMed]
- Milenkovic, V.M.; Rivera, A.; Horling, F.; Weber, B.H.F. Insertion and topology of normal and mutant bestrophin-1 in the endoplasmic reticulum membrane. J. Biol. Chem. 2007, 282, 1313–1321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsunenari, T.; Sun, H.; Williams, J.; Cahill, H.; Smallwood, P.; Yau, K.W.; Nathans, J. Structure-Function Analysis of the Bestrophin Family of Anion Channels. J. Biol. Chem. 2003, 278, 41114–41125. [Google Scholar] [CrossRef] [Green Version]
- Yang, T.; Liu, Q.; Kloss, B.; Bruni, R.; Kalathur, R.C.; Guo, Y.; Kloppmann, E.; Rost, B.; Colecraft, H.M.; Hendrickson, W.A. Structure and selectivity in bestrophin ion channels. Science 2014, 346, 355–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dickson, V.K.; Pedi, L.; Long, S.B. Structure and insights into the function of a Ca2+-activated Cl− channel. Nature 2014, 516, 213–218. [Google Scholar] [CrossRef] [Green Version]
- Singh Grewal, S.; Smith, J.J.; Carr, A.-J.F. Bestrophinopathies: Perspectives on clinical disease, Bestrophin-1 function and developing therapies. Ther. Adv. Ophthalmol. 2021, 13, 2515841421997191. [Google Scholar] [CrossRef]
- Boon, C.J.F.; Klevering, B.J.; Leroy, B.P.; Hoyng, C.B.; Keunen, J.E.E.; den Hollander, A.I. The spectrum of ocular phenotypes caused by mutations in the BEST1 gene. Prog. Retin. Eye Res. 2009, 28, 187–205. [Google Scholar] [CrossRef]
- Mullins, R.F.; Oh, K.T.; Heffron, E.; Hageman, G.S.; Stone, E.M. Late development of vitelliform lesions and flecks in a patient with best disease: Clinicopathologic correlation. Arch. Ophthalmol. 2005, 123, 1588–1594. [Google Scholar] [CrossRef] [Green Version]
- Lorenz, B.; Preising, M.N. Best’s disease. Overview of pathology and its causes. Der Ophthalmol. 2005, 102, 111–115. [Google Scholar] [CrossRef]
- Nowomiejska, K.; Nasser, F.; Stingl, K.; Schimpf-Linzenbold, S.; Biskup, S.; Brzozowska, A.; Rejdak, R.; Kohl, S.; Zrenner, E. Disease expression caused by different variants in the BEST1 gene: Genotype and phenotype findings in bestrophinopathies. Acta Ophthalmol. 2021, 100, e847–e858. [Google Scholar] [CrossRef]
- Marmorstein, A.D.; Cross, H.E.; Peachey, N.S. Functional roles of bestrophins in ocular epithelia. Prog. Retin. Eye Res. 2009, 28, 206–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Renner, A.B.; Tillack, H.; Kraus, H.; Kohl, S.; Wissinger, B.; Mohr, N.; Weber, B.H.F.; Kellner, U.; Foerster, M.H. Morphology and functional characteristics in adult vitelliform macular dystrophy. Retina 2004, 24, 929–939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casalino, G.; Khan, K.N.; Armengol, M.; Wright, G.; Pontikos, N.; Georgiou, M.; Webster, A.R.; Robson, A.G.; Grewal, P.S.; Michaelides, M. Autosomal Recessive Bestrophinopathy: Clinical Features, Natural History, and Genetic Findings in Preparation for Clinical Trials. Ophthalmology 2021, 128, 706–718. [Google Scholar] [CrossRef] [PubMed]
- Strauss, O. The retinal pigment epithelium in visual function. Physiol. Rev. 2005, 85, 845–881. [Google Scholar] [CrossRef] [Green Version]
- Kevany, B.M.; Palczewski, K. Phagocytosis of retinal rod and cone photoreceptors. Physiology 2010, 25, 8–15. [Google Scholar] [CrossRef] [Green Version]
- Wimmers, S.; Karl, M.O.; Strauss, O. Ion channels in the RPE. Prog. Retin. Eye Res. 2007, 26, 263–301. [Google Scholar] [CrossRef]
- Strauß, O.; Müller, C.; Reichhart, N.; Tamm, E.R.; Gomez, N.M. The role of bestrophin-1 in intracellular Ca2+ signaling. Adv. Exp. Med. Biol. 2014, 801, 113–119. [Google Scholar] [CrossRef]
- Nguyen, M.T.T.; Arnheiter, H. Signaling and transcriptional regulation in early mammalian eye development: A link between FGF and MITF. Development 2000, 127, 3581–3591. [Google Scholar] [CrossRef]
- Qu, Z.; Hartzell, H.C. Bestrophin Cl− channels are highly permeable to HCO3−. Am. J. Physiol.-Cell Physiol. 2008, 294, C1371–C1377. [Google Scholar] [CrossRef] [Green Version]
- Woo, D.H.; Han, K.S.; Shim, J.W.; Yoon, B.E.; Kim, E.; Bae, J.Y.; Oh, S.J.; Hwang, E.M.; Marmorstein, A.D.; Bae, Y.C.; et al. TREK-1 and Best1 channels mediate fast and slow glutamate release in astrocytes upon GPCR activation. Cell 2012, 151, 25–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.; Yoon, B.E.; Berglund, K.; Oh, S.J.; Park, H.; Shin, H.S.; Augustine, G.J.; Lee, C.J. Channel-mediated tonic GABA release from glia. Science 2010, 330, 790–796. [Google Scholar] [CrossRef] [PubMed]
- Marmorstein, A.D.; Johnson, A.A.; Bachman, L.A.; Andrews-Pfannkoch, C.; Knudsen, T.; Gilles, B.J.; Hill, M.; Gandhi, J.K.; Marmorstein, L.Y.; Pulido, J.S. Mutant Best1 Expression and Impaired Phagocytosis in an iPSC Model of Autosomal Recessive Bestrophinopathy. Sci. Rep. 2018, 8, 4487. [Google Scholar] [CrossRef] [PubMed]
- Nachtigal, A.-L.; Milenkovic, A.; Brandl, C.; Schulz, H.L.; Duerr, L.M.J.; Lang, G.E.; Reiff, C.; Herrmann, P.; Kellner, U.; Weber, B.H.F. Mutation-Dependent Pathomechanisms Determine the Phenotype in the Bestrophinopathies. Int. J. Mol. Sci. 2020, 21, 1597. [Google Scholar] [CrossRef] [Green Version]
- Uggenti, C.; Briant, K.; Streit, A.-K.; Thomson, S.; Koay, Y.H.; Baines, R.A.; Swanton, E.; Manson, F.D. Restoration of mutant bestrophin-1 expression, localisation and function in a polarised epithelial cell model. Dis. Model. Mech. 2016, 9, 1317–1328. [Google Scholar] [CrossRef] [Green Version]
- Sinha, D.; Steyer, B.; Shahi, P.K.; Mueller, K.P.; Valiauga, R.; Edwards, K.L.; Bacig, C.; Steltzer, S.S.; Srinivasan, S.; Abdeen, A.; et al. Human iPSC Modeling Reveals Mutation-Specific Responses to Gene Therapy in a Genotypically Diverse Dominant Maculopathy. Am. J. Hum. Genet. 2020, 107, 278–292. [Google Scholar] [CrossRef]
- Milenkovic, V.M.; Röhrl, E.; Weber, B.H.F.; Strauss, O. Disease-associated missense mutations in bestrophin-1 affect cellular trafficking and anion conductance. J. Cell Sci. 2011, 124, 2988–2996. [Google Scholar] [CrossRef] [Green Version]
- Johnson, A.A.; Lee, Y.S.; Chadburn, A.J.; Tammaro, P.; Manson, F.D.; Marmorstein, L.Y.; Marmorstein, A.D. Disease-causing mutations associated with four bestrophinopathies exhibit disparate effects on the localization, but not the oligomerization, of Bestrophin-1. Exp. Eye Res. 2014, 121, 74–85. [Google Scholar] [CrossRef] [Green Version]
- Doumanov, J.A.; Zeitz, C.; Dominguez Gimenez, P.; Audo, I.; Krishna, A.; Alfano, G.; Bellido Diaz, M.L.; Moskova-Doumanova, V.; Lancelot, M.E.; Sahel, J.A.; et al. Disease-causing mutations in BEST1 gene are associated with altered sorting of bestrophin-1 protein. Int. J. Mol. Sci. 2013, 14, 15121–15140. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Q.; Kong, Y.; Kittredge, A.; Li, Y.; Shen, Y.; Zhang, Y.; Tsang, S.H.; Yang, T. Distinct expression requirements and rescue strategies for BEST1 loss- and gain-of-function mutations. Elife 2021, 10, e67622. [Google Scholar] [CrossRef]
- Ji, C.; Kittredge, A.; Hopiavuori, A.; Ward, N.; Chen, S.; Fukuda, Y.; Zhang, Y.; Yang, T. Dual Ca2+-dependent gates in human Bestrophin1 underlie disease-causing mechanisms of gain-of-function mutations. Commun. Biol. 2019, 2, 240. [Google Scholar] [CrossRef] [PubMed]
- Ji, C.; Li, Y.; Kittredge, A.; Hopiavuori, A.; Ward, N.; Yao, P.; Fukuda, Y.; Zhang, Y.; Tsang, S.H.; Yang, T. Investigation and Restoration of BEST1 Activity in Patient-derived RPEs with Dominant Mutations. Sci. Rep. 2019, 9, 19026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Domingo-Prim, J.; Riera, M.; Abad-Morales, V.; Ruiz-Nogales, S.; Corcostegui, B.; Pomares, E. Generation of Best disease-derived induced pluripotent stem cell line (FRIMOi006-A) carrying a novel dominant mutation in BEST1 gene. Stem Cell Res. 2019, 40, 101570. [Google Scholar] [CrossRef] [PubMed]
- Regent, F.; Morizur, L.; Lesueur, L.; Habeler, W.; Plancheron, A.; Ben M’Barek, K.; Monville, C. Automation of human pluripotent stem cell differentiation toward retinal pigment epithelial cells for large-scale productions. Sci. Rep. 2019, 9, 10646. [Google Scholar] [CrossRef] [Green Version]
- Johnson, A.A.; Andrews-Pfannkoch, C.; Nelson, T.J.; Pulido, J.S.; Marmorstein, A.D. Disease modeling studies using induced pluripotent stem cells: Are we using enough controls? Regen. Med. 2017, 12, 899–903. [Google Scholar] [CrossRef]
- Domingo-Prim, J.; Riera, M.; Burés-Jelstrup, A.; Corcostegui, B.; Pomares, E. Establishment of an induced pluripotent stem cell line (FRIMOi005-A) derived from a retinitis pigmentosa patient carrying a dominant mutation in RHO gene. Stem Cell Res. 2019, 38, 101468. [Google Scholar] [CrossRef]
- Carr, A.J.; Vugler, A.A.; Yu, L.; Semo, M.; Coffey, P.; Moss, S.E.; Greenwood, J. The expression of retinal cell markers in human retinal pigment epithelial cells and their augmentation by the synthetic retinoid fenretinide. Mol. Vis. 2011, 17, 1701–1715. [Google Scholar]
- Vugler, A.; Carr, A.J.; Lawrence, J.; Chen, L.L.; Burrell, K.; Wright, A.; Lundh, P.; Semo, M.; Ahmado, A.; Gias, C.; et al. Elucidating the phenomenon of HESC-derived RPE: Anatomy of cell genesis, expansion and retinal transplantation. Exp. Neurol. 2008, 214, 347–361. [Google Scholar] [CrossRef]
- Yue, F.; Johkura, K.; Shirasawa, S.; Yokoyama, T.; Inoue, Y.; Tomotsune, D.; Sasaki, K. Differentiation of primate ES cells into retinal cells induced by ES cell-derived pigmented cells. Biochem. Biophys. Res. Commun. 2010, 394, 877–883. [Google Scholar] [CrossRef]
- Esumi, N.; Kachi, S.; Hackler, L.; Masuda, T.; Yang, Z.; Campochiaro, P.A.; Zack, D.J. BEST1 expression in the retinal pigment epithelium is modulated by OTX family members. Hum. Mol. Genet. 2009, 18, 128–141. [Google Scholar] [CrossRef] [Green Version]
- Marmorstein, A.D.; Marmorstein, L.Y.; Rayborn, M.; Wang, X.; Hollyfield, J.G.; Petrukhin, K. Bestrophin, the product of the Best vitelliform macular dystrophy gene (VMD2), localizes to the basolateral plasma membrane of the retinal pigment epithelium. Proc. Natl. Acad. Sci. USA 2000, 97, 12758–12763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, T.; Tian, C.; Xu, H.; Tang, X.; Huang, L.; Zhao, M. Disease-causing mutations associated with bestrophinopathies promote apoptosis in retinal pigment epithelium cells. Graefe’s Arch. Clin. Exp. Ophthalmol. 2020, 258, 2251–2261. [Google Scholar] [CrossRef] [PubMed]
- Miller, A.N.; Vaisey, G.; Long, S.B. Molecular mechanisms of gating in the calcium-activated chloride channel bestrophin. Elife 2019, 8, e43231. [Google Scholar] [CrossRef] [PubMed]
- Moshfegh, Y.; Velez, G.; Li, Y.; Bassuk, A.G.; Mahajan, V.B.; Tsang, S.H. BESTROPHIN1 mutations cause defective chloride conductance in patient stem cell-derived RPE. Hum. Mol. Genet. 2016, 25, 2672–2680. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Stanton, J.B.; Wu, J.; Yu, K.; Hartzell, H.C.; Peachey, N.S.; Marmorstein, L.Y.; Marmorstein, A.D. Suppression of Ca2+ signaling in a mouse model of Best disease. Hum. Mol. Genet. 2010, 19, 1108–1118. [Google Scholar] [CrossRef] [Green Version]
- Toulis, V.; García-Monclús, S.; de la Peña-Ramírez, C.; Arenas-Galnares, R.; Abril, J.F.; Todi, S.V.; Khan, N.; Garanto, A.; do Costa, M.C.; Marfany, G. The Deubiquitinating Enzyme Ataxin-3 Regulates Ciliogenesis and Phagocytosis in the Retina. Cell Rep. 2020, 33, 108360. [Google Scholar] [CrossRef]
- Williams, L.L.; Lew, H.M.; Shannon, B.T.; Singley, C.T.; Davidorf, F.H.; Jin, R.; Wolinsky, J.S. Phagocytosis of latex beads is defective in cultured human retinal pigment epithelial cells with persistent rubella virus infection. Am. J. Pathol. 1993, 142, 451–461. [Google Scholar]
- Kimura, H.; Ogura, Y.; Moritera, T.; Honda, Y.; Tabata, Y.; Ikada, Y. In vitro phagocytosis of polylactide microspheres by retinal pigment epithelial cells and intracellular drug release. Curr. Eye Res. 1994, 13, 353–360. [Google Scholar] [CrossRef]
- Ainscough, S.L.; Feigl, B.; Malda, J.; Harkin, D.G. Discovery and characterization of IGFBP-mediated endocytosis in the human retinal pigment epithelial cell line ARPE-19. Exp. Eye Res. 2009, 89, 629–637. [Google Scholar] [CrossRef]
- Klettner, A.; Tahmaz, N.; Dithmer, M.; Richert, E.; Roider, J. Effects of aflibercept on primary RPE cells: Toxicity, wound healing, uptake and phagocytosis. Br. J. Ophthalmol. 2014, 98, 1448–1452. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.-Q.; Fan, Y.-G.; Dang, Y.-L.; Liu, Y.-L.; Liu, H.; Li, L.-H. Down-regulation of protein kinase C alpha/ezrin signals in light-induced phagocytic crisis of retinal pigment epithelium cells. Int. J. Ophthalmol. 2017, 10, 1040–1045. [Google Scholar] [CrossRef] [PubMed]
- Rahman, N.; Georgiou, M.; Khan, K.N.; Michaelides, M. Macular dystrophies: Clinical and imaging features, molecular genetics and therapeutic options. Br. J. Ophthalmol. 2020, 104, 451–460. [Google Scholar] [CrossRef]
- Stenson, P.D.; Mort, M.; Ball, E.V.; Chapman, M.; Evans, K.; Azevedo, L.; Hayden, M.; Heywood, S.; Millar, D.S.; Phillips, A.D.; et al. The Human Gene Mutation Database (HGMD(®)): Optimizing its use in a clinical diagnostic or research setting. Hum. Genet. 2020, 139, 1197–1207. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.J.; Nommiste, B.; Carr, A.J.F. Bestrophin1: A Gene that Causes Many Diseases. Adv. Exp. Med. Biol. 2019, 1185, 419–423. [Google Scholar] [CrossRef] [PubMed]
- Yardley, J.; Leroy, B.P.; Hart-Holden, N.; Lafaut, B.A.; Loeys, B.; Messiaen, L.M.; Perveen, R.; Reddy, M.A.; Bhattacharya, S.S.; Traboulsi, E.; et al. Mutations of VMD2 splicing regulators cause nanophthalmos and autosomal dominant vitreoretinochoroidopathy (ADVIRC). Investig. Ophthalmol. Vis. Sci. 2004, 45, 3683–3689. [Google Scholar] [CrossRef] [Green Version]
- Hinkle, J.W.; Mahmoudzadeh, R.; Kuriyan, A.E. Cell-based therapies for retinal diseases: A review of clinical trials and direct to consumer “cell therapy” clinics. Stem Cell Res. Ther. 2021, 12, 538. [Google Scholar] [CrossRef]
- Andreazzoli, M.; Barravecchia, I.; De Cesari, C.; Angeloni, D.; Demontis, G.C. Inducible Pluripotent Stem Cells to Model and Treat Inherited Degenerative Diseases of the Outer Retina: 3D-Organoids Limitations and Bioengineering Solutions. Cells 2021, 10, 2489. [Google Scholar] [CrossRef]
- Vaisey, G.; Long, S.B. An allosteric mechanism of inactivation in the calcium-dependent chloride channel BEST1. J. Gen. Physiol. 2018, 150, 1484–1497. [Google Scholar] [CrossRef] [Green Version]
- Llavona, P.; Pinelli, M.; Mutarelli, M.; Marwah, V.S.; Schimpf-Linzenbold, S.; Thaler, S.; Yoeruek, E.; Vetter, J.; Kohl, S.; Wissinger, B. Allelic Expression Imbalance in the Human Retinal Transcriptome and Potential Impact on Inherited Retinal Diseases. Genes 2017, 8, 283. [Google Scholar] [CrossRef] [Green Version]
- Martinez Velazquez, L.A.; Ballios, B.G. The Next Generation of Molecular and Cellular Therapeutics for Inherited Retinal Disease. Int. J. Mol. Sci. 2021, 22, 11542. [Google Scholar] [CrossRef]
- Davidson, A.E.; Millar, I.D.; Urquhart, J.E.; Burgess-Mullan, R.; Shweikh, Y.; Parry, N.; O’Sullivan, J.; Maher, G.J.; McKibbin, M.; Downes, S.M.; et al. Missense mutations in a retinal pigment epithelium protein, bestrophin-1, cause retinitis pigmentosa. Am. J. Hum. Genet. 2009, 85, 581–592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, Q.; Prussia, A.; Yu, K.; Cui, Y.; Hartzell, H.C. Regulation of bestrophin Cl channels by calcium: Role of the C terminus. J. Gen. Physiol. 2008, 132, 681–692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qu, Z.; Cheng, W.; Cui, Y.; Cui, Y.; Zheng, J. Human disease-causing mutations disrupt an N-C-terminal interaction and channel function of bestrophin 1. J. Biol. Chem. 2009, 284, 16473–16481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaisey, G.; Miller, A.N.; Long, S.B. Distinct regions that control ion selectivity and calcium-dependent activation in the bestrophin ion channel. Proc. Natl. Acad. Sci. USA 2016, 113, E7399–E7408. [Google Scholar] [CrossRef] [Green Version]
- Cordes, M.; Bucichowski, P.; Alfaar, A.S.; Tsang, S.H.; Almedawar, S.; Reichhart, N.; Strauß, O. Inhibition of Ca2+ channel surface expression by mutant bestrophin-1 in RPE cells. FASEB J. 2020, 34, 4055–4071. [Google Scholar] [CrossRef] [Green Version]
- Johnson, A.A.; Lee, Y.S.; Brett Stanton, J.; Yu, K.; Hartzell, C.H.; Marmorstein, L.Y.; Marmorstein, A.D. Differential effects of best disease causing missense mutations on bestrophin-1 trafficking. Hum. Mol. Genet. 2013, 22, 4688–4697. [Google Scholar] [CrossRef] [Green Version]
- Marquardt, A.; Stöhr, H.; Passmore, L.A.; Krämer, F.; Rivera, A.; Weber, B.H. Mutations in a novel gene, VMD2, encoding a protein of unknown properties cause juvenile-onset vitelliform macular dystrophy (Best’s disease). Hum. Mol. Genet. 1998, 7, 1517–1525. [Google Scholar] [CrossRef] [Green Version]
- Tian, L.; Sun, T.; Xu, K.; Zhang, X.; Peng, X.; Li, Y. Screening of BEST1 Gene in a Chinese Cohort With Best Vitelliform Macular Dystrophy or Autosomal Recessive Bestrophinopathy. Investig. Ophthalmol. Vis. Sci. 2017, 58, 3366–3375. [Google Scholar] [CrossRef] [Green Version]
- White, K.; Marquardt, A.; Weber, B.H. VMD2 mutations in vitelliform macular dystrophy (Best disease) and other maculopathies. Hum. Mutat. 2000, 15, 301–308. [Google Scholar] [CrossRef]
- Lee, J.H.; Oh, J.O.; Lee, C.S. Induced Pluripotent Stem Cell Modeling of Best Disease and Autosomal Recessive Bestrophinopathy. Yonsei Med. J. 2020, 61, 816–825. [Google Scholar] [CrossRef]
- Lee, C.S.; Jun, I.; Choi, S.; Lee, J.H.; Lee, M.G.; Lee, S.C.; Kim, E.K. A Novel BEST1 Mutation in Autosomal Recessive Bestrophinopathy. Investig. Ophthalmol. Vis. Sci. 2015, 56, 8141–8150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lotery, A.J.; Munier, F.L.; Fishman, G.A.; Weleber, R.G.; Jacobson, S.G.; Affatigato, L.M.; Nichols, B.E.; Schorderet, D.F.; Sheffield, V.C.; Stone, E.M. Allelic variation in the VMD2 gene in best disease and age-related macular degeneration. Investig. Ophthalmol. Vis. Sci. 2000, 41, 1291–1296. [Google Scholar]
- Davidson, A.E.; Millar, I.D.; Burgess-Mullan, R.; Maher, G.J.; Urquhart, J.E.; Brown, P.D.; Black, G.C.M.; Manson, F.D.C. Functional characterization of bestrophin-1 missense mutations associated with autosomal recessive bestrophinopathy. Investig. Ophthalmol. Vis. Sci. 2011, 52, 3730–3736. [Google Scholar] [CrossRef] [Green Version]
- Krämer, F.; Mohr, N.; Kellner, U.; Rudolph, G.; Weber, B.H.F. Ten novel mutations in VMD2 associated with Best macular dystrophy (BMD). Hum. Mutat. 2003, 22, 418. [Google Scholar] [CrossRef] [PubMed]
- Bakall, B.; Marknell, T.; Ingvast, S.; Koisti, M.J.; Sandgren, O.; Li, W.; Bergen, A.A.; Andreasson, S.; Rosenberg, T.; Petrukhin, K.; et al. The mutation spectrum of the bestrophin protein—Functional implications. Hum. Genet. 1999, 104, 383–389. [Google Scholar] [CrossRef]
- Eksandh, L.; Bakall, B.; Bauer, B.; Wadelius, C.; Andréasson, S. Best’s vitelliform macular dystrophy caused by a new mutation (Val89Ala) in the VMD2 gene. Ophthalmic Genet. 2001, 22, 107–115. [Google Scholar] [CrossRef]
- Krämer, F.; White, K.; Pauleikhoff, D.; Gehrig, A.; Passmore, L.; Rivera, A.; Rudolph, G.; Kellner, U.; Andrassi, M.; Lorenz, B.; et al. Mutations in the VMD2 gene are associated with juvenile-onset vitelliform macular dystrophy (Best disease) and adult vitelliform macular dystrophy but not age-related macular degeneration. Eur. J. Hum. Genet. 2000, 8, 286–292. [Google Scholar] [CrossRef] [Green Version]
- Burgess, R.; Millar, I.D.; Leroy, B.P.; Urquhart, J.E.; Fearon, I.M.; De Baere, E.; Brown, P.D.; Robson, A.G.; Wright, G.A.; Kestelyn, P.; et al. Biallelic mutation of BEST1 causes a distinct retinopathy in humans. Am. J. Hum. Genet. 2008, 82, 19–31. [Google Scholar] [CrossRef] [Green Version]
- Marmorstein, A.D.; Kinnick, T.R.; Stanton, J.B.; Johnson, A.A.; Lynch, R.M.; Marmorstein, L.Y. Bestrophin-1 influences transepithelial electrical properties and Ca2+signaling in human retinal pigment epithelium. Mol. Vis. 2015, 21, 347–359. [Google Scholar]
- Pianta, M.J.; Aleman, T.S.; Cideciyan, A.V.; Sunness, J.S.; Li, Y.; Campochiaro, B.A.; Campochiaro, P.A.; Zack, D.J.; Stone, E.M.; Jacobson, S.G. In vivo micropathology of Best macular dystrophy with optical coherence tomography. Exp. Eye Res. 2003, 76, 203–211. [Google Scholar] [CrossRef]
- Johnson, A.A.; Bachman, L.A.; Gilles, B.J.; Cross, S.D.; Stelzig, K.E.; Resch, Z.T.; Marmorstein, L.Y.; Pulido, J.S.; Marmorstein, A.D. Autosomal recessive bestrophinopathy is not associated with the loss of bestrophin-1 anion channel function in a patient with a novel BEST1 mutation. Investig. Ophthalmol. Vis. Sci. 2015, 56, 4619–4630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Habibi, I.; Falfoul, Y.; Todorova, M.G.; Wyrsch, S.; Vaclavik, V.; Helfenstein, M.; Turki, A.; ElMatri, K.; ElMatri, L.; Schorderet, D.F. Clinical and Genetic Findings of Autosomal Recessive Bestrophinopathy (ARB). Genes 2019, 10, 953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iannaccone, A.; Kerr, N.C.; Kinnick, T.R.; Calzada, J.I.; Stone, E.M. Autosomal recessive best vitelliform macular dystrophy: Report of a family and management of early-onset neovascular complications. Arch. Ophthalmol. 2011, 129, 211–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, T.; Tian, C.; Hu, Q.; Liu, Z.; Zou, J.; Huang, L.; Zhao, M. Clinical and Mutation Analysis of Patients with Best Vitelliform Macular Dystrophy or Autosomal Recessive Bestrophinopathy in Chinese Population. Biomed Res. Int. 2018, 2018, 4582816. [Google Scholar] [CrossRef]
- Kinnick, T.R.; Mullins, R.F.; Dev, S.; Leys, M.; Mackey, D.A.; Kay, C.N.; Lam, B.L.; Fishman, G.A.; Traboulsi, E.; Iezzi, R.; et al. Autosomal recessive vitelliform macular dystrophy in a large cohort of vitelliform macular dystrophy patients. Retina 2011, 31, 581–595. [Google Scholar] [CrossRef]
- Singh, R.; Shen, W.; Kuai, D.; Martin, J.M.; Guo, X.; Smith, M.A.; Perez, E.T.; Phillips, M.J.; Simonett, J.M.; Wallace, K.A.; et al. iPS cell modeling of best disease: Insights into the pathophysiology of an inherited macular degeneration. Hum. Mol. Genet. 2013, 22, 593–607. [Google Scholar] [CrossRef] [Green Version]
- Pomares, E.; Burés-Jelstrup, A.; Ruiz-Nogales, S.; Corcóstegui, B.; González-Duarte, R.; Navarro, R. Nonsense-mediated decay as the molecular cause for autosomal recessive bestrophinopathy in two unrelated families. Investig. Ophthalmol. Vis. Sci. 2012, 53, 532–537. [Google Scholar] [CrossRef] [Green Version]
- Downs, K.; Zacks, D.N.; Caruso, R.; Karoukis, A.J.; Branham, K.; Yashar, B.M.; Haimann, M.H.; Trzupek, K.; Meltzer, M.; Blain, D.; et al. Molecular testing for hereditary retinal disease as part of clinical care. Arch. Ophthalmol. 2007, 125, 252–258. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Zhang, Y.; Xu, Y.; Kittredge, A.; Ward, N.; Chen, S.; Tsang, S.H.; Yang, T. Patient-specific mutations impair BESTROPHIN1’s essential role in mediating Ca2+-dependent Cl-currents in human RPE. eLife 2017, 6, e29914. [Google Scholar] [CrossRef] [Green Version]
- Wittström, E.; Ekvall, S.; Schatz, P.; Bondeson, M.-L.; Ponjavic, V.; Andréasson, S. Morphological and functional changes in multifocal vitelliform retinopathy and biallelic mutations in BEST1. Ophthalmic Genet. 2011, 32, 83–96. [Google Scholar] [CrossRef]
- Liu, J.; Taylor, R.L.; Baines, R.A.; Swanton, L.; Freeman, S.; Corneo, B.; Patel, A.; Marmorstein, A.; Knudsen, T.; Black, G.C.; et al. Small molecules restore bestrophin 1 expression and function of both dominant and recessive bestrophinopathies in patient-derived retinal pigment epithelium. Investig. Ophthalmol. Vis. Sci. 2020, 61, 28. [Google Scholar] [CrossRef] [PubMed]
- Carter, D.A.; Smart, M.J.K.; Letton, W.V.G.; Ramsden, C.M.; Nommiste, B.; Chen, L.L.; Fynes, K.; Muthiah, M.N.; Goh, P.; Lane, A.; et al. Mislocalisation of BEST1 in iPSC-derived retinal pigment epithelial cells from a family with autosomal dominant vitreoretinochoroidopathy (ADVIRC). Sci. Rep. 2016, 6, 33792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burgess, R.; MacLaren, R.E.; Davidson, A.E.; Urquhart, J.E.; Holder, G.E.; Robson, A.G.; Moore, A.T.; Keefe, R.O.; Black, G.C.M.; Manson, F.D.C. ADVIRC is caused by distinct mutations in BEST1 that alter pre-mRNA splicing. J. Med. Genet. 2009, 46, 620–625. [Google Scholar] [CrossRef] [PubMed]
- Yu, K.; Cui, Y.; Hartzell, H.C. The bestrophin mutation A243V, linked to adult-onset vitelliform macular dystrophy, impairs its chloride channel function. Investig. Ophthalmol. Vis. Sci. 2006, 47, 4956–4961. [Google Scholar] [CrossRef]
- Chung, M.M.; Oh, K.T.; Streb, L.M.; Kimura, A.E.; Stone, E.M. Visual outcome following subretinal hemorrhage in Best disease. Retina 2001, 21, 575–580. [Google Scholar] [CrossRef]
- Hufendiek, K.; Hufendiek, K.; Jägle, H.; Stöhr, H.; Book, M.; Spital, G.; Rustambayova, G.; Framme, C.; Weber, B.H.F.; Renner, A.B.; et al. Clinical heterogeneity in autosomal recessive bestrophinopathy with biallelic mutations in the BEST1 gene. Int. J. Mol. Sci. 2020, 21, 9353. [Google Scholar] [CrossRef]
- Guo, Y.; Wang, P.; Ma, J.H.; Cui, Z.; Yu, Q.; Liu, S.; Xue, Y.; Zhu, D.; Cao, J.; Li, Z.; et al. Modeling Retinitis Pigmentosa: Retinal Organoids Generated From the iPSCs of a Patient With the USH2A Mutation Show Early Developmental Abnormalities. Front. Cell. Neurosci. 2019, 13, 361. [Google Scholar] [CrossRef] [Green Version]
- Bonilha, V.L.; Bell, B.A.; DeBenedictis, M.J.; Hagstrom, S.A.; Fishman, G.A.; Hollyfield, J.G. Cellular Changes in Retinas From Patients With BEST1 Mutations. Front. Cell Dev. Biol. 2020, 8, 573330. [Google Scholar] [CrossRef]
- Scott, W.K.; Slifer, S.H.; Welch, J.K.; Schwartz, S.G.; Kovach, J.L.; Sadda, S.; Nittala, M.G.; Haines, J.L.; Pericak-Vance, M.A. Retinal pigment epithelium (RPE) pigmentary changes are associated with progression from early to advanced age-related macular degeneration (AMD) independent of CFH and ARMS2 risk genotypes. Investig. Ophthalmol. Vis. Sci. 2020, 61, 4189. [Google Scholar]
- Ke, Y.; Wang, C.; Zhang, J.; Zhong, X.; Wang, R.; Zeng, X.; Ba, X. The Role of PARPs in Inflammation—And Metabolic—Related Diseases: Molecular Mechanisms and Beyond. Cells 2019, 8, 1047. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Li, S.; Luo, X.; Song, Z.; Long, X.; Zhu, X. Knockdown of PARP6 or survivin promotes cell apoptosis and inhibits cell invasion of colorectal adenocarcinoma cells. Oncol. Rep. 2017, 37, 2245–2251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vermehren-Schmaedick, A.; Huang, J.Y.; Levinson, M.; Pomaville, M.B.; Reed, S.; Bellus, G.A.; Gilbert, F.; Keren, B.; Heron, D.; Haye, D.; et al. Characterization of PARP6 Function in Knockout Mice and Patients with Developmental Delay. Cells 2021, 10, 1289. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Grosskurth, S.E.; Cheung, T.; Petteruti, P.; Zhang, J.; Wang, X.; Wang, W.; Gharahdaghi, F.; Wu, J.; Su, N.; et al. Pharmacological Inhibition of PARP6 Triggers Multipolar Spindle Formation and Elicits Therapeutic Effects in Breast Cancer. Cancer Res. 2018, 78, 6691–6702. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; Zhang, Y.; Chu, M.; Wang, L.; Shen, H.; Zhang, Z.; Hu, J.; Yi, W.; Yang, W.; Ma, X. PARP6 acts as an oncogene and positively regulates Survivin in gastric cancer. Int. J. Clin. Exp. Pathol. 2018, 11, 2364–2371. [Google Scholar] [PubMed]
- Matsuda, H.; Yoshimura, N.; Matsumura, M.; Honda, Y. The retinal toxicity of befunolol and other adrenergic beta-blocking agents: Inhibition of phagocytic activity of cultured retinal pigment epithelial cells. Acta Ophthalmol. 1983, 61, 343–352. [Google Scholar] [CrossRef] [PubMed]
- Klettner, A.; Möhle, F.; Lucius, R.; Roider, J. Quantifying FITC-labeled latex beads opsonized with photoreceptor outer segment fragments: An easy and inexpensive method of investigating phagocytosis in retinal pigment epithelium cells. Ophthalmic Res. 2011, 46, 88–91. [Google Scholar] [CrossRef]
- Hewage, N.; Altman, D. A role for myosin VI in retinal pigment epithelium phagocytosis. Biochem. Biophys. Res. Commun. 2018, 504, 759–764. [Google Scholar] [CrossRef]
- Sodi, A.; Passerini, I.; Murro, V.; Caputo, R.; Bacci, G.M.; Bodoj, M.; Torricelli, F.; Menchini, U. BEST1 sequence variants in Italian patients with vitelliform macular dystrophy. Mol. Vis. 2012, 18, 2736–2748. [Google Scholar]
- Meunier, I.; Sénéchal, A.; Dhaenens, C.-M.; Arndt, C.; Puech, B.; Defoort-Dhellemmes, S.; Manes, G.; Chazalette, D.; Mazoir, E.; Bocquet, B.; et al. Systematic screening of BEST1 and PRPH2 in juvenile and adult vitelliform macular dystrophies: A rationale for molecular analysis. Ophthalmology 2011, 118, 1130–1136. [Google Scholar] [CrossRef]
- Marchant, D.; Gogat, K.; Boutboul, S.; Péquignot, M.; Sternberg, C.; Dureau, P.; Roche, O.; Uteza, Y.; Hache, J.C.; Puech, B.; et al. Identification of novel VMD2 gene mutations in patients with best vitelliform macular dystrophy. Hum. Mutat. 2001, 17, 235. [Google Scholar] [CrossRef]
- Wong, R.L.M.; Hou, P.; Choy, K.-W.; Chiang, S.W.Y.; Tam, P.O.S.; Li, H.; Chan, W.-M.; Lam, D.S.C.; Pang, C.-P.; Lai, T.Y.Y. Novel and homozygous BEST1 mutations in Chinese patients with Best vitelliform macular dystrophy. Retina 2010, 30, 820–827. [Google Scholar] [CrossRef]
- Guo, J.; Gao, F.; Tang, W.; Qi, Y.; Xuan, Y.; Liu, W.; Li, L.; Ye, X.; Xu, G.; Wu, J.; et al. Novel best1 mutations detected by next-generation sequencing in a chinese population with vitelliform macular dystrophy. Retina 2019, 39, 1613–1622. [Google Scholar] [CrossRef]
- Gao, F.-J.; Qi, Y.-H.; Hu, F.-Y.; Wang, D.-D.; Xu, P.; Guo, J.-L.; Li, J.-K.; Zhang, Y.-J.; Li, W.; Chen, F.; et al. Mutation spectrum of the bestrophin-1 gene in a large Chinese cohort with bestrophinopathy. Br. J. Ophthalmol. 2020, 104, 846–851. [Google Scholar] [CrossRef]
- Alapati, A.; Goetz, K.; Suk, J.; Navani, M.; Al-Tarouti, A.; Jayasundera, T.; Tumminia, S.J.; Lee, P.; Ayyagari, R. Molecular diagnostic testing by eyeGENE: Analysis of patients with hereditary retinal dystrophy phenotypes involving central vision loss. Investig. Ophthalmol. Vis. Sci. 2014, 55, 5510–5521. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.J.; Kaufman, S.; Packo, K.; Stöhr, H.; Weber, B.H.F.; Goldberg, M.F. Long-Term Macular Changes in the First Proband of Autosomal Dominant Vitreoretinochoroidopathy (ADVIRC) Due to a Newly Identified Mutation in BEST1. Ophthalmic Genet. 2016, 37, 102–108. [Google Scholar] [CrossRef]
- Ponjavic, V.; Eksandh, L.; Andréasson, S.; Sjöström, K.; Bakall, B.; Ingvast, S.; Wadelius, C.; Ehinger, B. Clinical expression of Best’s vitelliform macular dystrophy in Swedish families with mutations in the bestrophin gene. Ophthalmic Genet. 1999, 20, 251–257. [Google Scholar] [CrossRef]
- Xuan, Y.; Zhang, Y.; Zong, Y.; Wang, M.; Li, L.; Ye, X.; Liu, W.; Chen, J.; Sun, X.; Zhang, Y.; et al. The Clinical Features and Genetic Spectrum of a Large Cohort of Chinese Patients With Vitelliform Macular Dystrophies. Am. J. Ophthalmol. 2020, 216, 69–79. [Google Scholar] [CrossRef]
- Vincent, A.; McAlister, C.; Vandenhoven, C.; Héon, E. BEST1-related autosomal dominant vitreoretinochoroidopathy: A degenerative disease with a range of developmental ocular anomalies. Eye 2011, 25, 113–118. [Google Scholar] [CrossRef]
- Nguyen, T.T.; Poornachandra, B.; Verma, A.; Mehta, R.A.; Phalke, S.; Battu, R.; Ramprasad, V.L.; Peterson, A.S.; Ghosh, A.; Seshagiri, S. Next generation sequencing identifies novel disease-associated BEST1 mutations in Bestrophinopathy patients. Sci. Rep. 2018, 8, 10176. [Google Scholar] [CrossRef]
- Querques, G.; Zerbib, J.; Santacroce, R.; Margaglione, M.; Delphin, N.; Querques, L.; Rozet, J.M.; Kaplan, J.; Souied, E.H. The spectrum of subclinical best vitelliform macular dystrophy in subjects with mutations in BEST1 gene. Investig. Ophthalmol. Vis. Sci. 2011, 52, 4678–4684. [Google Scholar] [CrossRef] [Green Version]
- Querques, G.; Zerbib, J.; Santacroce, R.; Margaglione, M.; Delphin, N.; Rozet, J.-M.; Kaplan, J.; Martinelli, D.; Delle Noci, N.; Soubrane, G.; et al. Functional and clinical data of Best vitelliform macular dystrophy patients with mutations in the BEST1 gene. Mol. Vis. 2009, 15, 2960–2972. [Google Scholar] [PubMed]
- Schatz, P.; Sharon, D.; Al-Hamdani, S.; Andréasson, S.; Larsen, M. Retinal structure in young patients aged 10 years or less with Best vitelliform macular dystrophy. Graefe’s Arch. Clin. Exp. Ophthalmol. = Albr. Von Graefes Arch. Fur Klin. Exp. Ophthalmol. 2016, 254, 215–221. [Google Scholar] [CrossRef] [PubMed]
- Peng, R.-M.; Hong, J.; Jin, Y.; Sun, Y.-Z.; Sun, Y.-Q.; Zhang, P. Mertk gene expression and photoreceptor outer segment phagocytosis by cultured rat bone marrow mesenchymal stem cells. Mol. Vis. 2017, 23, 8–19. [Google Scholar] [PubMed]






| Variant | Aminoacidic Change | Phenotype |
|---|---|---|
| c.214T > G | p.Y72D | BVMD [85] |
| c.217A > C | p.I73L | BVMD [108] |
| c.217A > T | p.I73F | BVMD [109] |
| c.218T > A | p.I73N | BVMD [110] |
| c.219C > G | p.I73M | BVMD [68] |
| c.223C > T | p.L75F | BVMD [111] |
| c.224T > C | p.L75P | BVMD [112,113] |
| c.227T > A | p.I76N | BVMD [85,113] |
| c.228C > G | p.I76M | BVMD [110] |
| c.227T > C | p.I76T | BVMD [110] |
| c.229C > T | p.P77S | BVMD [33] |
| c.232_233 insT | p.S79FfsX153 | BVMD [113] |
| c.236C > A | p.S79Y | ARB [113] |
| c.239T > G | p.F80C | BVMD [108] |
| c.240C > A | p.F80L | BVMD [27,72] |
| c.238T > G | p.F80V | BVMD [108] |
| c.241G > A | p.V81M | BVMD [113] |
| c.241G > T | p.V81L | ARB [113] |
| c.244C > G | p.L82V | BVMD [27,75] |
| c.248G > C | p.G83A | BVMD [114] |
| c.248G > A | p.G83D | ADVIRC [115] |
| c.250T > G | p.F84V | BVMD [85] |
| c.253T > C | p.Y85H | BVMD [1,29,116] |
| c.254A > C | p.Y85S | BVMD [117] |
| c.256G > A | p.V86M | ADVIRC [55,66,93,118] |
| c.266T > C | p.V89A | BVMD [76] |
| c.272C > T | p.T91I | BVMD [72,119] |
| c.274C > T | p.R92C | BVMD [75,84,120] |
| c.274C > G | p.R92G | BVMD [121] |
| c.274C > A | p.R92S | BVMD [27,77] |
| c.275G > A | p.R92H | BVMD [110,122] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Navinés-Ferrer, A.; Ruiz-Nogales, S.; Navarro, R.; Pomares, E. Impaired Bestrophin Channel Activity in an iPSC-RPE Model of Best Vitelliform Macular Dystrophy (BVMD) from an Early Onset Patient Carrying the P77S Dominant Mutation. Int. J. Mol. Sci. 2022, 23, 7432. https://doi.org/10.3390/ijms23137432
Navinés-Ferrer A, Ruiz-Nogales S, Navarro R, Pomares E. Impaired Bestrophin Channel Activity in an iPSC-RPE Model of Best Vitelliform Macular Dystrophy (BVMD) from an Early Onset Patient Carrying the P77S Dominant Mutation. International Journal of Molecular Sciences. 2022; 23(13):7432. https://doi.org/10.3390/ijms23137432
Chicago/Turabian StyleNavinés-Ferrer, Arnau, Sheila Ruiz-Nogales, Rafael Navarro, and Esther Pomares. 2022. "Impaired Bestrophin Channel Activity in an iPSC-RPE Model of Best Vitelliform Macular Dystrophy (BVMD) from an Early Onset Patient Carrying the P77S Dominant Mutation" International Journal of Molecular Sciences 23, no. 13: 7432. https://doi.org/10.3390/ijms23137432