
Metabolic Reprogramming in Sickle Cell Diseases: Pathophysiology and Drug Discovery Opportunities

 ,
, 
Abstract
:1. Introduction
1.1. Pathophysiology of Sickle Cell Disease
1.2. Current Treatments of SCD
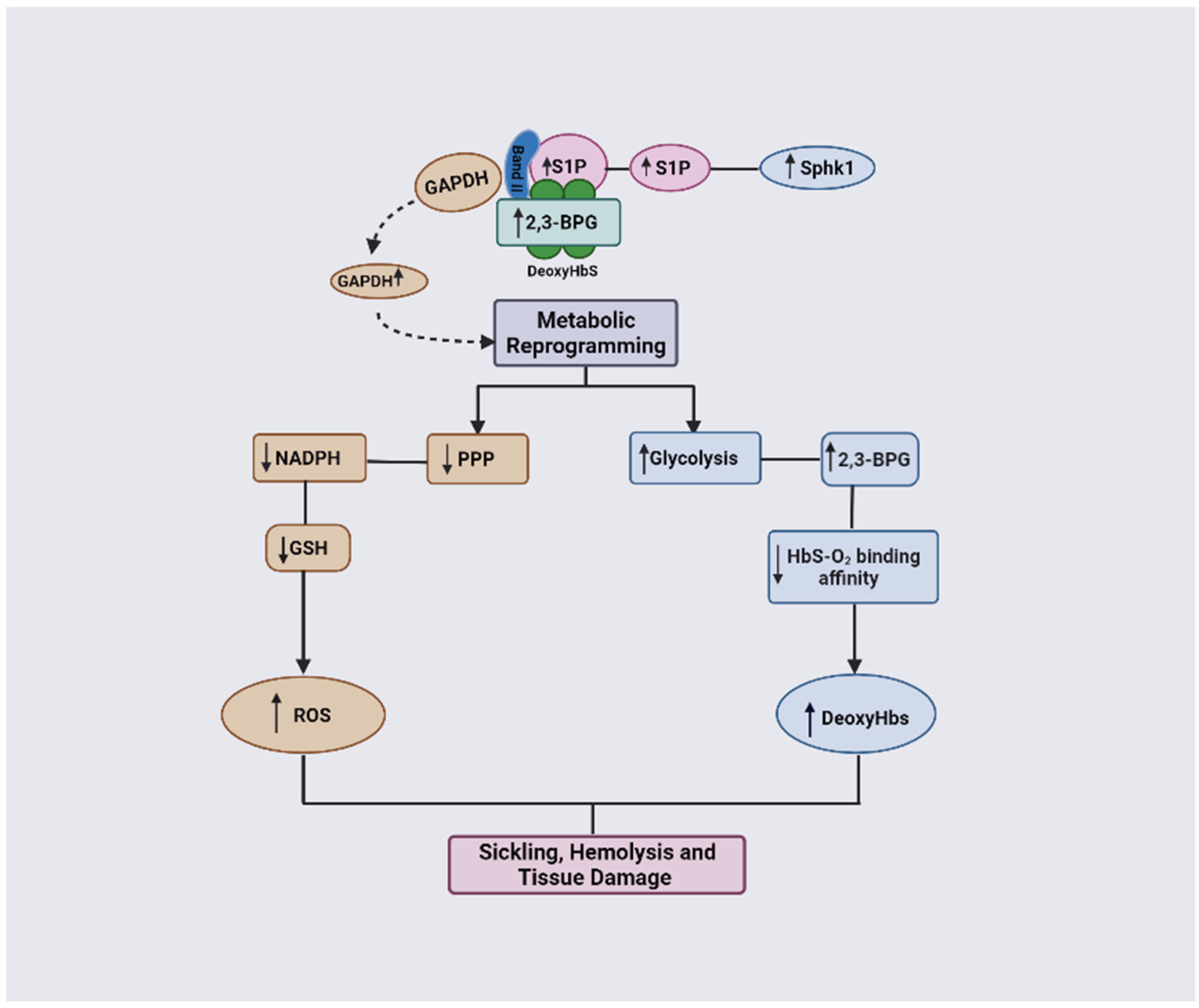
1.3. 2,3-BPG in SCD Pathogenesis and Potential Targets for SCD Therapeutics
2. Glucose Metabolism
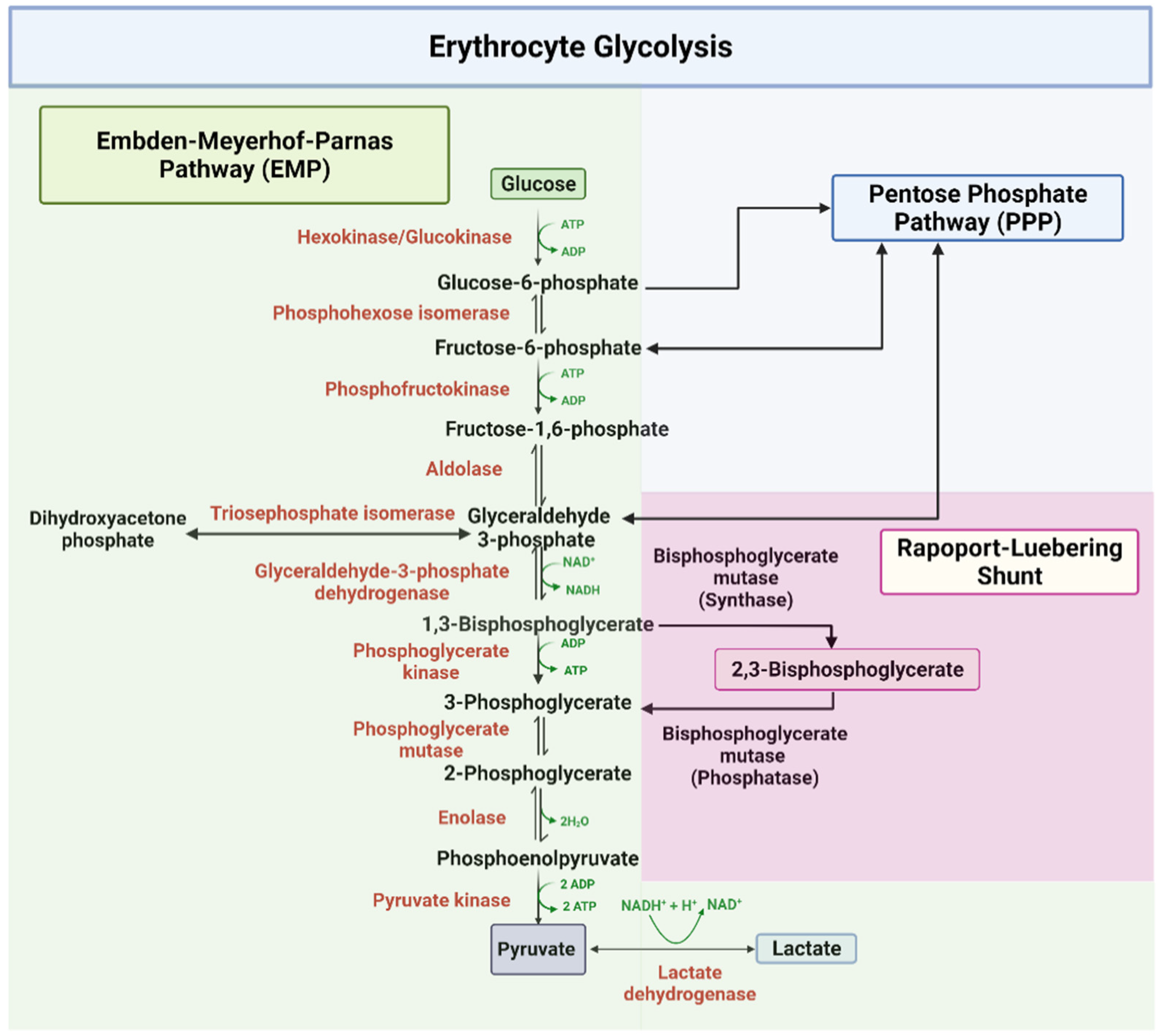
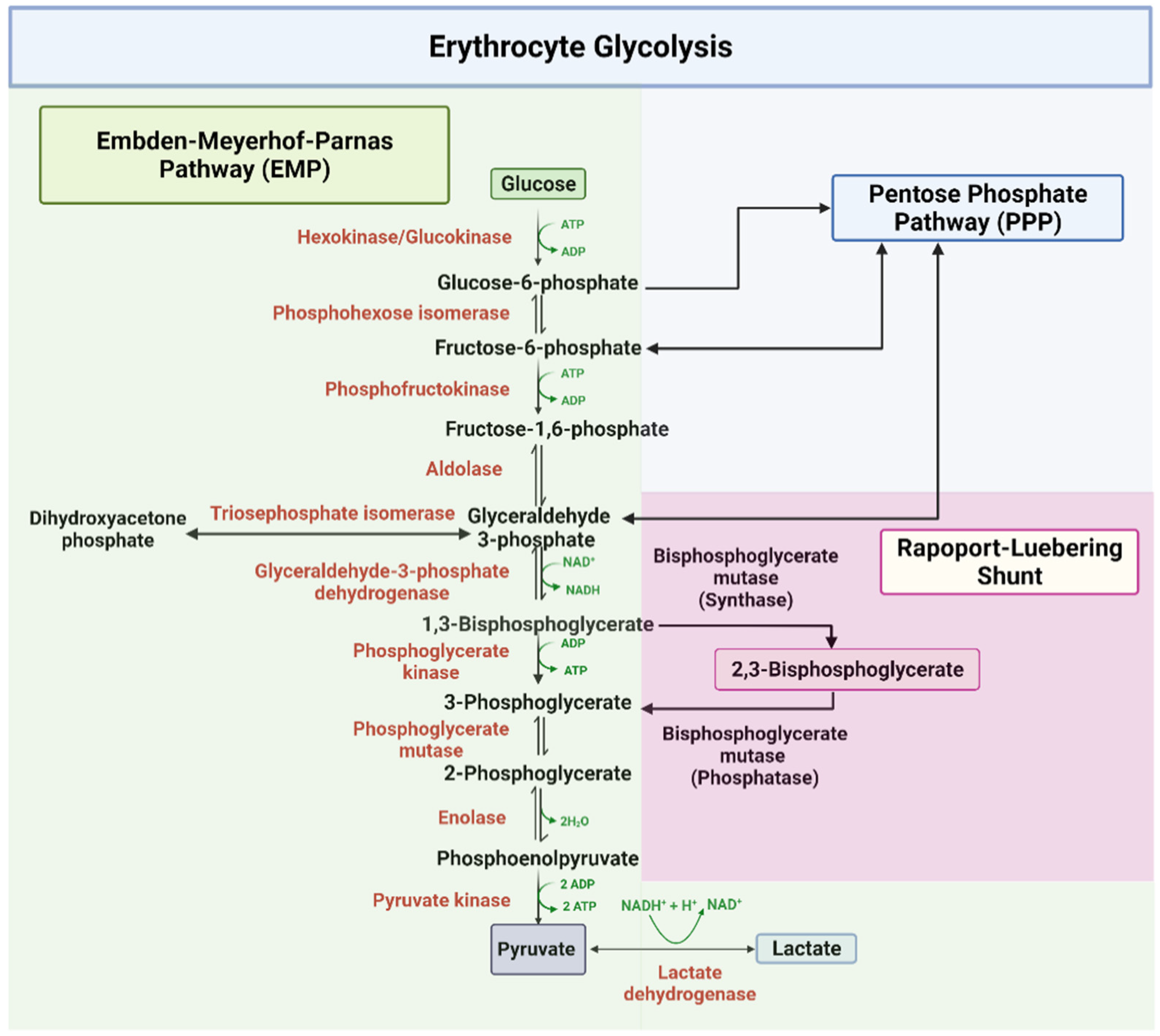
2.1. Embden–Meyerhof–Parnas Pathway (EMP)
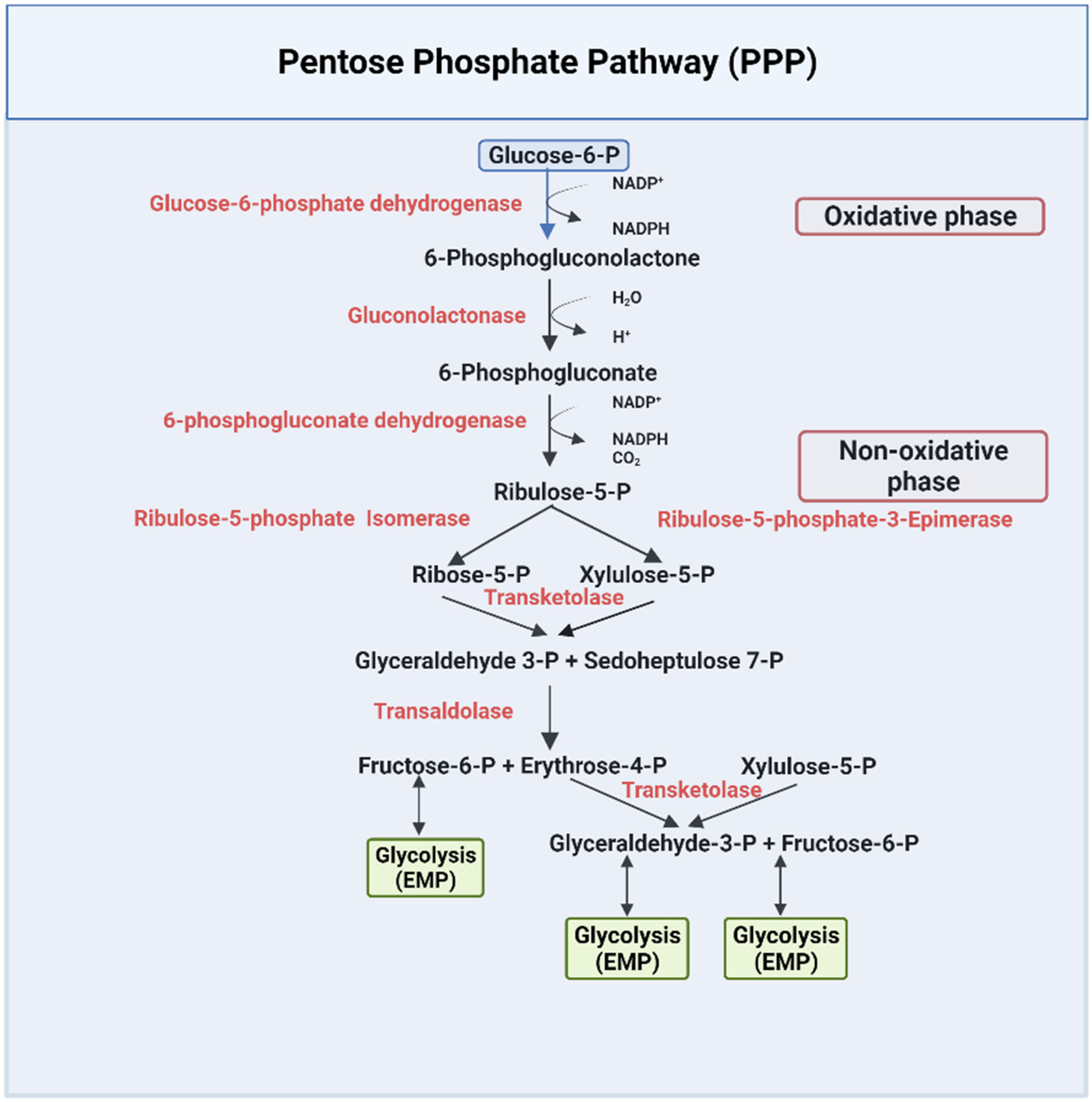
2.2. The Pentose Phosphate Pathway (PPP)
3. Glucose Metabolism and Pathophysiology of SCD
3.1. SCD and the Glucose Flux Switch toward Glycolysis Relative to the Pentose Phosphate Pathway
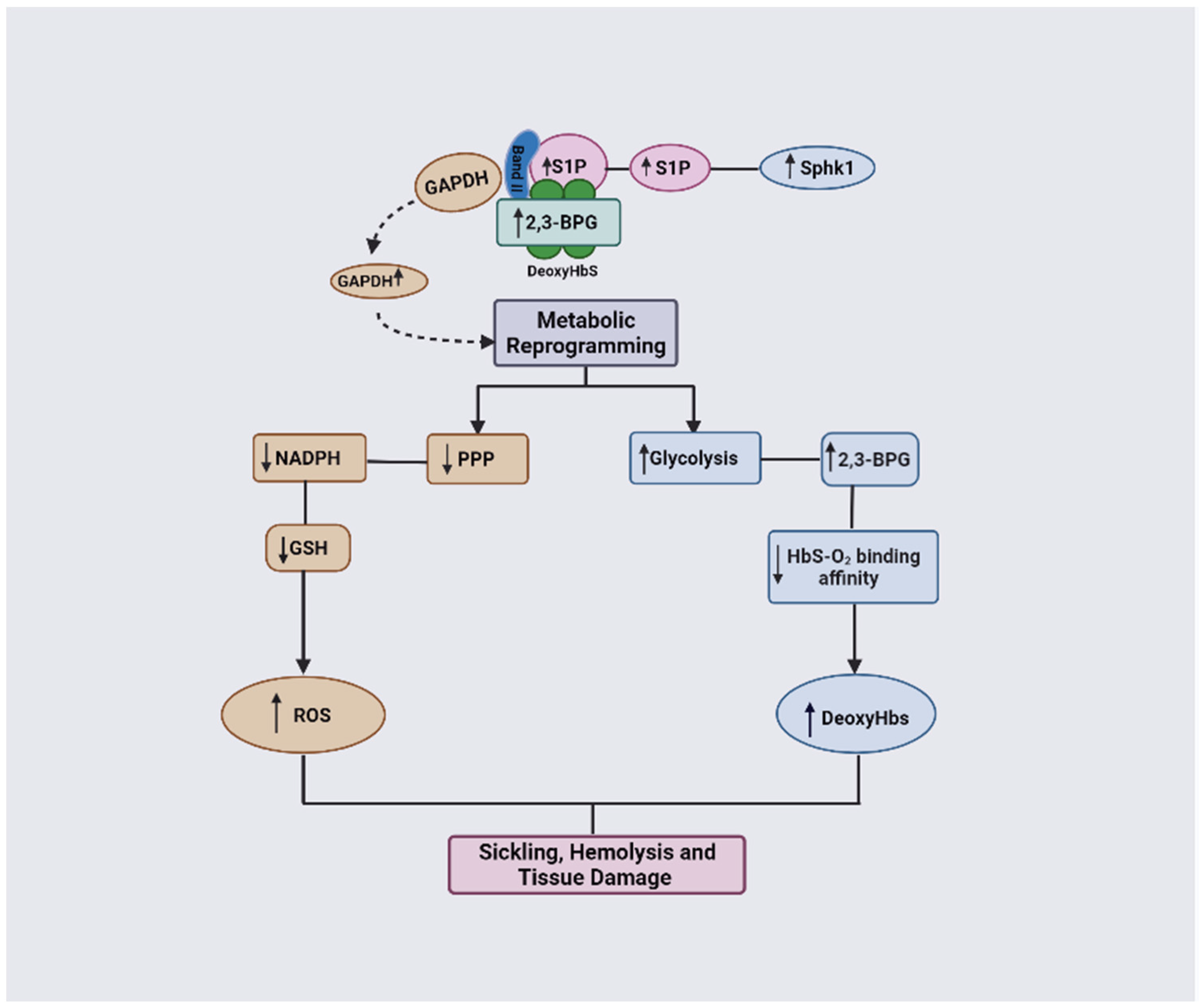
3.2. The Role of 2,3-BPG in SCD Pathophysiology
3.3. The Combinatorial Role of 2,3-BPG and S1P in SCD Pathophysiology
4. Drug Discovery Opportunities and Challenges
4.1. Glycolytic Enzymes
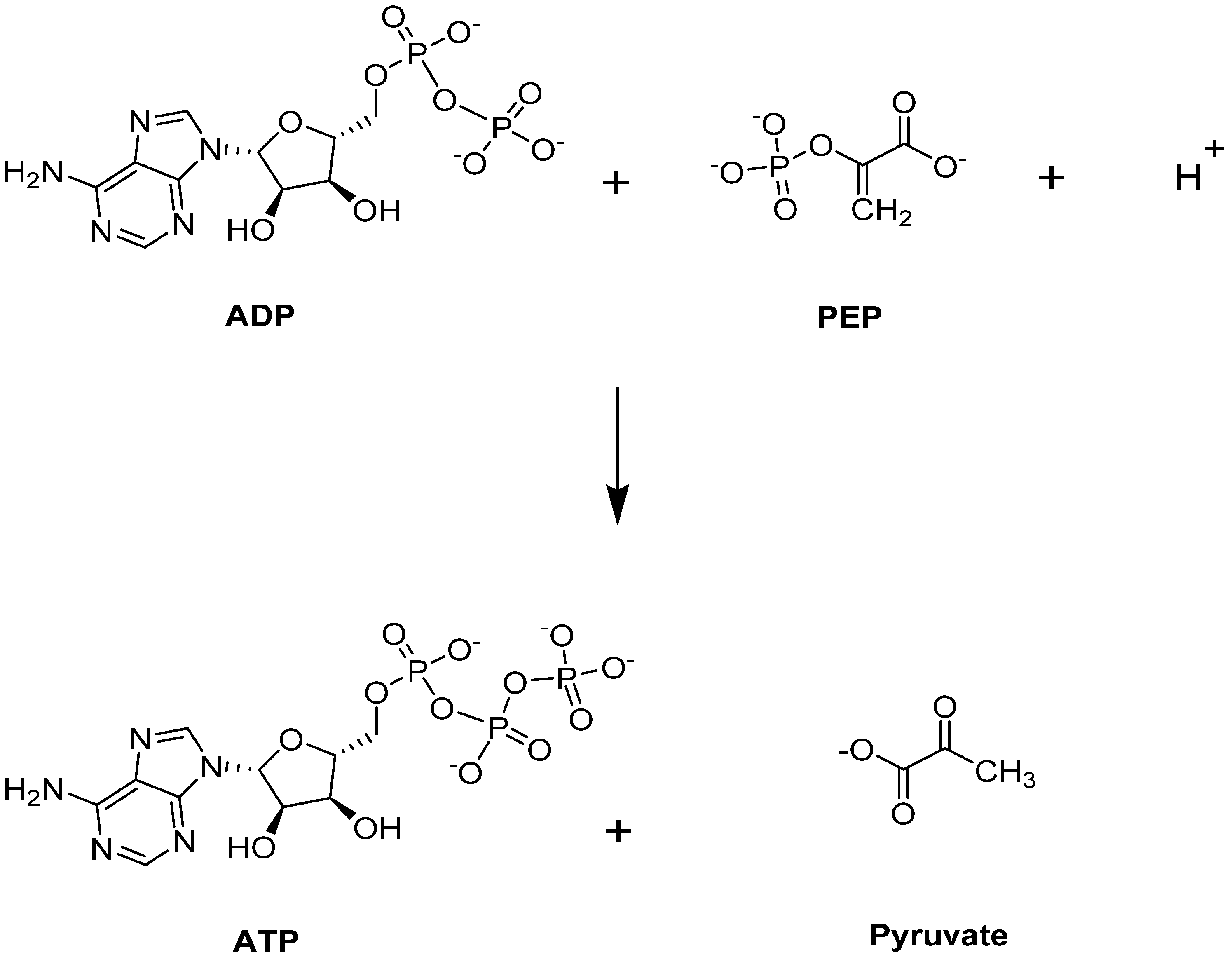
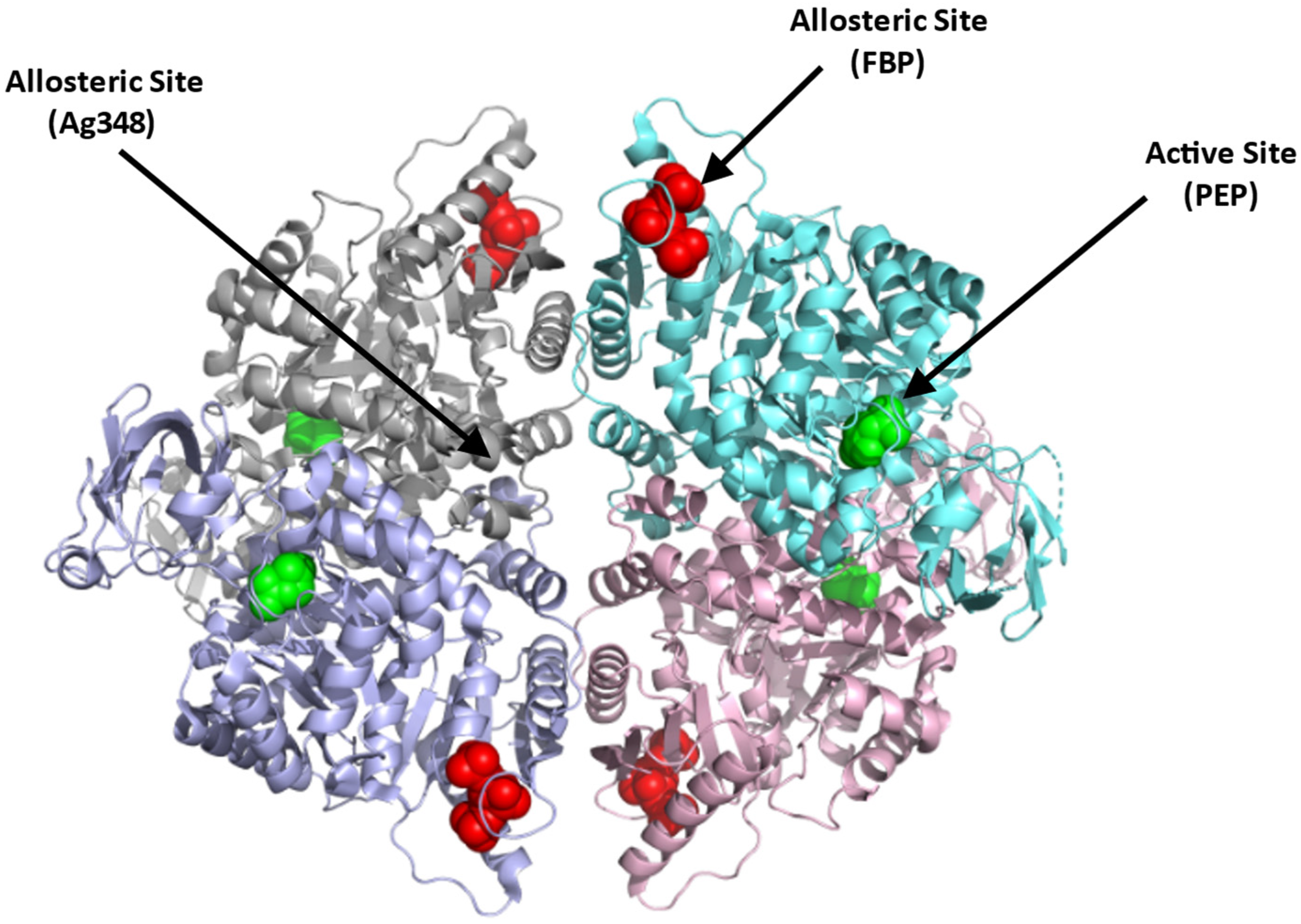
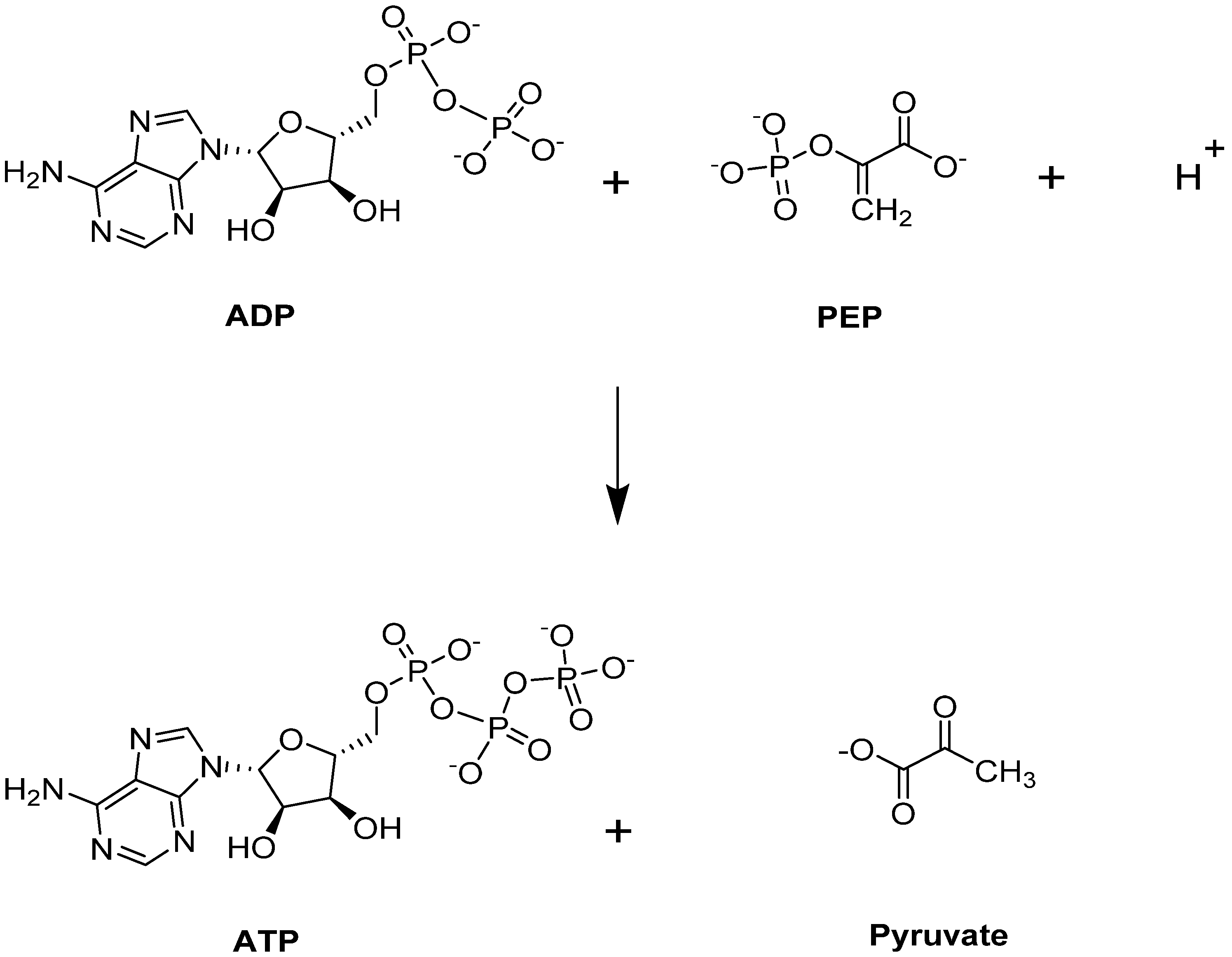
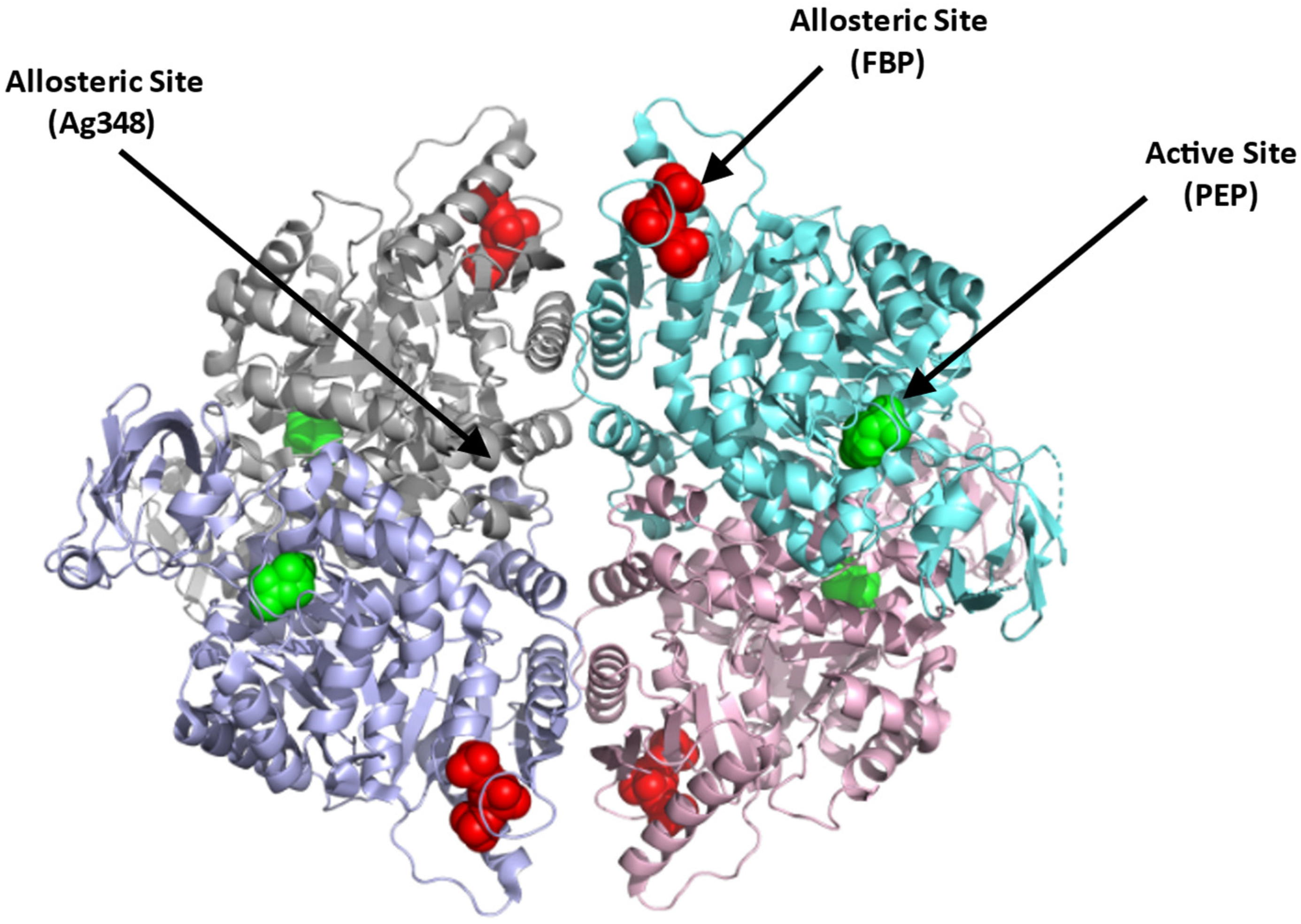
4.1.1. Erythrocyte Pyruvate Kinase (PKR)
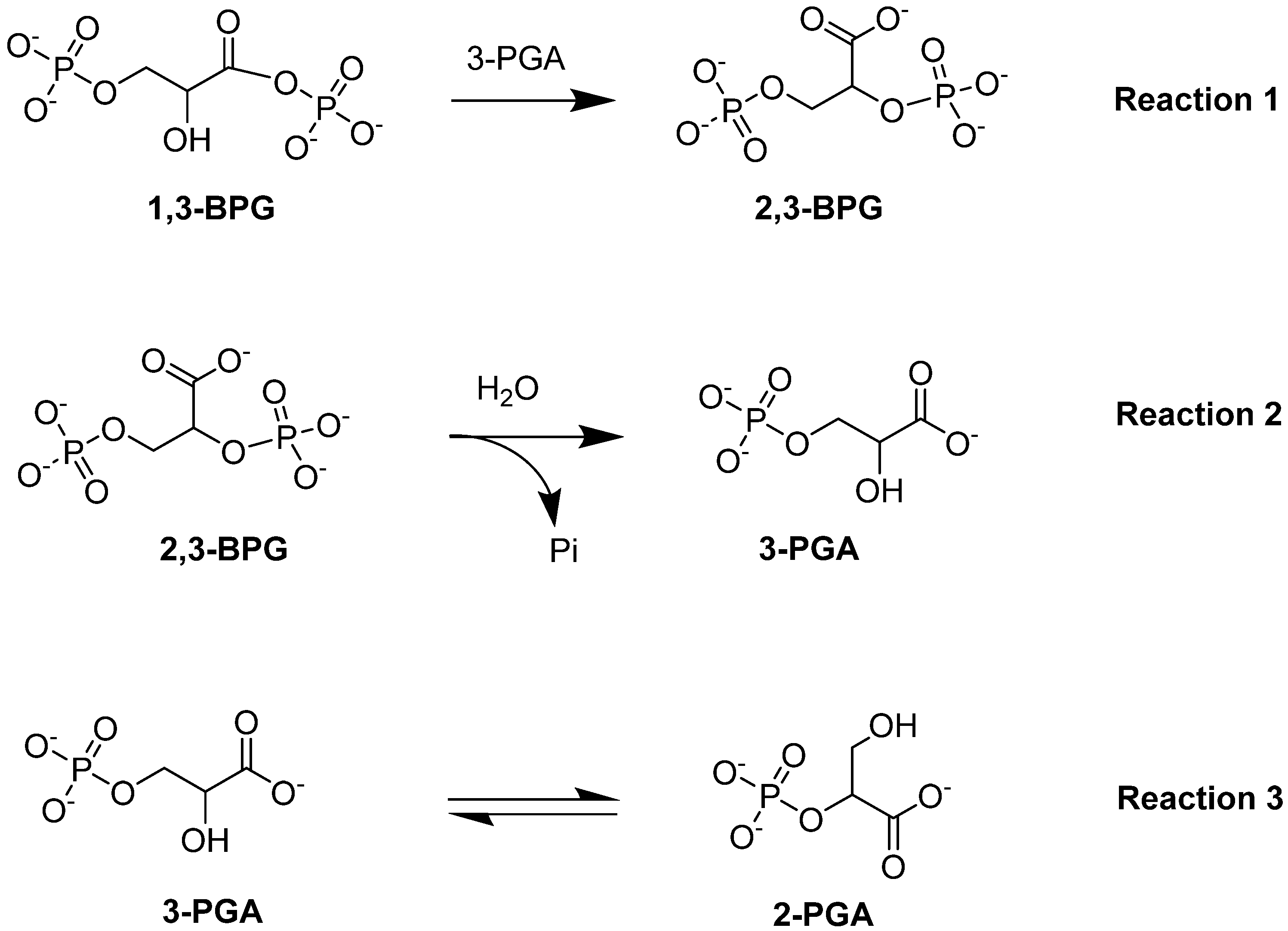
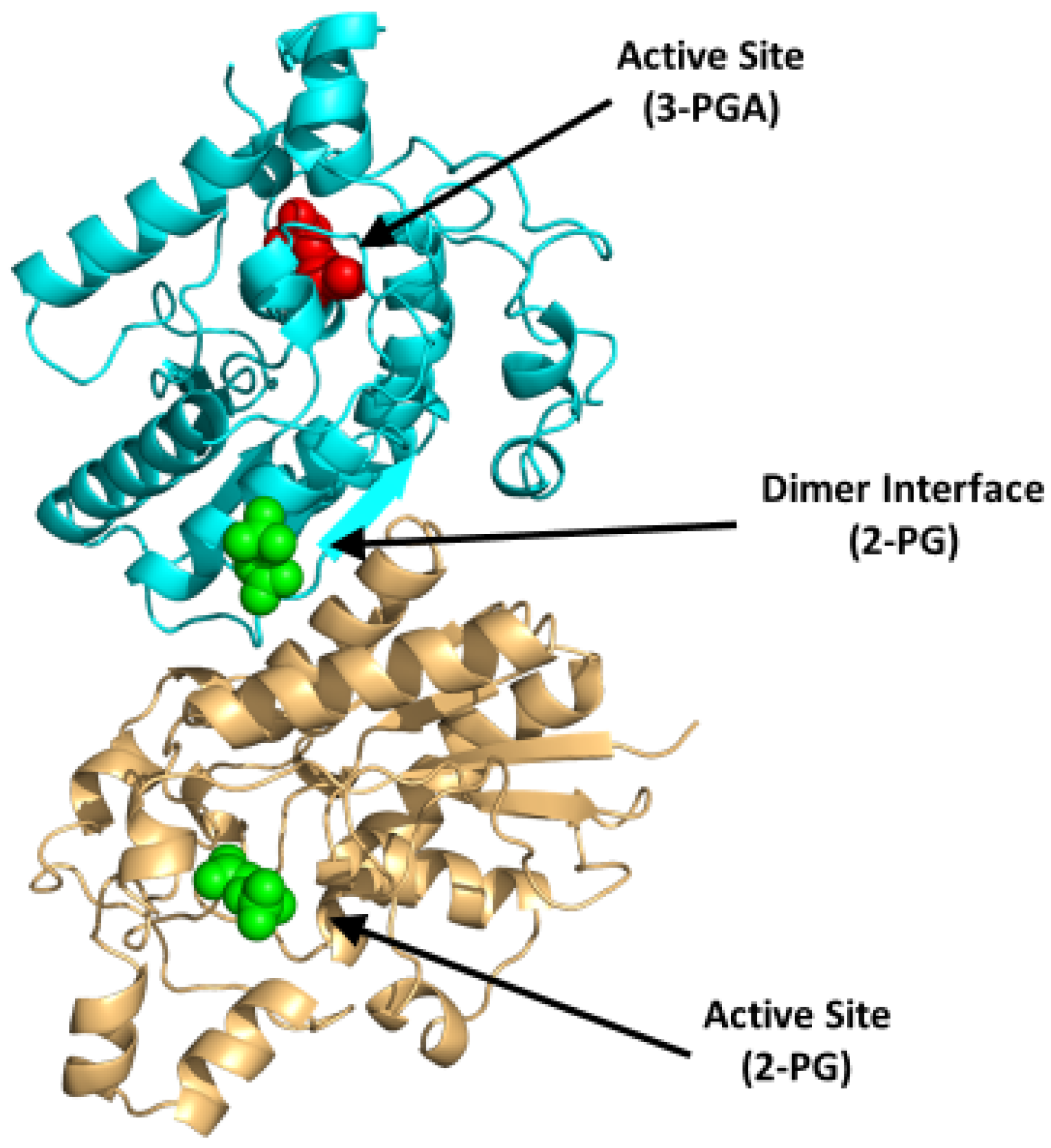
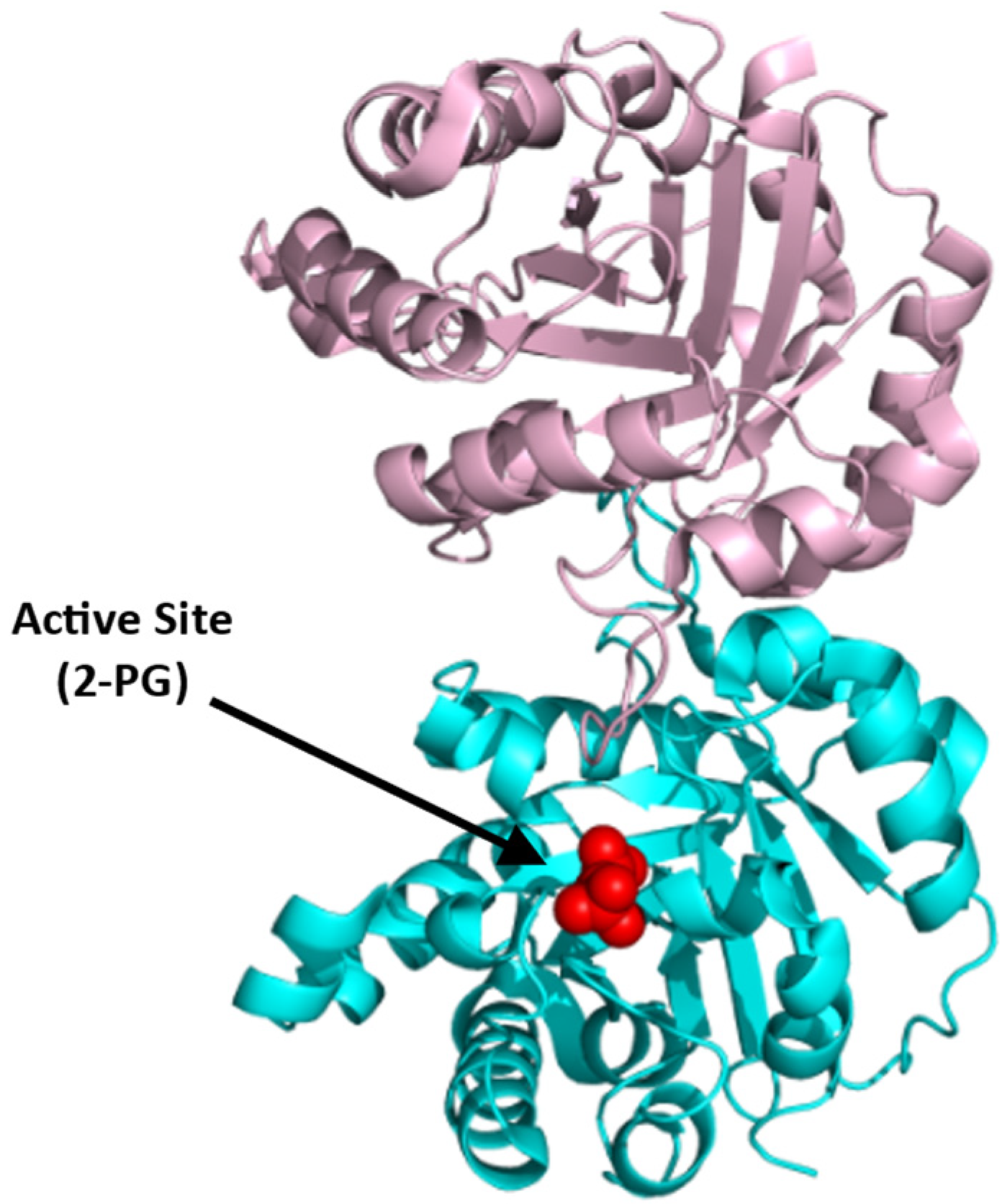
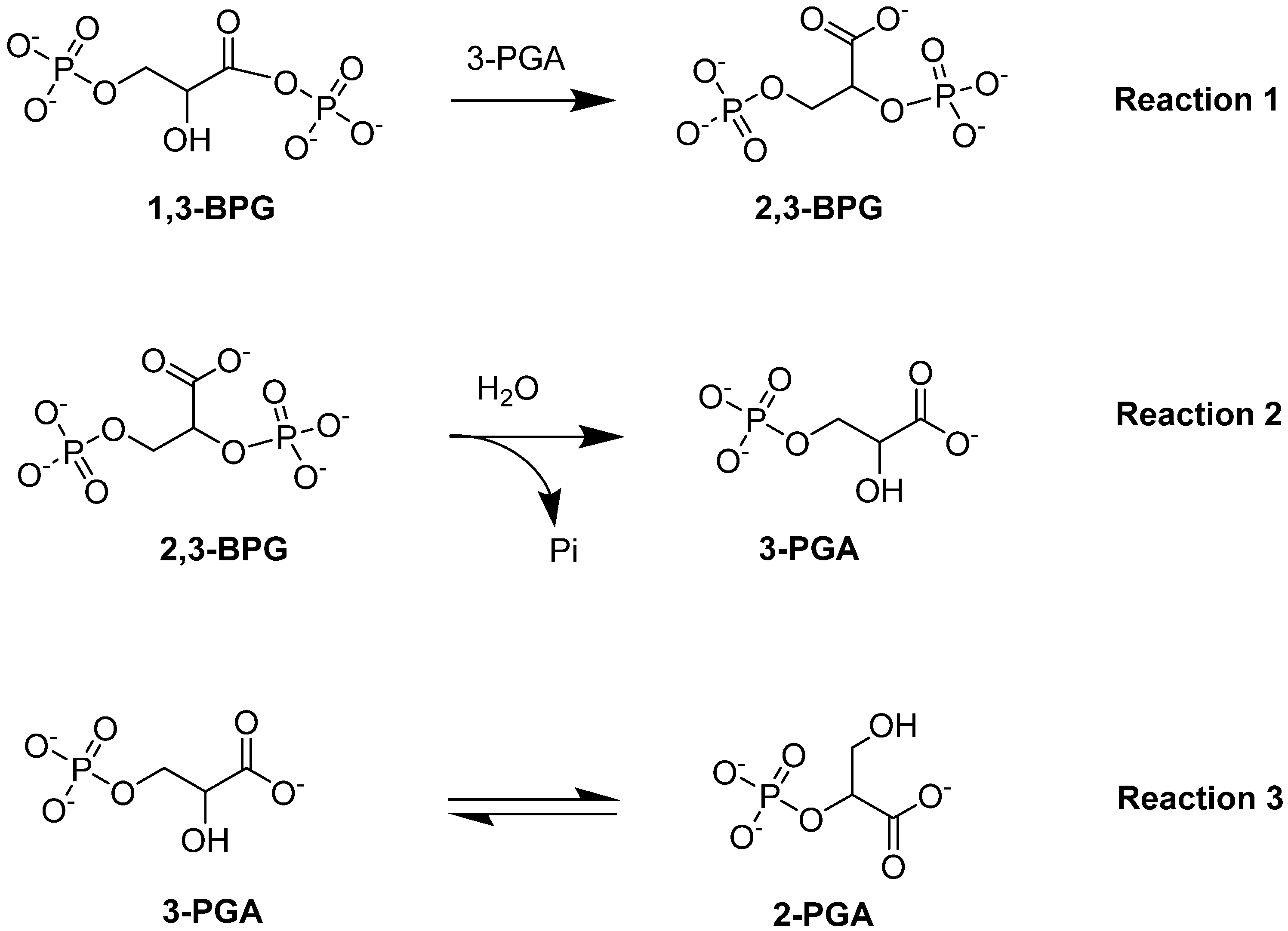
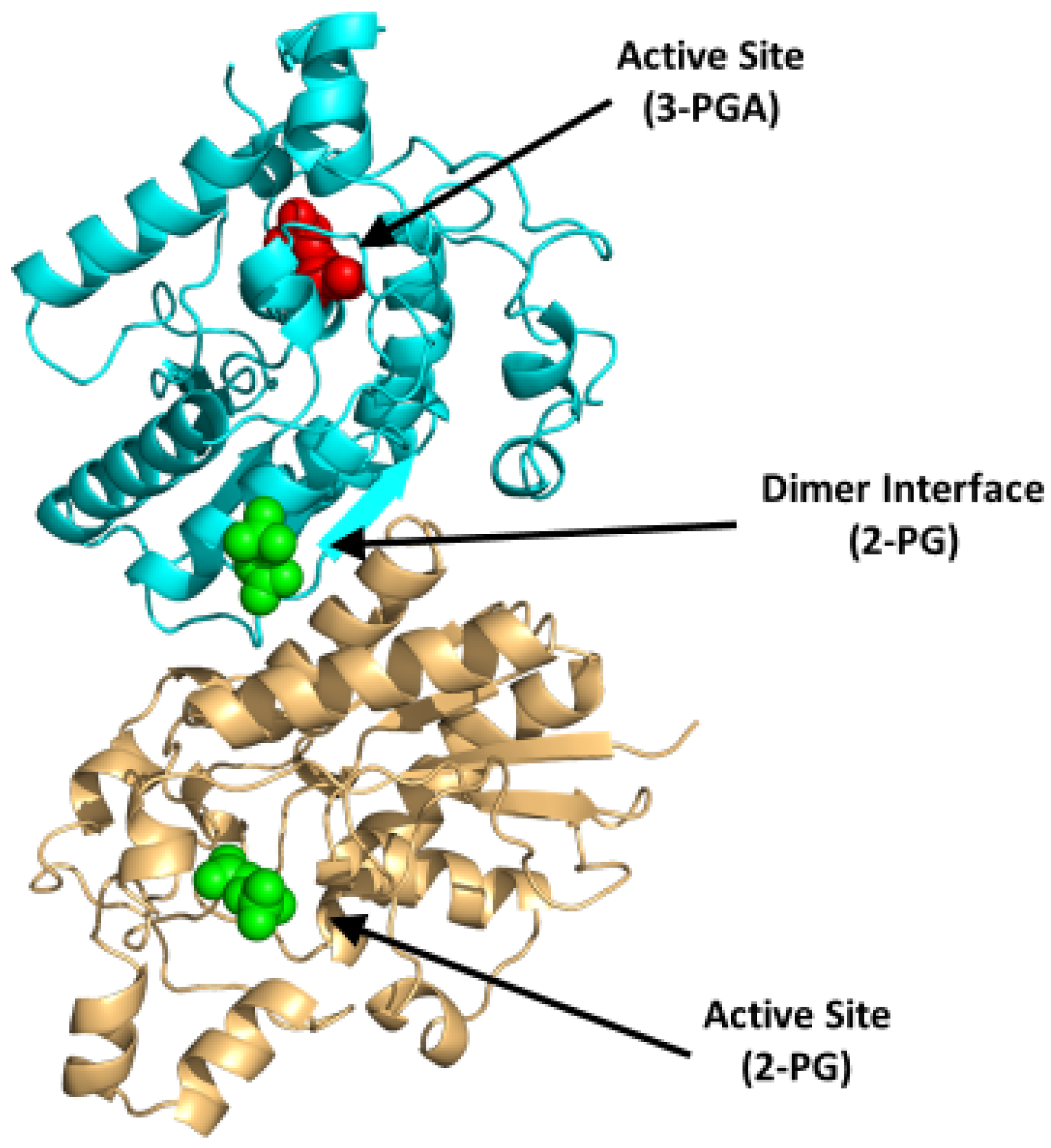
4.1.2. Bisphosphoglycerate Mutase (BPGM)
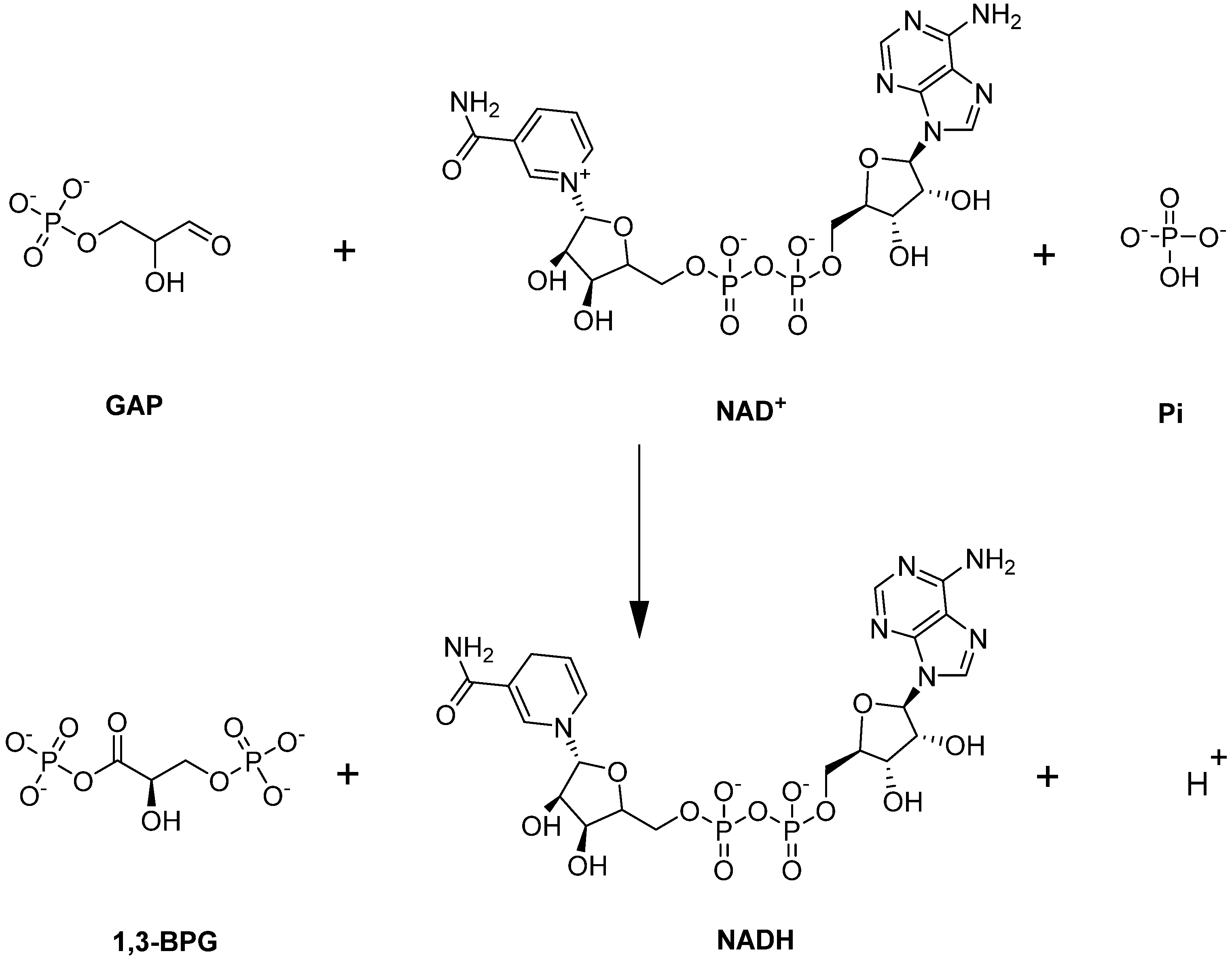
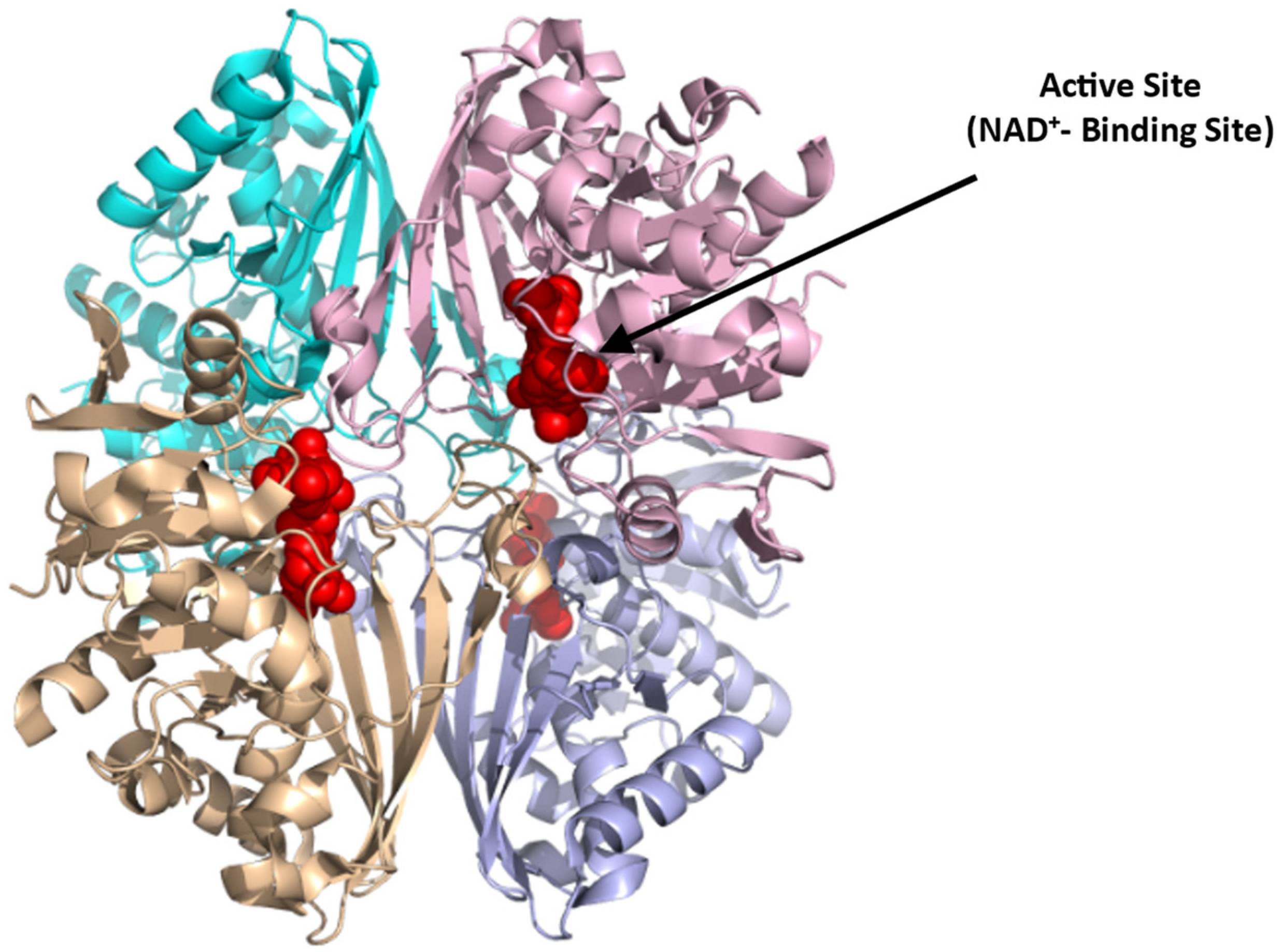
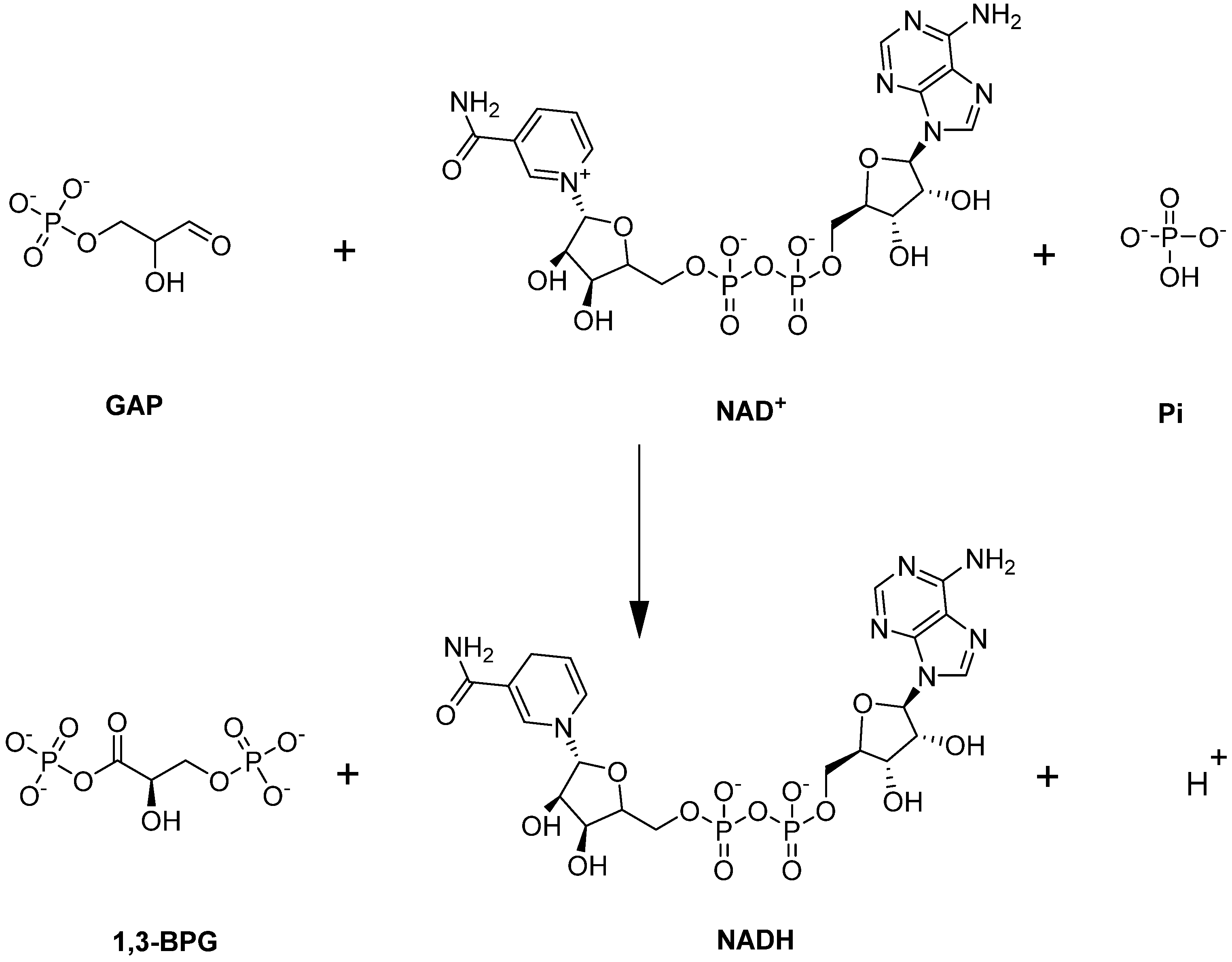
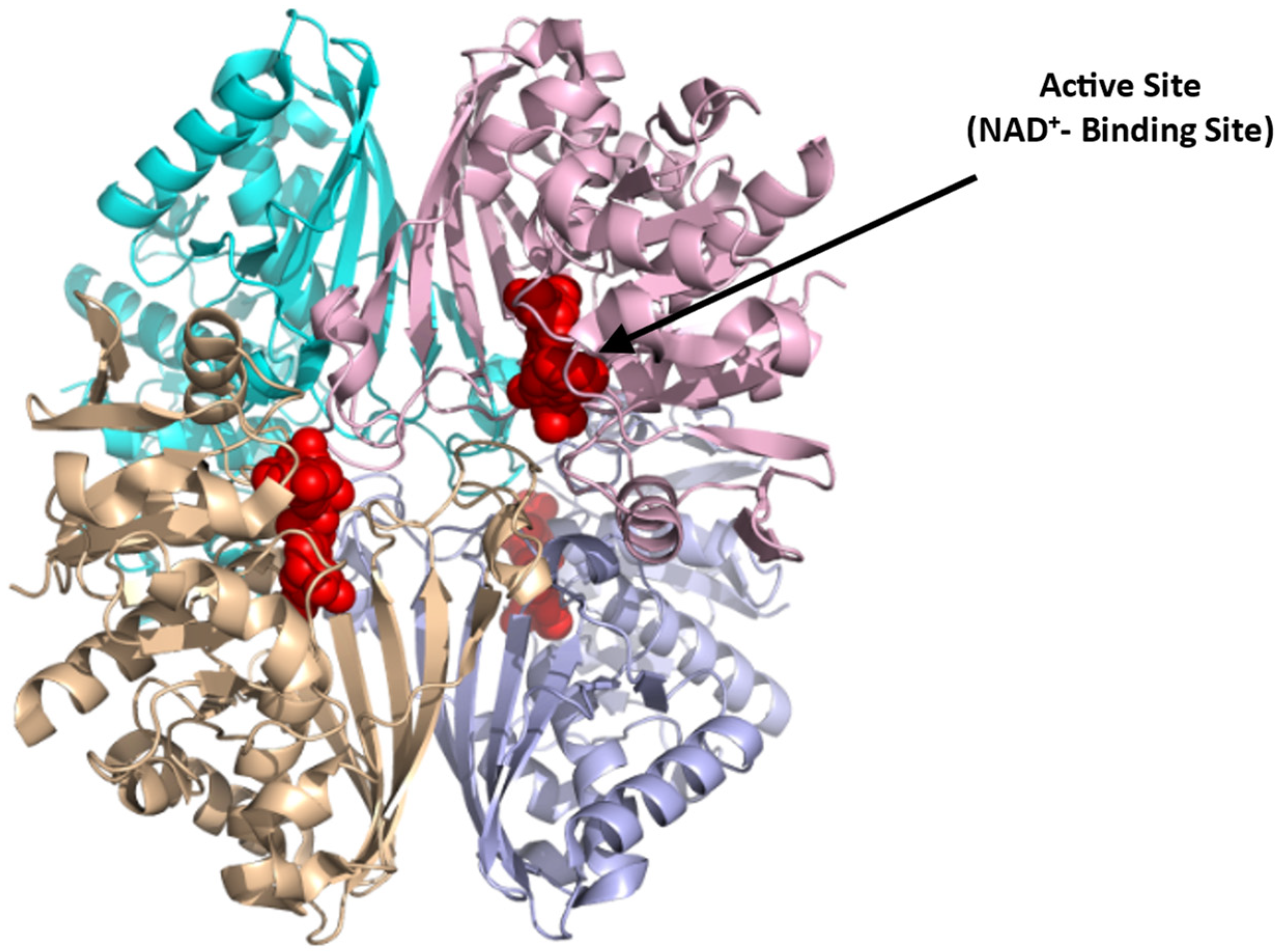
4.1.3. Glyceraldehyde-3-phosphate Dehydrogenase (GAPDH)
4.1.4. Triosephosphate Isomerase (TPI)
4.2. PPP Enzymes
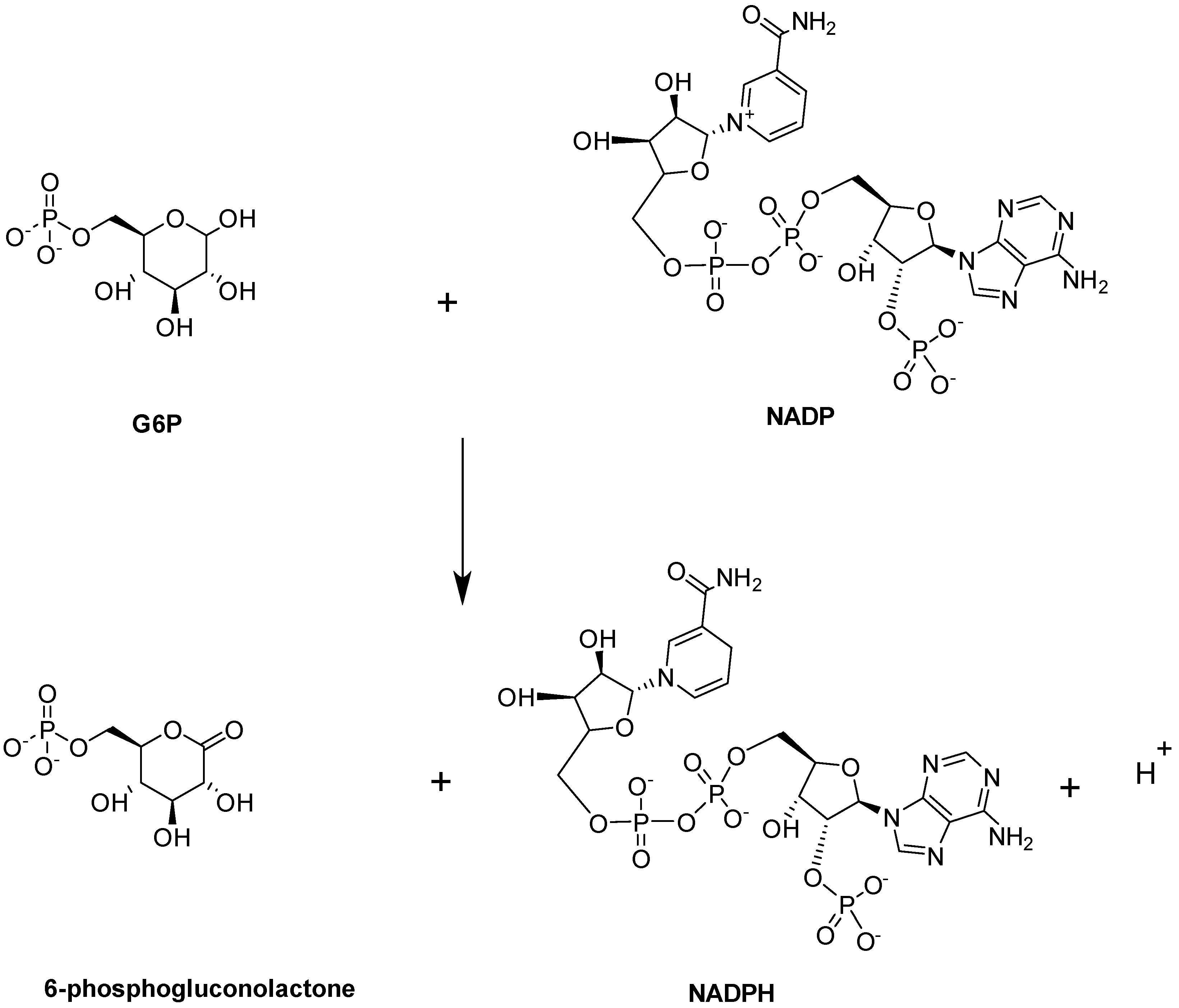
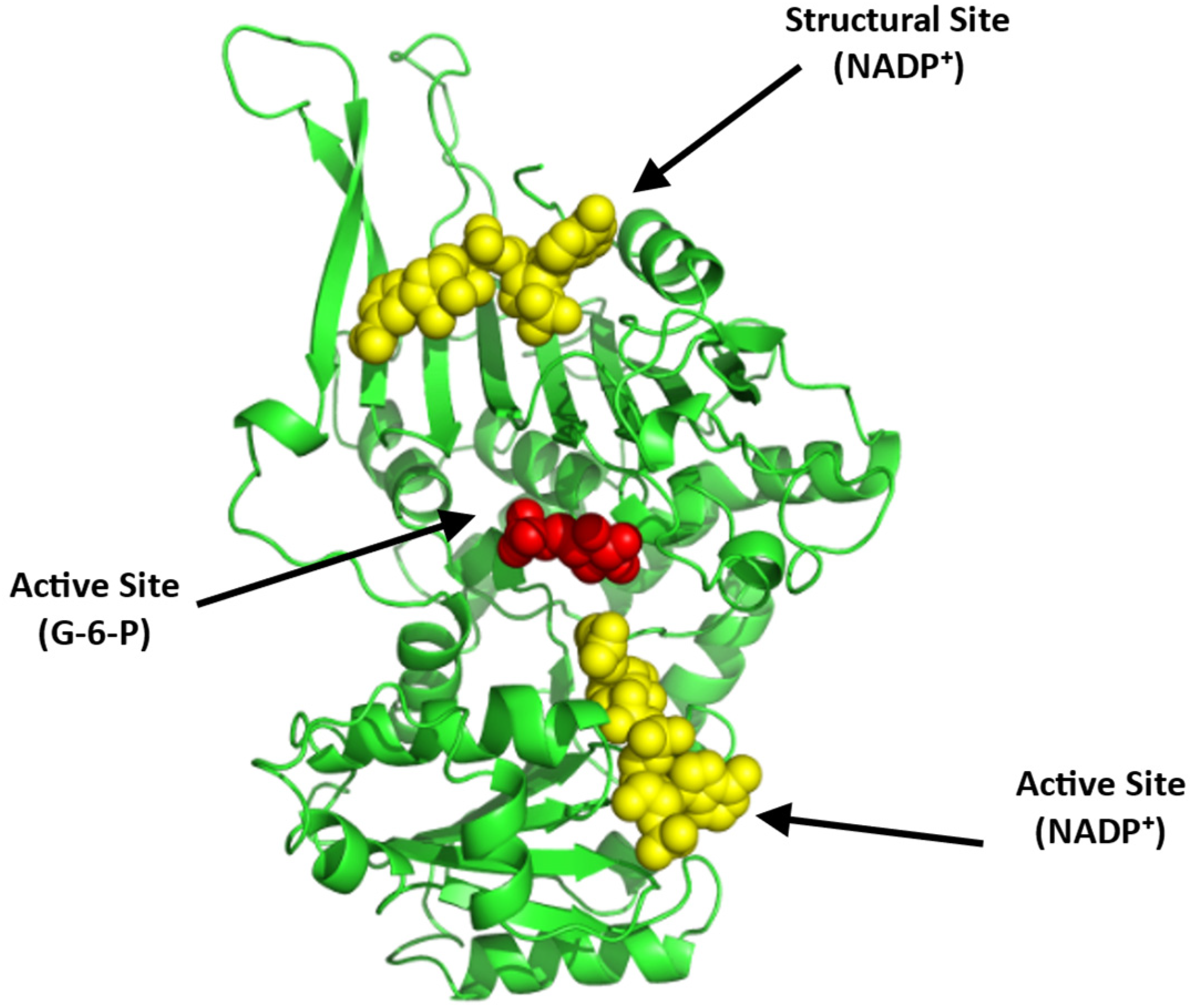
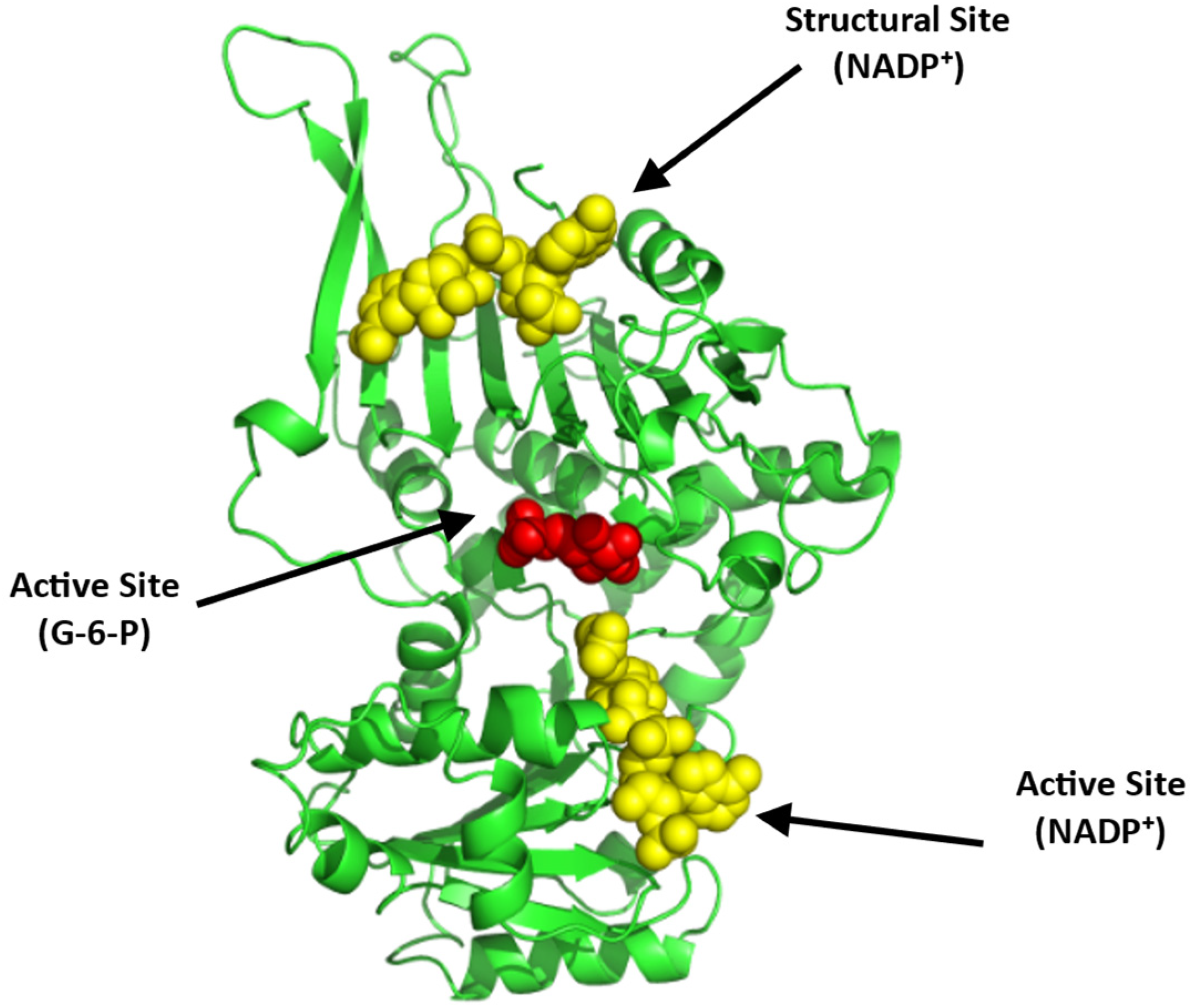
Glucose-6-phosphate Dehydrogenase (G6PD)
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Piel, F.B.; Hay, S.I.; Gupta, S.; Weatherall, D.J.; Williams, T.N. Global Burden of Sickle Cell Anaemia in Children under Five, 2010-2050: Modelling Based on Demographics, Excess Mortality, and Interventions. PLoS Med. 2013, 10, e1001484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Safo, M.K.; Aljahdali, A.; Burnett, J.; Abraham, D.J.; Abdulmalik, O. Therapeutic Strategies for the Treatment of Sickle Cell Disease. In Burger’s Medicinal Chemistry and Drug Discovery; American Cancer Society: Atlanta, GE, USA, 2021; pp. 1–31. [Google Scholar]
- Shah, F.; Dwivedi, M. Pathophysiology and Recent Therapeutic Insights of Sickle Cell Disease. Ann. Hematol. 2020, 99, 925–935. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, M.H.; Ghatge, M.S.; Safo, M.K. Hemoglobin: Structure, Function and Allostery. Subcell. Biochem. 2020, 94, 345–382. [Google Scholar] [CrossRef] [PubMed]
- Kister, J.; Kiger, L.; Francina, A.; Hanny, P.; Szymanowicz, A.; Blouquit, Y.; Promeé, D.; Galactéros, F.; Delaunay, J.; Wajcman, H. Hemoglobin Roanne [A94(G1) Asp å Glu]: A Variant of the A1β2 Interface with an Unexpected High Oxygen Affinity. Biochim. Biophys. Acta BBA-Protein Struct. Mol. Enzymol. 1995, 1246, 34–38. [Google Scholar] [CrossRef]
- Thom, C.S.; Dickson, C.F.; Gell, D.A.; Weiss, M.J. Hemoglobin Variants: Biochemical Properties and Clinical Correlates. Cold Spring Harb. Perspect. Med. 2013, 3, a011858. [Google Scholar] [CrossRef] [Green Version]
- Bunn, H.F. Pathogenesis and Treatment of Sickle Cell Disease. N. Engl. J. Med. 1997, 337, 762–769. [Google Scholar] [CrossRef]
- Ghatge, M.S.; Ahmed, M.H.; Omar, A.S.M.; Pagare, P.P.; Rosef, S.; Kellogg, G.E.; Abdulmalik, O.; Safo, M.K. Crystal Structure of Carbonmonoxy Sickle Hemoglobin in R-State Conformation. J. Struct. Biol. 2016, 194, 446–450. [Google Scholar] [CrossRef] [Green Version]
- Benesch, R.E.; Kwong, S.; Edalji, R.; Benesch, R. Alpha Chain Mutations with Opposite Effects on the Gelation of Hemoglobin S-PubMed. J. Biol. Chem. 1979, 254, 8169–8172. [Google Scholar] [CrossRef]
- Rhoda, M.D.; Martin, J.; Blouquit, Y.; Garel, M.C.; Edelstein, S.J.; Rosa, J. Sickle Cell Hemoglobin Fiber Formation Strongly Inhibited by the Stanleyville II Mutation (Alpha 78 Asn Leads to Lys). Biochem. Biophys. Res. Commun. 1983, 111, 8–13. [Google Scholar] [CrossRef]
- Cartier, A.; Hla, T. Sphingosine 1-Phosphate: Lipid Signaling in Pathology and Therapy. Science 2019, 366, eaar5551. [Google Scholar] [CrossRef]
- Sun, K.; D’alessandro, A.; Ahmed, M.H.; Zhang, Y.; Song, A.; Ko, T.-P.; Nemkov, T.; Reisz, J.A.; Wu, H.; Adebiyi, M.; et al. Structural and Functional Insight of Sphingosine 1-Phosphate-Mediated Pathogenic Metabolic Reprogramming in Sickle Cell Disease. Sci. Rep. 2017, 7, 15281. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Berka, V.; Song, A.; Sun, K.; Wang, W.; Zhang, W.; Ning, C.; Li, C.; Zhang, Q.; Bogdanov, M.; et al. Elevated Sphingosine-1-Phosphate Promotes Sickling and Sickle Cell Disease Progression. J. Clin. Investig. 2014, 124, 2750–2761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torrance, J.; Jacobs, P.; Restrepo, A.; Eschbach, J.; Lenfant, C.; Finch, C.A. Intraerythrocytic Adaptation to Anemia. N. Engl. J. Med. 1970, 283, 165–169. [Google Scholar] [CrossRef] [PubMed]
- Poillon, W.N.; Kim, B.C. 2,3-Diphosphoglycerate and Intracellular PH as Interdependent Determinants of the Physiologic Solubility of Deoxyhemoglobin S. Blood 1990, 76, 1028–1036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poillon, W.N.; Kim, B.C.; Welty, E.V.; Walder, J.A. The Effect of 2,3-Diphosphoglycerate on the Solubility of Deoxyhemoglobin S. Arch. Biochem. Biophys. 1986, 249, 301–305. [Google Scholar] [CrossRef]
- Poillon, W.; Kim, B.; Labotka, R.; Hicks, C.; Kark, J. Antisickling Effects of 2,3-Diphosphoglycerate Depletion. Blood 1995, 85, 3289–3296. [Google Scholar] [CrossRef]
- Safo, M.K.; Kato, G.J. Therapeutic Strategies to Alter the Oxygen Affinity of Sickle Hemoglobin. Hematol. Oncol. Clin. N. Am. 2014, 28, 217–231. [Google Scholar] [CrossRef] [Green Version]
- Aliyu, Z.Y.; Gordeuk, V.; Sachdev, V.; Babadoko, A.; Mamman, A.I.; Akpanpe, P.; Attah, E.; Suleiman, Y.; Aliyu, N.; Yusuf, J.; et al. Prevalence and Risk Factors for Pulmonary Artery Systolic Hypertension among Sickle Cell Disease Patients in Nigeria. Am. J. Hematol. 2008, 83, 485–490. [Google Scholar] [CrossRef] [Green Version]
- De Franceschi, L. Pathophisiology of Sickle Cell Disease and New Drugs for the Treatment. Mediterr. J. Hematol. Infect. Dis. 2009, 1, e2009024. [Google Scholar] [CrossRef]
- Akinsheye, I.; Klings, E.S. Sickle Cell Anemia and Vascular Dysfunction: The Nitric Oxide Connection. J. Cell. Physiol. 2010, 224, 620–625. [Google Scholar] [CrossRef]
- Platt, O.S. Hydroxyurea for the Treatment of Sickle Cell Anemia. N. Engl. J. Med. 2008, 358, 1362–1369. [Google Scholar] [CrossRef] [Green Version]
- Blair, H.A. Crizanlizumab: First Approval. Drugs 2020, 80, 79–84. [Google Scholar] [CrossRef] [PubMed]
- Metcalf, B.; Chuang, C.; Dufu, K.; Patel, M.P.; Silva-Garcia, A.; Johnson, C.; Lu, Q.; Partridge, J.R.; Patskovska, L.; Patskovsky, Y.; et al. Discovery of GBT440, an Orally Bioavailable R-State Stabilizer of Sickle Cell Hemoglobin. ACS Med. Chem. Lett. 2017, 8, 321–326. [Google Scholar] [CrossRef] [PubMed]
- Quinn, C.T. L-Glutamine for Sickle Cell Anemia: More Questions than Answers. Blood 2018, 132, 689–693. [Google Scholar] [CrossRef]
- Vichinsky, E.; Hoppe, C.C.; Ataga, K.I.; Ware, R.E.; Nduba, V.; El-Beshlawy, A.; Hassab, H.; Achebe, M.M.; Alkindi, S.; Brown, R.C.; et al. A Phase 3 Randomized Trial of Voxelotor in Sickle Cell Disease. N. Engl. J. Med. 2019, 381, 509–519. [Google Scholar] [CrossRef] [PubMed]
- Yenamandra, A.; Marjoncu, D. Voxelotor: A Hemoglobin S Polymerization Inhibitor for the Treatment of Sickle Cell Disease. J. Adv. Pract. Oncol. 2020, 11, 873–877. [Google Scholar] [CrossRef]
- Darbari, D.S.; Sheehan, V.A.; Ballas, S.K. The Vaso-Occlusive Pain Crisis in Sickle Cell Disease: Definition, Pathophysiology, and Management. Eur. J. Haematol. 2020, 105, 237–246. [Google Scholar] [CrossRef]
- McGann, P.T.; Ware, R.E. Hydroxyurea Therapy for Sickle Cell Anemia. Expert Opin. Drug Saf. 2015, 14, 1749–1758. [Google Scholar] [CrossRef] [Green Version]
- Brandow, A.M.; Panepinto, J.A. Hydroxyurea Use in Sickle Cell Disease: The Battle with Low Prescription Rates, Poor Patient Compliance and Fears of Toxicities. Expert Rev. Hematol. 2010, 3, 255–260. [Google Scholar] [CrossRef] [Green Version]
- Sinha, C.B.; Bakshi, N.; Ross, D.; Krishnamurti, L. From Trust to Skepticism: An in-Depth Analysis across Age Groups of Adults with Sickle Cell Disease on Their Perspectives Regarding Hydroxyurea. PLoS ONE 2018, 13, e0199375. [Google Scholar] [CrossRef]
- Angelucci, E.; Matthes-Martin, S.; Baronciani, D.; Bernaudin, F.; Bonanomi, S.; Cappellini, M.D.; Dalle, J.-H.; Di Bartolomeo, P.; de Heredia, C.D.; Dickerhoff, R.; et al. Hematopoietic Stem Cell Transplantation in Thalassemia Major and Sickle Cell Disease: Indications and Management Recommendations from an International Expert Panel. Haematologica 2014, 99, 811–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Talano, J.-A.; Cairo, M.S. Hematopoietic Stem Cell Transplantation for Sickle Cell Disease: State of the Science. Eur. J. Haematol. 2015, 94, 391–399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Demirci, S.; Uchida, N.; Tisdale, J.F. Gene Therapy for Sickle Cell Disease: An Update. Cytotherapy 2018, 20, 899–910. [Google Scholar] [CrossRef] [PubMed]
- Ribeil, J.-A.; Hacein-Bey-Abina, S.; Payen, E.; Magnani, A.; Semeraro, M.; Magrin, E.; Caccavelli, L.; Neven, B.; Bourget, P.; El Nemer, W.; et al. Gene Therapy in a Patient with Sickle Cell Disease. N. Engl. J. Med. 2017, 376, 848–855. [Google Scholar] [CrossRef]
- Tasan, I.; Jain, S.; Zhao, H. Use of Genome-Editing Tools to Treat Sickle Cell Disease. Hum. Genet. 2016, 135, 1011–1028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eaton, W.A.; Bunn, H.F. Treating Sickle Cell Disease by Targeting HbS Polymerization. Blood 2017, 129, 2719–2726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Alessandro, A.; Xia, Y. Erythrocyte Adaptive Metabolic Reprogramming under Physiological and Pathological Hypoxia. Curr. Opin. Hematol. 2020, 27, 155–162. [Google Scholar] [CrossRef]
- Grace, R.F.; Glader, B. Red Blood Cell Enzyme Disorders. Pediatr. Clin. N. Am. 2018, 65, 579–595. [Google Scholar] [CrossRef]
- van Wijk, R.; van Solinge, W.W. The Energy-Less Red Blood Cell Is Lost: Erythrocyte Enzyme Abnormalities of Glycolysis. Blood 2005, 106, 4034–4042. [Google Scholar] [CrossRef]
- Engelking, L.R. Chapter 24—Introduction to Glycolysis (The Embden-Meyerhoff Pathway (EMP)). In Textbook of Veterinary Physiological Chemistry, 3rd ed.; Engelking, L.R., Ed.; Academic Press: Boston, MA, USA, 2015; pp. 153–158. ISBN 978-0-12-391909-0. [Google Scholar]
- Chaudhry, R.; Varacallo, M. Biochemistry, Glycolysis. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Aziz, H.; Mohiuddin, S.S. Biochemistry, Hexose Monophosphate Pathway. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Huisjes, R.; Bogdanova, A.; van Solinge, W.W.; Schiffelers, R.M.; Kaestner, L.; van Wijk, R. Squeezing for Life—Properties of Red Blood Cell Deformability. Front. Physiol. 2018, 9, 656. [Google Scholar] [CrossRef]
- Kumari, A. Glycolysis. In Sweet Biochemistry; Academic Press: Cambridge, MA, USA, 2018; pp. 1–5. ISBN 978-0-12-814453-4. [Google Scholar]
- Martinov, M.V.; Plotnikov, A.G.; Vitvitsky, V.M.; Ataullakhanov, F.I. Deficiencies of Glycolytic Enzymes as a Possible Cause of Hemolytic Anemia. Biochim. Biophys. Acta BBA-Gen. Subj. 2000, 1474, 75–87. [Google Scholar] [CrossRef]
- Rapoport, S.; Luebering, J. The formation of 2,3-diphosphoglycerate in rabbit erythrocytes: The existence of a diphosphoglycerate mutase. J. Biol. Chem. 1950, 183, 507–516. [Google Scholar] [CrossRef]
- Alfarouk, K.O.; Ahmed, S.B.M.; Elliott, R.L.; Benoit, A.; Alqahtani, S.S.; Ibrahim, M.E.; Bashir, A.H.H.; Alhoufie, S.T.S.; Elhassan, G.O.; Wales, C.C.; et al. The Pentose Phosphate Pathway Dynamics in Cancer and Its Dependency on Intracellular PH. Metabolites 2020, 10, 285. [Google Scholar] [CrossRef] [PubMed]
- Pastore, A.; Piemonte, F.; Locatelli, M.; Lo Russo, A.; Gaeta, L.M.; Tozzi, G.; Federici, G. Determination of Blood Total, Reduced, and Oxidized Glutathione in Pediatric Subjects. Clin. Chem. 2001, 47, 1467–1469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pompella, A.; Visvikis, A.; Paolicchi, A.; Tata, V.D.; Casini, A.F. The Changing Faces of Glutathione, a Cellular Protagonist. Biochem. Pharmacol. 2003, 66, 1499–1503. [Google Scholar] [CrossRef]
- Chang, J.C.; van der Hoeven, L.H.; Haddox, C.H. Glutathione Reductase in the Red Blood Cells. Ann. Clin. Lab. Sci. 1978, 8, 23–29. [Google Scholar]
- Kuhn, V.; Diederich, L.; Keller, T.C.S.; Kramer, C.M.; Lückstädt, W.; Panknin, C.; Suvorava, T.; Isakson, B.E.; Kelm, M.; Cortese-Krott, M.M. Red Blood Cell Function and Dysfunction: Redox Regulation, Nitric Oxide Metabolism, Anemia. Antioxid. Redox Signal. 2017, 26, 718–742. [Google Scholar] [CrossRef] [Green Version]
- Rogers, S.C.; Said, A.; Corcuera, D.; McLaughlin, D.; Kell, P.; Doctor, A. Hypoxia Limits Antioxidant Capacity in Red Blood Cells by Altering Glycolytic Pathway Dominance. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2009, 23, 3159–3170. [Google Scholar] [CrossRef] [Green Version]
- Chu, H.; McKenna, M.M.; Krump, N.A.; Zheng, S.; Mendelsohn, L.; Thein, S.L.; Garrett, L.J.; Bodine, D.M.; Low, P.S. Reversible Binding of Hemoglobin to Band 3 Constitutes the Molecular Switch That Mediates O2 Regulation of Erythrocyte Properties. Blood 2016, 128, 2708–2716. [Google Scholar] [CrossRef]
- Vona, R.; Sposi, N.M.; Mattia, L.; Gambardella, L.; Straface, E.; Pietraforte, D. Sickle Cell Disease: Role of Oxidative Stress and Antioxidant Therapy. Antioxidants 2021, 10, 296. [Google Scholar] [CrossRef]
- Silva, D.G.H.; Belini Junior, E.; de Almeida, E.A.; Bonini-Domingos, C.R. Oxidative Stress in Sickle Cell Disease: An Overview of Erythrocyte Redox Metabolism and Current Antioxidant Therapeutic Strategies. Free Radic. Biol. Med. 2013, 65, 1101–1109. [Google Scholar] [CrossRef] [PubMed]
- Kruger, N.J.; von Schaewen, A. The Oxidative Pentose Phosphate Pathway: Structure and Organisation. Curr. Opin. Plant Biol. 2003, 6, 236–246. [Google Scholar] [CrossRef]
- Safo, M.K.; Bruno, S. Allosteric Effectors of Hemoglobin: Past, Present and Future. In Chemistry and Biochemistry of Oxygen Therapeutics; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2011; pp. 285–300. ISBN 978-1-119-97542-7. [Google Scholar]
- Grasso, J.A.; Sullivan, A.L.; Sullivan, L.W. Ultrastructural Studies of the Bone Marrow in Sickle Cell Anaemia. II. The Morphology of Erythropoietic Cells and Their Response to Deoxygenation in Vitro. Br. J. Haematol. 1975, 31, 381–389. [Google Scholar] [CrossRef]
- Connor, J.; Pak, C.C.; Schroit, A.J. Exposure of Phosphatidylserine in the Outer Leaflet of Human Red Blood Cells. Relationship to Cell Density, Cell Age, and Clearance by Mononuclear Cells. J. Biol. Chem. 1994, 269, 2399–2404. [Google Scholar] [CrossRef]
- Cohen-Solal, M.; Préhu, C.; Wajcman, H.; Poyart, C.; Bardakdjian-Michau, J.; Kister, J.; Promé, D.; Valentin, C.; BAchir, D.; Galactéros, F. A New Sickle Cell Disease Phenotype Associating Hb S Trait, Severe Pyruvate Kinase Deficiency (PK Conakry), and an A2 Globin Gene Variant (Hb Conakry): A New Sickle Cell Disease Phenotype. Br. J. Haematol. 1998, 103, 950–956. [Google Scholar] [CrossRef]
- Alli, N.; Coetzee, M.; Louw, V.; van Rensburg, B.; Rossouw, G.; Thompson, L.; Pissard, S.; Thein, S.L. Sickle Cell Disease in a Carrier with Pyruvate Kinase Deficiency. Hematology 2008, 13, 369–372. [Google Scholar] [CrossRef]
- Gupta, V.; Bamezai, R.N.K. Human Pyruvate Kinase M2: A Multifunctional Protein: Multifunctional Human PKM2. Protein Sci. 2010, 19, 2031–2044. [Google Scholar] [CrossRef] [Green Version]
- Grace, R.F.; Zanella, A.; Neufeld, E.J.; Morton, D.H.; Eber, S.; Yaish, H.; Glader, B. Erythrocyte Pyruvate Kinase Deficiency: 2015 Status Report. Am. J. Hematol. 2015, 90, 825–830. [Google Scholar] [CrossRef] [Green Version]
- Valentini, G.; Chiarelli, L.; Fortin, R.; Speranza, M.L.; Galizzi, A.; Mattevi, A. The Allosteric Regulation of Pyruvate Kinase. J. Biol. Chem. 2000, 275, 18145–18152. [Google Scholar] [CrossRef] [Green Version]
- Valentini, G.; Chiarelli, L.R.; Fortin, R.; Dolzan, M.; Galizzi, A.; Abraham, D.J.; Wang, C.; Bianchi, P.; Zanella, A.; Mattevi, A. Structure and Function of Human Erythrocyte Pyruvate Kinase. Molecular Basis of Nonspherocytic Hemolytic Anemia. J. Biol. Chem. 2002, 277, 23807–23814. [Google Scholar] [CrossRef] [Green Version]
- Nathan, D.G.; Oski, F.A.; Miller, D.R.; Gardner, F.H. Life-Span and Organ Sequestration of the Red Cells in Pyruvate Kinase Deficiency. N. Engl. J. Med. 1968, 278, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Mentzer, W.C.; Baehner, R.L.; Schmidt-Schönbein, H.; Robinson, S.H.; Nathan, D.G. Selective Reticulocyte Destruction in Erythrocyte Pyruvate Kinase Deficiency. J. Clin. Investig. 1971, 50, 688–699. [Google Scholar] [CrossRef]
- Glader, B.E. Salicylate-Induced Injury of Pyruvate-Kinase-Deficient Erythrocytes. N. Engl. J. Med. 1976, 294, 916–918. [Google Scholar] [CrossRef]
- Kung, C.; Hixon, J.; Kosinski, P.A.; Cianchetta, G.; Histen, G.; Chen, Y.; Hill, C.; Gross, S.; Si, Y.; Johnson, K.; et al. AG-348 Enhances Pyruvate Kinase Activity in Red Blood Cells from Patients with Pyruvate Kinase Deficiency. Blood 2017, 130, 1347–1356. [Google Scholar] [CrossRef] [PubMed]
- Kalfa, T.A.; Kuypers, F.A.; Telen, M.J.; Malik, P.; Konstantinidis, D.G.; Estepp, J.H.; Kim, H.J.; Saraf, S.L.; Wilson, L.; Ribadeneira, M.D.; et al. Phase 1 Single (SAD) and Multiple Ascending Dose (MAD) Studies of the Safety, Tolerability, Pharmacokinetics (PK) and Pharmacodynamics (PD) of FT-4202, an Allosteric Activator of Pyruvate Kinase-R, in Healthy and Sickle Cell Disease Subjects. Blood 2019, 134, 616. [Google Scholar] [CrossRef]
- McFarlane, J.S.; Ronnebaum, T.A.; Meneely, K.M.; Chilton, A.; Fenton, A.W.; Lamb, A.L. Changes in the Allosteric Site of Human Liver Pyruvate Kinase upon Activator Binding Include the Breakage of an Intersubunit Cation-π Bond. Acta Crystallogr. Sect. F Struct. Biol. Commun. 2019, 75, 461–469. [Google Scholar] [CrossRef] [Green Version]
- Pritlove, D.C.; Gu, M.; Boyd, C.A.R.; Randeva, H.S.; Vatish, M. Novel Placental Expression of 2,3-Bisphosphoglycerate Mutase. Placenta 2006, 27, 924–927. [Google Scholar] [CrossRef]
- Joulin, V.; Garel, M.C.; Le Boulch, P.; Valentin, C.; Rosa, R.; Rosa, J.; Cohen-Solal, M. Isolation and Characterization of the Human 2,3-Bisphosphoglycerate Mutase Gene. J. Biol. Chem. 1988, 263, 15785–15790. [Google Scholar] [CrossRef]
- Aljahdali, A.S.; Musayev, F.N.; Burgner, J.W.; Ghatge, M.S.; Shekar, V.; Zhang, Y.; Omar, A.M.; Safo, M.K. Molecular Insight into 2-Phosphoglycolate Activation of the Phosphatase Activity of Bisphosphoglycerate Mutase. Acta Crystallogr. Sect. Struct. Biol. 2022, 78, 472–482. [Google Scholar] [CrossRef]
- Garel, M.-C.; Joulin, V.; Le Boulchf, P.; Calvin, M.-C.; Prehu, M.-O.; Arous, N.; Longinll, R.; Rosa, R.; Rosa, J.; Cohen-Solal, M. Human Bisphosphoglycerate Mutase. J. Biol. Chem. 1989, 264, 18966–18972. [Google Scholar] [CrossRef]
- Rose, Z.B.; Liebowitz, J. 2,3-Diphosphoglycerate Phosphatase from Human Erythrocytes. General Properties and Activation by Anions. J. Biol. Chem. 1970, 245, 3232–3241. [Google Scholar] [CrossRef]
- Wang, Y.; Wei, Z.; Bian, Q.; Cheng, Z.; Wan, M.; Liu, L.; Gong, W. Crystal Structure of Human Bisphosphoglycerate Mutase. J. Biol. Chem. 2004, 279, 39132–39138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reynolds, C.H. Activation of Human Erythrocyte 2,3-Bisphosphoglycerate Phosphatase at Physiological Concentrations of Substrate. Arch. Biochem. Biophys. 1986, 250, 106–111. [Google Scholar] [CrossRef]
- Knee, K.M.; Barakat, A.; Tomlinson, L.; Ramaiah, L.; Wenzel, Z.; Kapinos, B.; Ahn, Y.; Lintner, N.G.; Field, S.D.; Jasuja, R.; et al. Sickle Cell Disease Model Mice Lacking 2,3-Dpg Show Reduced RBC Sickling and Improvements in Markers of Hemolytic Anemia. Blood 2020, 136, 27–28. [Google Scholar] [CrossRef]
- Garel, M.C.; Arous, N.; Calvin, M.C.; Craescu, C.T.; Rosa, J.; Rosa, R. A Recombinant Bisphosphoglycerate Mutase Variant with Acid Phosphatase Homology Degrades 2,3-Diphosphoglycerate. Proc. Natl. Acad. Sci. USA 1994, 91, 3593–3597. [Google Scholar] [CrossRef] [Green Version]
- Tarze, A.; Deniaud, A.; Le Bras, M.; Maillier, E.; Molle, D.; Larochette, N.; Zamzami, N.; Jan, G.; Kroemer, G.; Brenner, C. GAPDH, a Novel Regulator of the pro-Apoptotic Mitochondrial Membrane Permeabilization. Oncogene 2007, 26, 2606–2620. [Google Scholar] [CrossRef] [Green Version]
- Muronetz, V.I.; Melnikova, A.K.; Barinova, K.V.; Schmalhausen, E.V. Inhibitors of Glyceraldehyde-3-Phosphate Dehydrogenase and Unexpected Effects of Its Reduced Activity. Biochem. Mosc. 2019, 84, 1268–1279. [Google Scholar] [CrossRef]
- Gizi, A.; Papassotiriou, I.; Apostolakou, F.; Lazaropoulou, C.; Papastamataki, M.; Kanavaki, I.; Kalotychou, V.; Goussetis, E.; Kattamis, A.; Rombos, I.; et al. Assessment of Oxidative Stress in Patients with Sickle Cell Disease: The Glutathione System and the Oxidant–Antioxidant Status. Blood Cells. Mol. Dis. 2011, 46, 220–225. [Google Scholar] [CrossRef]
- Jenkins, J.L.; Tanner, J.J. IUCr High-Resolution Structure of Human d-Glyceraldehyde-3-Phosphate Dehydrogenase. Acta Crystallogr. Sect. D Biol. Crystallogr. 2006, 62, 290–301. [Google Scholar] [CrossRef]
- Li, T.; Tan, X.; Yang, R.; Miao, Y.; Zhang, M.; Xi, Y.; Guo, R.; Zheng, M.; Li, B. Discovery of Novel Glyceraldehyde-3-Phosphate Dehydrogenase Inhibitor via Docking-Based Virtual Screening. Bioorganic Chem. 2020, 96, 103620. [Google Scholar] [CrossRef]
- Verlinde, C.L.; Callens, M.; Van Calenbergh, S.; Van Aerschot, A.; Herdewijn, P.; Hannaert, V.; Michels, P.A.; Opperdoes, F.R.; Hol, W.G. Selective Inhibition of Trypanosomal Glyceraldehyde-3-Phosphate Dehydrogenase by Protein Structure-Based Design: Toward New Drugs for the Treatment of Sleeping Sickness. J. Med. Chem. 1994, 37, 3605–3613. [Google Scholar] [CrossRef] [PubMed]
- Liberti, M.V.; Dai, Z.; Wardell, S.E.; Baccile, J.A.; Liu, X.; Gao, X.; Baldi, R.; Mehrmohamadi, M.; Johnson, M.O.; Madhukar, N.S.; et al. A Predictive Model for Selective Targeting of the Warburg Effect through GAPDH Inhibition with a Natural Product. Cell Metab. 2017, 26, 648–659.e8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ganapathy-Kanniappan, S. Evolution of GAPDH as a Druggable Target of Tumor Glycolysis? Expert Opin. Ther. Targets 2018, 22, 295–298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orosz, F.; Oláh, J.; Ovádi, J. Triosephosphate Isomerase Deficiency: Facts and Doubts. IUBMB Life 2006, 58, 703–715. [Google Scholar] [CrossRef] [PubMed]
- Schneider, A.S. Triosephosphate Isomerase Deficiency: Historical Perspectives and Molecular Aspects. Best Pract. Res. Clin. Haematol. 2000, 13, 119–140. [Google Scholar] [CrossRef]
- Lo, T.W.; Westwood, M.E.; McLellan, A.C.; Selwood, T.; Thornalley, P.J. Binding and Modification of Proteins by Methylglyoxal under Physiological Conditions. A Kinetic and Mechanistic Study with N Alpha-Acetylarginine, N Alpha-Acetylcysteine, and N Alpha-Acetyllysine, and Bovine Serum Albumin. J. Biol. Chem. 1994, 269, 32299–32305. [Google Scholar] [CrossRef]
- Ralser, M.; Wamelink, M.M.; Kowald, A.; Gerisch, B.; Heeren, G.; Struys, E.A.; Klipp, E.; Jakobs, C.; Breitenbach, M.; Lehrach, H.; et al. Dynamic Rerouting of the Carbohydrate Flux Is Key to Counteracting Oxidative Stress. J. Biol. 2007, 6, 10. [Google Scholar] [CrossRef] [Green Version]
- Mande, S.C.; Mainfroid, V.; Kalk, K.H.; Goraj, K.; Martial, J.A.; Hol, W.G. Crystal Structure of Recombinant Human Triosephosphate Isomerase at 2.8 A Resolution. Triosephosphate Isomerase-Related Human Genetic Disorders and Comparison with the Trypanosomal Enzyme. Protein Sci. Publ. Protein Soc. 1994, 3, 810–821. [Google Scholar] [CrossRef] [Green Version]
- Marsh, L.; Shah, K. A Novel Inhibitor of Mammalian Triosephosphate Isomerase Found by an In Silico Approach. Int. J. Med. Chem. 2014, 2014, e469125. [Google Scholar] [CrossRef]
- Beltran-Hortelano, I.; Alcolea, V.; Font, M.; Pérez-Silanes, S. Examination of Multiple Trypanosoma Cruzi Targets in a New Drug Discovery Approach for Chagas Disease. Bioorg. Med. Chem. 2022, 58, 116577. [Google Scholar] [CrossRef]
- Mehta, A.; Mason, P.J.; Vulliamy, T.J. Glucose-6-Phosphate Dehydrogenase Deficiency. Best Pract. Res. Clin. Haematol. 2000, 13, 21–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nnamezie Igwilo, H.; Salawu, L.; Adeoye Adedeji, T. The Impact of Glucose-6-Phosphate Dehydrogenase Deficiency on the Frequency of Vasoocclusive Crisis in Patients with Sickle Cell Anemia. Plasmatology 2021, 15, 1–10. [Google Scholar] [CrossRef]
- Antwi-Baffour, S.; Adjei, J.K.; Forson, P.O.; Akakpo, S.; Kyeremeh, R.; Seidu, M.A. Comorbidity of Glucose-6-Phosphate Dehydrogenase Deficiency and Sickle Cell Disease Exert Significant Effect on RBC Indices. Anemia 2019, 2019, 3179173. [Google Scholar] [CrossRef] [PubMed]
- Kotaka, M.; Gover, S.; Vandeputte-Rutten, L.; Au, S.W.N.; Lam, V.M.S.; Adams, M.J. Structural Studies of Glucose-6-Phosphate and NADP+ Binding to Human Glucose-6-Phosphate Dehydrogenase. Acta Crystallogr. D Biol. Crystallogr. 2005, 61, 495–504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hwang, S.; Mruk, K.; Rahighi, S.; Raub, A.G.; Chen, C.-H.; Dorn, L.E.; Horikoshi, N.; Wakatsuki, S.; Chen, J.K.; Mochly-Rosen, D. Correcting Glucose-6-Phosphate Dehydrogenase Deficiency with a Small-Molecule Activator. Nat. Commun. 2018, 9, 4045. [Google Scholar] [CrossRef]
- Raub, A.G.; Hwang, S.; Horikoshi, N.; Cunningham, A.D.; Rahighi, S.; Wakatsuki, S.; Mochly-Rosen, D. Small-Molecule Activators of Glucose-6-Phosphate Dehydrogenase (G6PD) Bridging the Dimer Interface. ChemMedChem 2019, 14, 1321–1324. [Google Scholar] [CrossRef]
- Wood, K.C.; Hsu, L.L.; Gladwin, M.T. Sickle Cell Disease Vasculopathy: A State of Nitric Oxide Resistance. Free Radic. Biol. Med. 2008, 44, 1506–1528. [Google Scholar] [CrossRef]
- Sivilotti, M.L.A. Oxidant Stress and Haemolysis of the Human Erythrocyte. Toxicol. Rev. 2004, 23, 169–188. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alramadhani, D.; Aljahdali, A.S.; Abdulmalik, O.; Pierce, B.D.; Safo, M.K. Metabolic Reprogramming in Sickle Cell Diseases: Pathophysiology and Drug Discovery Opportunities. Int. J. Mol. Sci. 2022, 23, 7448. https://doi.org/10.3390/ijms23137448
Alramadhani D, Aljahdali AS, Abdulmalik O, Pierce BD, Safo MK. Metabolic Reprogramming in Sickle Cell Diseases: Pathophysiology and Drug Discovery Opportunities. International Journal of Molecular Sciences. 2022; 23(13):7448. https://doi.org/10.3390/ijms23137448
Chicago/Turabian StyleAlramadhani, Dina, Anfal S. Aljahdali, Osheiza Abdulmalik, B. Daniel Pierce, and Martin K. Safo. 2022. "Metabolic Reprogramming in Sickle Cell Diseases: Pathophysiology and Drug Discovery Opportunities" International Journal of Molecular Sciences 23, no. 13: 7448. https://doi.org/10.3390/ijms23137448
APA StyleAlramadhani, D., Aljahdali, A. S., Abdulmalik, O., Pierce, B. D., & Safo, M. K. (2022). Metabolic Reprogramming in Sickle Cell Diseases: Pathophysiology and Drug Discovery Opportunities. International Journal of Molecular Sciences, 23(13), 7448. https://doi.org/10.3390/ijms23137448