
Modelling Autism Spectrum Disorder (ASD) and Attention-Deficit/Hyperactivity Disorder (ADHD) Using Mice and Zebrafish



Abstract
1. Introduction
1.1. Overview and Epidemiology of Autism Spectrum Disorder (ASD)
1.1.1. Overview
- Difficulty communicating and interacting with others.
- Limited interests and repetitive behaviours.
- Troubles in functioning at school, work, and in society.
1.1.2. Prevalence
1.1.3. Signs and Symptoms of ASD
1.1.4. Causes and Risk Factors
1.1.5. ASD Diagnosis
1.1.6. Treatments of ASD
1.2. Overview and Epidemiology of Attention-Deficit/Hyperactivity Disorder (ADHD)
1.2.1. Overview
- Inattention: difficulty doing a task and staying focused and organized.
- Hyperactivity: moving constantly, including in inappropriate situations, or demonstrating excessive fidgets, taps, or talks. In adults, hyperactivity is manifested by extreme restlessness or talking too much.
- Impulsivity: acting without thinking or difficulty with self-control. Importantly, it can be manifested by a desire for immediate rewards or the incapacity to wait for gratification.
1.2.2. Prevalence
1.2.3. Signs and Symptoms of ADHD
1.2.4. Diagnosis of ADHD
1.2.5. Risk Factors
1.2.6. Treatment of ADHD
1.3. Key Differences, Similarities and Conditions That Can Be Mistaken for ASD or ADHD
1.4. Molecular Biology and Mechanisms Underlaying ASD and ADHD
1.4.1. ASD
1.4.2. ADHD
2. Current Behavioural Tests of ASD and ADHD in Research
- Strong similarity to human phenotype.
- Same biological phenomena that are responsible for the disease in humans.
- Similar response to potential treatments used in humans.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Disorders | Core Areas Affected | Behavioural Tests | |
|---|---|---|---|
| Mouse | Zebrafish | ||
| ASD | Socialization | Novel partner preference test/Social approach test [65,66,67,68] Reciprocal social interaction test [3] Juvenile play test [68] | Social preference test [69] Shoaling test [69] Social interaction test [70] |
| Nonsocial behaviours (repetitive behaviour, motor alterations and limited range of activities) | Self-grooming test [66,67,68] Repetitive novel object test [71,72] Open-field test [65,68] Social transmission of food preference [68] Predator avoidance test | Open field test [73,74,75,76,77] T-maze test [77] Predator avoidance test [78,79] | |
| Communication | Social transmission of food preference test [68] Impaired vocalization test [80,81] | Not available to date | |
| ADHD | Attention and learning deficits | Y-maze spontaneous alternation test [66] Barnes maze test [65,66] | Five-choice serial reaction time task (5-CSRTT) [82,83] T-maze test [77] Inhibition avoidance task [84] |
| Hyperactivity-Impulsivity | Open field test [66] | Open field test [74,75,76] T-maze test [77] Five-choice serial reaction time task (5-CSRTT) [82,83] Novel tank test [85,86,87] | |
| Aggressiveness | Resident–Intruder Paradigm [65,88,89] | Mirror test [69,70,90] | |
2.1. Behavioural Tests in Mice
2.1.1. The Social Approach Test
2.1.2. The Reciprocal Social Interaction Test
2.1.3. Juvenile Play
2.1.4. Repetitive Grooming Test
2.1.5. Repetitive Novel Object Test
2.1.6. The Social Transmission of Food Preference
2.1.7. The Resident–Intruder Paradigm
2.1.8. The Y-Maze Spontaneous Alternation Test
2.1.9. The Barnes Maze Test
2.1.10. The Impaired Vocalization Test
2.2. Behavioural Tests in Zebrafish
2.2.1. The Social Preference Test
2.2.2. Shoaling Test
2.2.3. Five-Choice Serial Reaction Time Task (5-CSRTT)
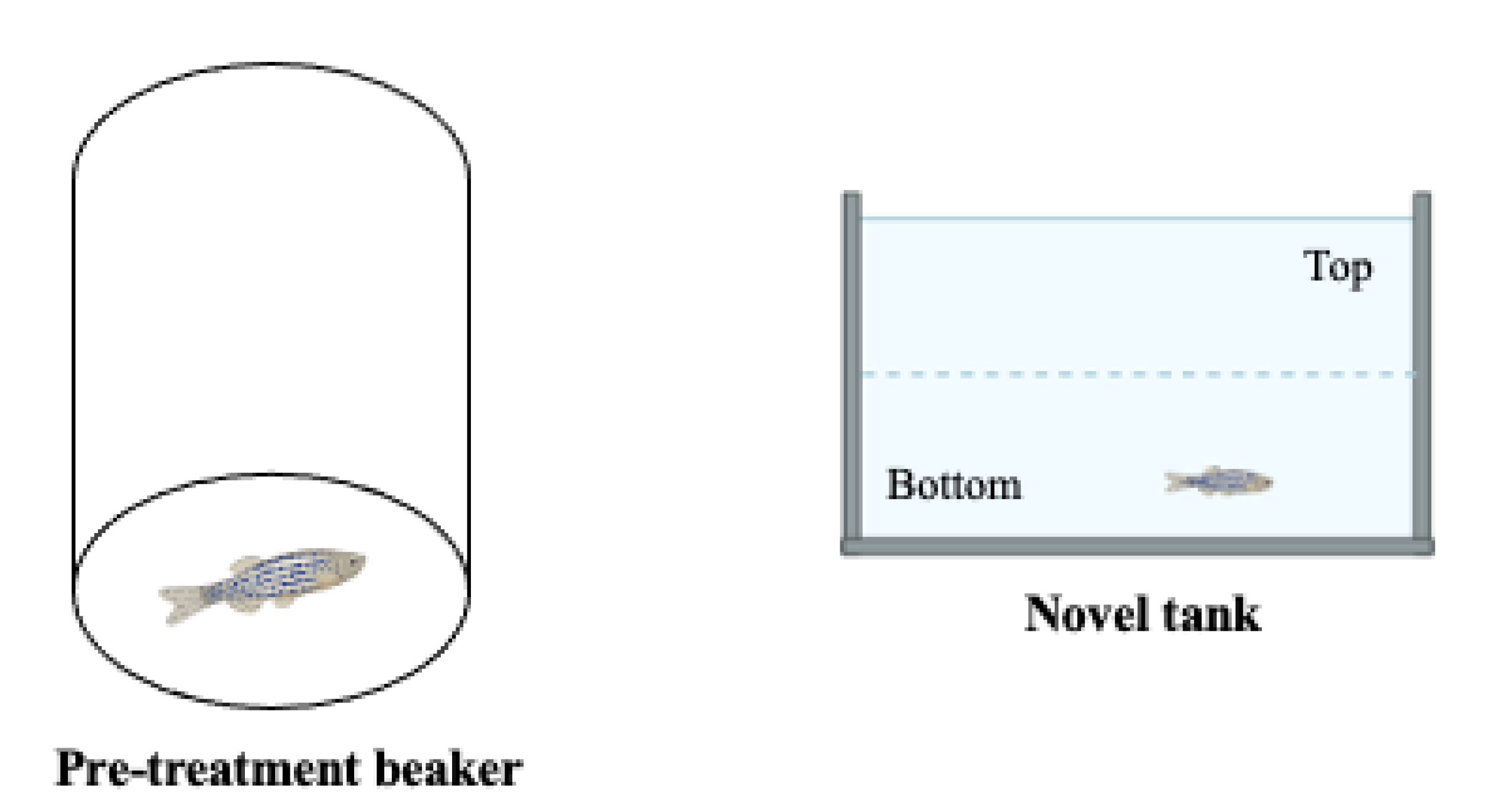
2.2.4. The Novel Tank Test
2.2.5. The Mirror-Attack Test
2.3. Behavioural Tests Common to Mice and Zebrafish
2.3.1. The Open Field Test
2.3.2. The Predator Avoidance Test
2.3.3. The T-Maze Test
3. Mouse Models of ASD and ADHD Research
- Face validity: mimic the fundamental behavioural deficits found in ASD or ADHD individuals;
- Construct validity: conform to the proposed pathophysiology or known therapeutics of ASD or ADHD;
- Predictive validity: predict unknown aspects of ASD or ADHD such as its genetics, neurobiology, or therapeutics.
3.1. Genetic Mouse Models of ASD
3.1.1. Black and Tan Brachyury (BTBR) T+ tf/J Mice
3.1.2. The Shank3 Knockout Mice
3.1.3. Fragile X Syndrome
3.1.4. The E3 Ubiquitin-Protein Ligase (Ube3a) Gene and 15q11-13 Duplication Maternal/Paternal
3.1.5. The Contactin-Associated Protein-like 2 (Cntnap2) Gene
3.1.6. Rett Syndrome
| Genes | Phenotypes | References |
|---|---|---|
| Actin like 6B (Actl6b) | Social and memory impairments, repetitive behaviours, hyperactivity | [119] |
| Activity dependent neuroprotector homeobox (Adpn) | Increased lethality, deficits in social memory, developmental alterations | [120,121,122] |
| Autophagy and beclin 1 regulator 1 (Ambra1) | Deficits in communication and social interactions, increased repetitive behaviours, reduced ultrasound communication in adults and pups, behaviour differences in male and female | [123] |
| Ankyrin repeat and sterile alpha motif domain containing 1B (Anks1b) | Social deficits, hyperactivity, and sensorimotor dysfunction | [124] |
| Rho GTPase activating protein 32 (Arhgap32) | Reduction in γ-aminobutyric acid type A receptor (GABAAR) levels and impaired GABAAR-mediated synaptic transmission | [125] |
| Rho guanine nucleotide exchange factor 10 (Arhgef10) | Impaired social interaction, hyperactivity, and decreased depression-like and anxiety-like behaviour | [126] |
| AT-rich interaction domain 1B (Arid1b) | Social behaviour impairment, altered vocalization, anxiety-like behaviour, neuroanatomical abnormalities | [127,128] |
| ASH1 like histone lysine methyltransferase (Ash1l) | Delayed eye development, increased lethality, infertility, dysfunction in immune response | [129,130] |
| ATPase phospholipid transporting 8A1 (Atp8a1) | Deficits in social behaviours | [131] |
| Ataxin1 (Atxn1) | Hyperactivity, impaired learning and memory, abnormal maturation and maintenance of upper-layer cortical neurons | [132] |
| Arginine vasopressin receptor 1B (Avpr1b) | Impaired social recognition, reduced pup ultrasonic vocalization | [133,134] |
| Cell cycle associated protein 1 (Caprin1) | Reduced sociality in a home cage and weak preference for social novelty | [135] |
| Coiled-coil and C2 domain containing 1A (Cc2d1a) | Reduced sociability, hyperactivity, anxiety, and excessive grooming | [135] |
| Chromodomain helicase DNA binding protein 2 (Chd2) | Developmental delay and increased mortality, decreased performance in object recognition test, reduced spatial working memory | [136,137] |
| Chromodomain helicase DNA binding protein 8 (Chd8) | Deficits in brain development, increased anxiety and repetitive behaviours, alteration in memory | [138,139,140,141,142] |
| Capicua transcriptional repressor (Cic) | Alteration in cortical and hippocampal morphology, reduced socialization | [143] |
| Contactin associated protein 2 (Cntnap2) | Delayed development, increased locomotor activity, impaired social interaction, and nest-building behaviours, increased epileptic behaviours | [144,145,146] |
| DEAD-box helicase 3 X-linked (Ddx3x) | Hyperactivity, anxiety-like behaviours, cognitive impairments in contextual fear memory but not novel object recognition memory, and motor deficits | [143] |
| Disco interacting protein 2 homolog A (Dip2a) | Excessive repetitive behaviours and defects in social novelty | [147] |
| DLG associated protein 1 (Dlgap1) | Post-synaptic density disruption and reduced sociability | [148] |
| Engrailed homeobox 2 (En2) | Reduced social interaction | [149,150] |
| Fibroblast growth factor 17 (Fgf17) | Reduced pup ultrasonic vocalization, lack of preference for social novelty, reduced reciprocal social interaction | [151] |
| Fragile X messenger ribonucleoprotein 1 (Fmr1) | Increased social approach, reduced repetitive behaviours, reduced anxiety, and normal locomotor activity | [108,152,153,154] |
| Forkhead box P2 (Foxp2) | Reduced pup ultrasonic vocalization, abnormality in Purkinje cells, severe motor impairments, premature death | [155,156,157] |
| Gamma-aminobutyric acid type A receptor subunit beta3 (Gabrb3) | Altered brain morphology, decreased sociability, reduced interneurons, increased seizures and anxiety, lack of preference for social novelty and impaired nest-building behaviour | [158,159,160,161] |
| Integrin subunit beta 3 (Itgb3) | Lack of preference for social novelty, and increased grooming behaviours | [162] |
| Lysine methyltransferase 5B (Kmt5b) | Deficits in neonatal reflexes and sociability, repetitive grooming, changes in thermal pain sensing, decreased depression and anxiety, increased fear, slower extinction learning, and lower body weight, length, and brain size | [163] |
| Methyl-CpG binding protein 2 (Mecp2) | Increased social avoidance, abnormal locomotor coordination, deficits in sociability and cognition | [116,164,165,166,167] |
| MET proto-oncogene, receptor tyrosine kinase (Met) | Deficits in cognitive function, hippocampal dysfunction | [168] |
| MicroRNA 137 (Mir137) | Dysregulated synaptic plasticity, repetitive behaviour, and impaired learning and social behaviour | [169] |
| Neuronal growth regulator 1 (Negr1) | Reversal learning deficits in the Morris water maze and increased susceptibility to pentylenetetrazol (PTZ)-induced seizures | [170] |
| Neuronal differentiation 2 (Neurod2) | Social interaction deficits, stereotypies, hyperactivity, occasionally spontaneous seizures | [171] |
| Neurite extension and migration factor (Nexmif) | Reduced sociability and communication, repetitive grooming behaviours, and deficits in learning and memory | [172] |
| Neuroligin 1 (Nlgn1) | Increased repetitive self-grooming, reduced pup ultrasonic vocalization, sociability, and reciprocal social interaction | [173,174,175,176] |
| Oxytocin receptor (Oxtr) | Impaired social behaviours, reduced pup ultrasonic vocalization | [177,178,179] |
| Protocadherin 19 (Pcdh19) | impaired behaviours including activity defects under stress conditions | [180] |
| Pogo transposable element derived with ZNF domain (Pogz) | Impaired social interaction | [181] |
| Phosphatase and tensin homolog (Pten) | High lethality, alteration in brain morphology, increased brain cells apoptosis, decreased Purkinje cells number, altered coordination and social memory and reduced sociability | [63,182,183,184,185] |
| RAB39B, member RAS oncogene family (Rab39b) | Cortical neurogenesis impairment and macrocephaly | [186] |
| Reelin (Reln) | Deficits in brain development, impaired coordination, and abnormal metabolism of neurotransmitters | [187,188] |
| Bifunctional polyamine/amino acid permease SAM3 (Sam3) | Impaired responses to social novelty, defects in social communication, and increased repetitive behaviour | [189] |
| Sodium voltage-gated channel alpha subunit 2 (Scn2a) | Increased cells apoptosis, seizures, hyperactivity, increased anxiety, and rearing | [190,191] |
| SUMO specific peptidase 1 (Senp1) | Social deficits and repetitive behaviours but normal learning and memory ability | [192] |
| SET domain containing 5 (Setd5) | Impairments in cognitive tasks, enhanced long-term potentiation, delayed ontogenetic profile of ultrasonic vocalization, behavioural inflexibility | [193] |
| SH3 and multiple ankyrin repeat domains 2 (Shank2) | Increased anxiety, hyperactivity, and repetitive behaviours, reduced social interaction and decreased social memory | [194,195,196] |
| SH3 and multiple ankyrin repeat domains 3 (Shank3) | Learning and sensory deficits, and impaired locomotor activity | [197] |
| TAO kinase 2 (Taok2) | Deficits in brain development, impaired memory, deficits in cortical layering, dendrite, and synapse formation, reduced excitatory neurotransmission and abnormalities in neural connectivity | [198] |
| T-box brain transcription factor 1 (Tbr1) | Increased anxiety and aggressiveness, reduced neural connections | [199,200] |
| Ubiquitin protein ligase E3A (Ube3a) | Low sociability, ultrasonic vocalization increased (pups) and decreased (adults) and impaired reversal learning | [201] |
| Urocortin 3 (Ucn3) | Abnormally low preference for novel conspecifics | [202] |
| UPF2 regulator of nonsense mediated mRNA decay (Upf2) | Impaired nonsense-mediated decay, memory deficits, abnormal long-term potentiation, increased social and communication deficits | [203] |
| UPF3B regulator of nonsense mediated mRNA decay (Upf3b) | Abnormal sleeping patterns, deficits in neural progenitors’ differentiation, impaired startle response | [204] |
3.2. Pharmacological Mouse Models of ASD
3.3. Genetic Mouse Models of ADHD
3.3.1. The Dopamine Transporter Knockout Mouse (DAT-KO)
3.3.2. Coloboma Mutant Mouse
3.3.3. Acallosal Mouse Strain I/LnJ
3.3.4. The Thyroid Hormone Receptor Beta 1 (Thrb1) Transgenic Mouse
3.3.5. α-Synuclein-Deficient Mice
3.4. Pharmacological and Environmental Mouse Models of ADHD
3.4.1. Juvenile Mouse with a Neonatal 6-Hydroxydopamine-Induced Brain Lesion
3.4.2. Exposure to Chemicals
3.4.3. Maternally Stressed Mice
4. Zebrafish Models of ASD and ADHD in Research
4.1. Genetic Zebrafish Models of ASD
4.2. Pharmacological Zebrafish Models of ASD
4.2.1. Valproic Acid
4.2.2. Other Drugs
4.3. Genetic Zebrafish Models of ADHD
4.4. Pharmacological Zebrafish Models of ADHD
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- DSM Cautionary Statement for Forensic Use of DSM-5. In Diagnostic and Statistical Manual of Mental Disorders, 5th ed.; Fernandes, J., Ed.; Climepsi Editores: Lisboa, Portugal, 2014; p. 991. ISBN 9780890425541. [Google Scholar]
- CDC Autism Prevalence Studies|Data|Centers for Disease Control and Prevention. Available online: https://data.cdc.gov/Public-Health-Surveillance/autism-prevalence-studies/9mw4-6adp (accessed on 20 April 2022).
- Lyall, K.; Croen, L.; Daniels, J.; Fallin, M.D.; Ladd-Acosta, C.; Lee, B.K.; Park, B.Y.; Snyder, N.W.; Schendel, D.; Volk, H.; et al. The Changing Epidemiology of Autism Spectrum Disorders. Annu. Rev. Public Health 2017, 38, 81–102. [Google Scholar] [CrossRef] [PubMed]
- IHME Prevalence of Autistic Spectrum Disorder. 2017. Available online: https://ourworldindata.org/grapher/prevalence-of-autistic-spectrum (accessed on 20 April 2022).
- IHME GBD Results Tool|GHDx. Available online: https://ghdx.healthdata.org/gbd-results-tool (accessed on 20 April 2022).
- NIMH Autism Spectrum Disorder. Available online: https://www.nimh.nih.gov/health/topics/autism-spectrum-disorders-asd (accessed on 20 April 2022).
- Autism Speaks. Autism and Health: Advances in Understanding and Treating the Health Conditions That Frequently Accompany Autism; Autism Speaks: New York, NY, USA, 2017. [Google Scholar]
- Colvert, E.; Tick, B.; McEwen, F.; Stewart, C.; Curran, S.R.; Woodhouse, E.; Gillan, N.; Hallett, V.; Lietz, S.; Garnett, T.; et al. Heritability of Autism Spectrum Disorder in a UK Population-Based Twin Sample. JAMA Psychiatry 2015, 72, 415–423. [Google Scholar] [CrossRef] [PubMed]
- Durkin, M.S.; Maenner, M.J.; Newschaffer, C.J.; Lee, L.C.; Cunniff, C.M.; Daniels, J.L.; Kirby, R.S.; Leavitt, L.; Miller, L.; Zahorodny, W.; et al. Advanced Parental Age and the Risk of Autism Spectrum Disorder. Am. J. Epidemiol. 2008, 168, 1268–1276. [Google Scholar] [CrossRef] [PubMed]
- Hallmayer, J.; Cleveland, S.; Torres, A.; Phillips, J.; Cohen, B.; Torigoe, T.; Miller, J.; Fedele, A.; Collins, J.; Smith, K.; et al. Genetic Heritability and Shared Environmental Factors Among Twin Pairs with Autism. Arch. Gen. Psychiatry 2011, 68, 1095–1102. [Google Scholar] [CrossRef] [PubMed]
- Ronald, A.; Happé, F.; Bolton, P.; Butcher, L.M.; Price, T.S.; Wheelwright, S.; Baron-Cohen, S.; Plomin, R. Genetic Heterogeneity between the Three Components of the Autism Spectrum: A Twin Study. J. Am. Acad. Child Adolesc. Psychiatry 2006, 45, 691–699. [Google Scholar] [CrossRef]
- Ozonoff, S.; Young, G.S.; Carter, A.; Messinger, D.; Yirmiya, N.; Zwaigenbaum, L.; Bryson, S.; Carver, L.J.; Constantino, J.N.; Dobkins, K.; et al. Recurrence Risk for Autism Spectrum Disorders: A Baby Siblings Research Consortium Study. Pediatrics 2011, 128, e488–e495. [Google Scholar] [CrossRef]
- Sumi, S.; Taniai, H.; Miyachi, T.; Tanemura, M. Sibling Risk of Pervasive Developmental Disorder Estimated by Means of an Epidemiologic Survey in Nagoya, Japan. J. Hum. Genet. 2006, 51, 518–522. [Google Scholar] [CrossRef][Green Version]
- Rosenberg, R.E.; Law, J.K.; Yenokyan, G.; McGready, J.; Kaufmann, W.E.; Law, P.A. Characteristics and Concordance of Autism Spectrum Disorders among 277 Twin Pairs. Arch. Pediatr. Adolesc. Med. 2009, 163, 907–914. [Google Scholar] [CrossRef]
- Taniai, H.; Nishiyama, T.; Miyachi, T.; Imaeda, M.; Sumi, S. Genetic Influences on the Broad Spectrum of Autism: Study of Proband-Ascertained Twins. Am. J. Med. Genet. B. Neuropsychiatr. Genet. 2008, 147B, 844–849. [Google Scholar] [CrossRef]
- Moss, J.; Chris, O. Autism in Genetic Syndromes: Implications for Assessment and Intervention; Cerebra Centre for Neurodevelopmental Disorders: Birmingham, UK, 2012. [Google Scholar]
- Garvía Peñuelas, B. Down Syndrome and Autism Spectrum Disorder. Int. Med. Rev. Down Syndr. 2017, 21, 1–2. [Google Scholar] [CrossRef]
- Hagerman, R.; Hoem, G.; Hagerman, P. Fragile X and Autism: Intertwined at the Molecular Level Leading to Targeted Treatments. Mol. Autism 2010, 1, 12. [Google Scholar] [CrossRef]
- Jain, A.; Marshall, J.; Buikema, A.; Bancroft, T.; Kelly, J.P.; Newschaffer, C.J. Autism Occurrence by MMR Vaccine Status among US Children with Older Siblings with and without Autism. JAMA 2015, 313, 1534. [Google Scholar] [CrossRef]
- Uno, Y.; Uchiyama, T.; Kurosawa, M.; Aleksic, B.; Ozaki, N. Early Exposure to the Combined Measles-Mumps-Rubella Vaccine and Thimerosal-Containing Vaccines and Risk of Autism Spectrum Disorder. Vaccine 2015, 33, 2511–2516. [Google Scholar] [CrossRef]
- Gadad, B.S.; Li, W.; Yazdani, U.; Grady, S.; Johnson, T.; Hammond, J.; Gunn, H.; Curtis, B.; English, C.; Yutuc, V.; et al. Administration of Thimerosal-Containing Vaccines to Infant Rhesus Macaques Does Not Result in Autism-like Behavior or Neuropathology. Proc. Natl. Acad. Sci. USA 2015, 112, 12498–12503. [Google Scholar] [CrossRef]
- Zerbo, O.; Qian, Y.; Yoshida, C.; Fireman, B.H.; Klein, N.P.; Croen, L.A. Association Between Influenza Infection and Vaccination during Pregnancy and Risk of Autism Spectrum Disorder. JAMA Pediatr. 2017, 171. [Google Scholar] [CrossRef]
- CDC What Is Autism Spectrum Disorder? Available online: https://www.cdc.gov/ncbddd/autism/facts.html (accessed on 16 May 2022).
- CDC Diagnostic Criteria|Autism Spectrum Disorder (ASD)|NCBDDD|CDC. Available online: https://www.cdc.gov/ncbddd/autism/hcp-dsm.html (accessed on 16 May 2022).
- WebMD Autism Therapies. Available online: https://www.webmd.com/brain/autism/therapies-to-help-with-autism (accessed on 20 April 2022).
- Cooper, K.; Loades, M.E.; Russell, A. Adapting Psychological Therapies for Autism—Therapist Experience, Skills and Confidence. Res. Autism Spectr. Disord. 2018, 45, 43. [Google Scholar] [CrossRef]
- AltogetherAutism Cognitive Behavioural Therapy Modifications for Those on the Autism Spectrum—Altogether Autism. Available online: https://www.altogetherautism.org.nz/cognitive-behavioural-therapy-modifications-for-those-on-the-autism-spectrum/ (accessed on 20 April 2022).
- Spain, D.; Sin, J.; Chalder, T.; Murphy, D.; Happé, F. Cognitive Behaviour Therapy for Adults with Autism Spectrum Disorders and Psychiatric Co-Morbidity: A Review. Res. Autism Spectr. Disord. 2015, 9, 151–162. [Google Scholar] [CrossRef]
- Fayyad, J.; Sampson, N.A.; Hwang, I.; Adamowski, T.; Aguilar-Gaxiola, S.; Al-Hamzawi, A.; Andrade, L.H.S.G.; Borges, G.; de Girolamo, G.; Florescu, S.; et al. The Descriptive Epidemiology of DSM-IV Adult ADHD in the World Health Organization World Mental Health Surveys. Atten. Defic. Hyperact. Disord. 2017, 9, 47–65. [Google Scholar] [CrossRef]
- Ebejer, J.L.; Medland, S.E.; van der Werf, J.; Gondro, C.; Henders, A.K.; Lynskey, M.; Martin, N.G.; Duffy, D.L. Attention Deficit Hyperactivity Disorder in Australian Adults: Prevalence, Persistence, Conduct Problems and Disadvantage. PLoS ONE 2012, 7, e47404. [Google Scholar] [CrossRef]
- Volkow, N.D.; Swanson, J.M. Adult Attention Deficit–Hyperactivity Disorder. N. Engl. J. Med. 2013, 369, 1935–1944. [Google Scholar] [CrossRef]
- McClernon, F.J.; Kollins, S.H. ADHD and Smoking. Ann. N. Y. Acad. Sci. 2008, 1141, 131–147. [Google Scholar] [CrossRef]
- Jung, Y.; Hsieh, L.S.; Lee, A.M.; Zhou, Z.; Coman, D.; Heath, C.J.; Hyder, F.; Mineur, Y.S.; Yuan, Q.; Goldman, D.; et al. An Epigenetic Mechanism Mediates Developmental Nicotine Effects on Neuronal Structure and Behavior. Nat. Neurosci. 2016, 19, 905–914. [Google Scholar] [CrossRef]
- Rahmani, Z.; Fayyazi Bordbar, M.R.; Dibaj, M.; Alimardani, M.; Moghbeli, M. Genetic and Molecular Biology of Autism Spectrum Disorder among Middle East Population: A Review. Hum. Genom. 2021, 15, 17. [Google Scholar] [CrossRef]
- Noroozi, R.; Taheri, M.; Movafagh, A.; Mirfakhraie, R.; Solgi, G.; Sayad, A.; Mazdeh, M.; Darvish, H. Glutamate Receptor, Metabotropic 7 (GRM7) Gene Variations and Susceptibility to Autism: A Case–Control Study. Autism Res. 2016, 9, 1161–1168. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Pan, C. Role of Metabotropic Glutamate Receptor 7 in Autism Spectrum Disorders: A Pilot Study. Life Sci. 2013, 92, 149–153. [Google Scholar] [CrossRef] [PubMed]
- Hosseinpour, M.; Mashayekhi, F.; Bidabadi, E.; Salehi, Z. Neuropilin-2 Rs849563 Gene Variations and Susceptibility to Autism in Iranian Population: A Case-Control Study. Metab. Brain Dis. 2017, 32, 1471–1474. [Google Scholar] [CrossRef] [PubMed]
- Giulivi, C.; Zhang, Y.F.; Omanska-Klusek, A.; Ross-Inta, C.; Wong, S.; Hertz-Picciotto, I.; Tassone, F.; Pessah, I.N. Mitochondrial Dysfunction in Autism. JAMA 2010, 304, 2389–2396. [Google Scholar] [CrossRef] [PubMed]
- Idriss, H.T.; Naismith, J.H. TNFα and the TNF Receptor Superfamily: Structure-Function Relationship(s). Microsc. Res. Technol. 2000, 50, 184–195. [Google Scholar] [CrossRef]
- Eftekharian, M.M.; Ghafouri-Fard, S.; Noroozi, R.; Omrani, M.D.; Arsang-jang, S.; Ganji, M.; Gharzi, V.; Noroozi, H.; Komaki, A.; Mazdeh, M.; et al. Cytokine Profile in Autistic Patients. Cytokine 2018, 108, 120–126. [Google Scholar] [CrossRef]
- Vaht, M.; Kiive, E.; Veidebaum, T.; Harro, J. A Functional Vesicular Monoamine Transporter 1 (VMAT1) Gene Variant Is Associated with Affect and the Prevalence of Anxiety, Affective, and Alcohol Use Disorders in a Longitudinal Population-Representative Birth Cohort Study. Int. J. Neuropsychopharmacol. 2016, 19, pyw013. [Google Scholar] [CrossRef]
- Uitterlinden, A.G.; Fang, Y.; Van Meurs, J.B.J.; Pols, H.A.P.; Van Leeuwen, J.P.T.M. Genetics and Biology of Vitamin D Receptor Polymorphisms. Gene 2004, 338, 143–156. [Google Scholar] [CrossRef]
- Emamalizadeh, B.; Jamshidi, J.; Movafagh, A.; Ohadi, M.; Khaniani, M.S.; Kazeminasab, S.; Biglarian, A.; Taghavi, S.; Motallebi, M.; Fazeli, A.; et al. RIT2 Polymorphisms: Is There a Differential Association? Mol. Neurobiol. 2016, 54, 2234–2240. [Google Scholar] [CrossRef]
- Mihai Bădescu, G.; Fîlfan, M.; Sandu, R.E.; Surugiu, R.; Ciobanu, O.; Popa-Wagner, A. Molecular Mechanisms Underlying Neurodevelopmental Disorders, ADHD and Autism. Rom. J. Morphol. Embryol. 2016, 57, 361–366. [Google Scholar]
- Hoogman, M.; Onnink, M.; Cools, R.; Aarts, E.; Kan, C.; Arias Vasquez, A.; Buitelaar, J.; Franke, B. The Dopamine Transporter Haplotype and Reward-Related Striatal Responses in Adult ADHD. Eur. Neuropsychopharmacol. 2013, 23, 469–478. [Google Scholar] [CrossRef]
- Binkovitz, L.; Thacker, P. What Does Molecular Imaging Reveal about the Causes of ADHD and the Potential for Better Management? Curr. Psychiatr. 2015, 14, 34. [Google Scholar]
- Volkow, N.D.; Wang, G.J.; Fowler, J.S.; Gatley, S.J.; Logan, J.; Ding, Y.S.; Hitzemann, R.; Pappas, N. Dopamine Transporter Occupancies in the Human Brain Induced by Therapeutic Doses of Oral Methylphenidate. Am. J. Psychiatry 1998, 155, 1325–1331. [Google Scholar] [CrossRef]
- Li, J.J.; Lee, S.S. Interaction of Dopamine Transporter Gene and Observed Parenting Behaviors on Attention-Deficit/Hyperactivity Disorder: A Structural Equation Modeling Approach. J. Clin. Child Adolesc. Psychol. 2013, 42, 174–186. [Google Scholar] [CrossRef]
- Volkow, N.D.; Wang, G.-J.; Kollins, S.H.; Wigal, T.L.; Newcorn, J.H.; Telang, F.; Fowler, J.S.; Zhu, W.; Logan, J.; Ma, Y.; et al. Evaluating Dopamine Reward Pathway in ADHD. JAMA 2009, 302, 1084. [Google Scholar] [CrossRef]
- Lou, H.C.; Rosa, P.; Pryds, O.; Karrebæk, H.; Lunding, J.; Cumming, P.; Gjedde, A. ADHD: Increased Dopamine Receptor Availability Linked to Attention Deficit and Low Neonatal Cerebral Blood Flow. Dev. Med. Child Neurol. 2004, 46, 179–183. [Google Scholar] [CrossRef]
- Baird, A.L.; Coogan, A.N.; Siddiqui, A.; Donev, R.M.; Thome, J. Adult Attention-Deficit Hyperactivity Disorder Is Associated with Alterations in Circadian Rhythms at the Behavioural, Endocrine and Molecular Levels. Mol. Psychiatry 2012, 17, 988–995. [Google Scholar] [CrossRef]
- Ji, E.-S.; Kim, C.-J.; Park, J.H.; Bahn, G.H. Duration-Dependence of the Effect of Treadmill Exercise on Hyperactivity in Attention Deficit Hyperactivity Disorder Rats. J. Exerc. Rehabil. 2014, 10, 75–80. [Google Scholar] [CrossRef] [PubMed]
- Dibner, C.; Schibler, U.; Albrecht, U. The Mammalian Circadian Timing System: Organization and Coordination of Central and Peripheral Clocks. Annu. Rev. Physiol. 2010, 72, 517–549. [Google Scholar] [CrossRef] [PubMed]
- Cryan, J.F.; Holmes, A. Model Organisms: The Ascent of Mouse: Advances in Modelling Human Depression and Anxiety. Nat. Rev. Drug Discov. 2005, 4, 775–790. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.H.; Bylykbashi, E.; Chatila, Z.K.; Lee, S.W.; Pulli, B.; Clemenson, G.D.; Kim, E.; Rompala, A.; Oram, M.K.; Asselin, C.; et al. Induced Adult Neurogenesis plus BDNF Mimicks the Effects of Exercise on Cognition in an Alzheimer’s Mouse Model. Science 2018, 361, 975. [Google Scholar] [CrossRef]
- Kodera, K.; Matsui, H. Zebrafish, Medaka and Turquoise Killifish for Understanding Human Neurodegenerative/Neurodevelopmental Disorders. Int. J. Mol. Sci. 2022, 23, 1399. [Google Scholar] [CrossRef]
- Matsui, H.; Sugie, A. An Optimized Method for Counting Dopaminergic Neurons in Zebrafish. PLoS ONE 2017, 12, e0184363. [Google Scholar] [CrossRef]
- Gerlai, R. Reproducibility and Replicability in Zebrafish Behavioral Neuroscience Research. Pharmacol. Biochem. Behav. 2019, 178, 30–38. [Google Scholar] [CrossRef]
- Fabian, P.; Tseng, K.C.; Smeeton, J.; Lancman, J.J.; Duc Si Dong, P.; Cerny, R.; Gage Crump, J. Lineage Analysis Reveals an Endodermal Contribution to the Vertebrate Pituitary. Science 2020, 370, 463. [Google Scholar] [CrossRef]
- Wang, W.; Hu, C.-K.; Zeng, A.; Alegre, D.; Hu, D.; Gotting, K.; Ortega Granillo, A.; Wang, Y.; Robb, S.; Schnittker, R.; et al. Changes in Regeneration-Responsive Enhancers Shape Regenerative Capacities in Vertebrates. Science 2020, 369, eaaz3090. [Google Scholar] [CrossRef]
- Pensado-López, A.; Veiga-Rúa, S.; Carracedo, Á.; Allegue, C.; Sánchez, L. Experimental Models to Study Autism Spectrum Disorders: HiPSCs, Rodents and Zebrafish. Genes 2020, 11, 1376. [Google Scholar] [CrossRef]
- Wöhr, M.; Scattoni, M.L. Behavioural Methods Used in Rodent Models of Autism Spectrum Disorders: Current Standards and New Developments. Behav. Brain Res. 2013, 251, 5–17. [Google Scholar] [CrossRef]
- Silverman, J.L.; Yang, M.; Lord, C.; Crawley, J.N. Behavioural Phenotyping Assays for Mouse Models of Autism. Nat. Rev. Neurosci. 2010, 11, 490–502. [Google Scholar] [CrossRef]
- Davids, E.; Zhang, K.; Tarazi, F.I.; Baldessarini, R.J. Animal Models of Attention-Deficit Hyperactivity Disorder. Brain Res. Rev. 2003, 42, 1–21. [Google Scholar] [CrossRef]
- Mortimer, N.; Ganster, T.; O’Leary, A.; Popp, S.; Freudenberg, F.; Reif, A.; Soler Artigas, M.; Ribasés, M.; Ramos-Quiroga, J.A.; Lesch, K.P.; et al. Dissociation of Impulsivity and Aggression in Mice Deficient for the ADHD Risk Gene Adgrl3: Evidence for Dopamine Transporter Dysregulation. Neuropharmacology 2019, 156, 107557. [Google Scholar] [CrossRef]
- Jhun, M.; Panwar, A.; Cordner, R.; Irvin, D.K.; Veiga, L.; Yeager, N.; Pechnick, R.N.; Schubloom, H.; Black, K.L.; Wheeler, C.J. CD103 Deficiency Promotes Autism (ASD) and Attention-Deficit Hyperactivity Disorder (ADHD) Behavioral Spectra and Reduces Age-Related Cognitive Decline. Front. Neurol. 2020, 11, 1794. [Google Scholar] [CrossRef]
- Moy, S.S.; Nadler, J.J.; Perez, A.; Barbaro, R.P.; Johns, J.M.; Magnuson, T.R.; Piven, J.; Crawley, J.N. Sociability and Preference for Social Novelty in Five Inbred Strains: An Approach to Assess Autistic-like Behavior in Mice. Genes Brain Behav. 2004, 3, 287–302. [Google Scholar] [CrossRef]
- McFarlane, H.G.; Kusek, G.K.; Yang, M.; Phoenix, J.L.; Bolivar, V.J.; Crawley, J.N. Autism-like Behavioral Phenotypes in BTBR T+tf/J Mice. Genes Brain Behav. 2008, 7, 152–163. [Google Scholar] [CrossRef]
- Moretz, J.A.; Martins, E.P.; Robison, B.D. The Effects of Early and Adult Social Environment on Zebrafish (Danio rerio) Behavior. Environ. Biol. Fishes 2007, 80, 91–101. [Google Scholar] [CrossRef]
- Gerlai, R.; Lahav, M.; Guo, S.; Rosenthal, A. Drinks like a Fish: Zebra Fish (Danio rerio) as a Behavior Genetic Model to Study Alcohol Effects. Pharmacol. Biochem. Behav. 2000, 67, 773–782. [Google Scholar] [CrossRef]
- Pearson, B.L.; Pobbe, R.L.H.; Defensor, E.B.; Oasay, L.; Bolivar, V.J.; Blanchard, D.C.; Blanchard, R.J. Motor and Cognitive Stereotypies in the BTBR T+tf/J Mouse Model of Autism. Genes Brain Behav. 2011, 10, 228–235. [Google Scholar] [CrossRef]
- Reynolds, S.; Urruela, M.; Devine, D.P. Effects of Environmental Enrichment on Repetitive Behaviors in the BTBR T+tf/J Mouse Model of Autism. Autism Res. 2013, 6, 337–343. [Google Scholar] [CrossRef] [PubMed]
- MacPhail, R.C.; Brooks, J.; Hunter, D.L.; Padnos, B.; Irons, T.D.; Padilla, S. Locomotion in Larval Zebrafish: Influence of Time of Day, Lighting and Ethanol. Neurotoxicology 2009, 30, 52–58. [Google Scholar] [CrossRef]
- Lange, M.; Norton, W.; Coolen, M.; Chaminade, M.; Merker, S.; Proft, F.; Schmitt, A.; Vernier, P.; Lesch, K.-P.; Bally-Cuif, L. The ADHD-Susceptibility Gene Lphn3.1 Modulates Dopaminergic Neuron Formation and Locomotor Activity during Zebrafish Development. Mol. Psychiatry 2012, 17, 946–954. [Google Scholar] [CrossRef] [PubMed]
- Ingebretson, J.J.; Masino, M.A. Quantification of Locomotor Activity in Larval Zebrafish: Considerations for the Design of High-Throughput Behavioral Studies. Front. Neural Circuits 2013, 7, 109. [Google Scholar] [CrossRef] [PubMed]
- Ulhaq, M.; Örn, S.; Carlsson, G.; Morrison, D.A.; Norrgren, L. Locomotor Behavior in Zebrafish (Danio rerio) Larvae Exposed to Perfluoroalkyl Acids. Aquat. Toxicol. 2013, 144–145, 332–340. [Google Scholar] [CrossRef] [PubMed]
- Grossman, L.; Utterback, E.; Stewart, A.; Gaikwad, S.; Chung, K.M.; Suciu, C.; Wong, K.; Elegante, M.; Elkhayat, S.; Tan, J.; et al. Characterization of Behavioral and Endocrine Effects of LSD on Zebrafish. Behav. Brain Res. 2010, 214, 277–284. [Google Scholar] [CrossRef]
- Spinello, C.; Yang, Y.; Macrì, S.; Porfiri, M. Zebrafish Adjust Their Behavior in Response to an Interactive Robotic Predator. Front. Robot. AI 2019, 6, 38. [Google Scholar] [CrossRef]
- Amaral, V.C.S.; Santos Gomes, K.; Nunes-de-Souza, R.L. Increased Corticosterone Levels in Mice Subjected to the Rat Exposure Test. Horm. Behav. 2010, 57, 128–133. [Google Scholar] [CrossRef]
- Roullet, F.I.; Wöhr, M.; Crawley, J.N. Female Urine-Induced Male Mice Ultrasonic Vocalizations, but Not Scent-Marking, Is Modulated by Social Experience. Behav. Brain Res. 2011, 216, 19–28. [Google Scholar] [CrossRef]
- Wöhr, M.; Roullet, F.I.; Crawley, J.N. Reduced Scent Marking and Ultrasonic Vocalizations in the BTBR T+tf/J Mouse Model of Autism. Genes Brain Behav. 2011, 10, 35–43. [Google Scholar] [CrossRef]
- Parker, M.O.; Brock, A.J.; Sudwarts, A.; Brennan, C.H. Atomoxetine Reduces Anticipatory Responding in a 5-Choice Serial Reaction Time Task for Adult Zebrafish. Psychopharmacology 2014, 231, 2671–2679. [Google Scholar] [CrossRef]
- Parker, M.O.; Ife, D.; Ma, J.; Pancholi, M.; Smeraldi, F.; Straw, C.; Brennan, C.H. Development and Automation of a Test of Impulse Control in Zebrafish. Front. Syst. Neurosci. 2013, 7, 65. [Google Scholar] [CrossRef]
- Wiprich, M.T.; Zanandrea, R.; Altenhofen, S.; Bonan, C.D. Influence of 3-Nitropropionic Acid on Physiological and Behavioral Responses in Zebrafish Larvae and Adults. Comp. Biochem. Physiol. Part C Toxicol. Pharmacol. 2020, 234, 108772. [Google Scholar] [CrossRef]
- Blaser, R.E.; Rosemberg, D.B. Measures of Anxiety in Zebrafish (Danio rerio): Dissociation of Black/White Preference and Novel Tank Test. PLoS ONE 2012, 7, e36931. [Google Scholar] [CrossRef]
- Egan, R.J.; Bergner, C.L.; Hart, P.C.; Cachat, J.M.; Canavello, P.R.; Elegante, M.F.; Elkhayat, S.I.; Bartels, B.K.; Tien, A.K.; Tien, D.H.; et al. Understanding Behavioral and Physiological Phenotypes of Stress and Anxiety in Zebrafish. Behav. Brain Res. 2009, 205, 38–44. [Google Scholar] [CrossRef]
- Maximino, C.; Lima, M.G.; de Jesus Oliveira Batista, E.; Oliveira, K.R.H.M.; Herculano, A.M. Interaction between 5-HT1B Receptors and Nitric Oxide in Zebrafish Responses to Novelty. Neurosci. Lett. 2015, 588, 54–56. [Google Scholar] [CrossRef]
- Jager, A.; Kanters, D.; Geers, F.; Buitelaar, J.K.; Kozicz, T.; Glennon, J.C. Methylphenidate Dose-Dependently Affects Aggression and Improves Fear Extinction and Anxiety in BALB/CJ Mice. Front. Psychiatry 2019, 10, 768. [Google Scholar] [CrossRef]
- Koolhaas, J.M.; Coppens, C.M.; de Boer, S.F.; Buwalda, B.; Meerlo, P.; Timmermans, P.J.A. The Resident-Intruder Paradigm: A Standardized Test for Aggression, Violence and Social Stress. J. Vis. Exp. 2013, 77, e4367. [Google Scholar] [CrossRef]
- Zimmermann, F.F.; Gaspary, K.V.; Leite, C.E.; De Paula Cognato, G.; Bonan, C.D. Embryological Exposure to Valproic Acid Induces Social Interaction Deficits in Zebrafish (Danio rerio): A Developmental Behavior Analysis. Neurotoxicol. Teratol. 2015, 52, 36–41. [Google Scholar] [CrossRef]
- Buske, C.; Gerlai, R. Shoaling Develops with Age in Zebrafish (Danio rerio). Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2011, 35, 1409–1415. [Google Scholar] [CrossRef]
- Dougnon, G.; Ito, M. Sedative Effects of the Essential Oil from the Leaves of Lantana Camara Occurring in the Republic of Benin via Inhalation in Mice. J. Nat. Med. 2020, 74, 159–169. [Google Scholar] [CrossRef]
- Dougnon, G.; Ito, M. Essential Oil from the Leaves of Chromolaena Odorata, and Sesquiterpene Caryophyllene Oxide Induce Sedative Activity in Mice. Pharmaceuticals 2021, 14, 651. [Google Scholar] [CrossRef]
- Dougnon, G.; Ito, M. Role of Ascaridole and P-Cymene in the Sleep-Promoting Effects of Dysphania Ambrosioides Essential Oil via the GABAergic System in a DdY Mouse Inhalation Model. J. Nat. Prod. 2021, 84, 91–100. [Google Scholar] [CrossRef]
- Baker, K.B.; Wray, S.P.; Ritter, R.; Mason, S.; Lanthorn, T.H.; Savelieva, K.V. Male and Female Fmr1 Knockout Mice on C57 Albino Background Exhibit Spatial Learning and Memory Impairments. Genes Brain. Behav. 2010, 9, 562–574. [Google Scholar] [CrossRef]
- Magara, F.; Ricceri, L.; Wolfer, D.P.; Lipp, H.P. The Acallosal Mouse Strain I/LnJ: A Putative Model of ADHD? Neurosci. Biobehav. Rev. 2000, 24, 45–50. [Google Scholar] [CrossRef]
- Abrahams, B.S.; Arking, D.E.; Campbell, D.B.; Mefford, H.C.; Morrow, E.M.; Weiss, L.A.; Menashe, I.; Wadkins, T.; Banerjee-Basu, S.; Packer, A. SFARI Gene 2.0: A Community-Driven Knowledgebase for the Autism Spectrum Disorders (ASDs). Mol. Autism 2013, 4, 36. [Google Scholar] [CrossRef]
- Meyza, K.Z.; Defensor, E.B.; Jensen, A.L.; Corley, M.J.; Pearson, B.L.; Pobbe, R.L.H.; Bolivar, V.J.; Blanchard, D.C.; Blanchard, R.J. The BTBR T+tf/J Mouse Model for Autism Spectrum Disorders–in Search of Biomarkers. Behav. Brain Res. 2013, 251, 25–34. [Google Scholar] [CrossRef]
- Scattoni, M.L.; Gandhy, S.U.; Ricceri, L.; Crawley, J.N. Unusual Repertoire of Vocalizations in the BTBR T+tf/J Mouse Model of Autism. PLoS ONE 2008, 3, e3067. [Google Scholar] [CrossRef]
- UniProt Find Your Protein. Available online: https://www.uniprot.org/ (accessed on 30 June 2022).
- Bozdagi, O.; Sakurai, T.; Papapetrou, D.; Wang, X.; Dickstein, D.L.; Takahashi, N.; Kajiwara, Y.; Yang, M.; Katz, A.M.; Scattoni, M.; et al. Haploinsufficiency of the Autism-Associated Shank3 Gene Leads to Deficits in Synaptic Function, Social Interaction, and Social Communication. Mol. Autism 2010, 1, 15. [Google Scholar] [CrossRef]
- Peça, J.; Feliciano, C.; Ting, J.T.; Wang, W.; Wells, M.F.; Venkatraman, T.N.; Lascola, C.D.; Fu, Z.; Feng, G. Shank3 Mutant Mice Display Autistic-like Behaviours and Striatal Dysfunction. Nature 2011, 472, 437–442. [Google Scholar] [CrossRef]
- Crawley, J.N. Translational Animal Models of Autism and Neurodevelopmental Disorders. Dialogues Clin. Neurosci. 2012, 14, 293. [Google Scholar] [CrossRef] [PubMed]
- Pieretti, M.; Zhang, F.; Fu, Y.H.; Warren, S.T.; Oostra, B.A.; Caskey, C.T.; Nelson, D.L. Absence of Expression of the FMR-1 Gene in Fragile X Syndrome. Cell 1991, 66, 817–822. [Google Scholar] [CrossRef]
- Fu, Y.H.; Kuhl, D.P.A.; Pizzuti, A.; Pieretti, M.; Sutcliffe, J.S.; Richards, S.; Verkert, A.J.M.H.; Holden, J.J.A.; Fenwick, R.G.; Warren, S.T.; et al. Variation of the CGG Repeat at the Fragile X Site Results in Genetic Instability: Resolution of the Sherman Paradox. Cell 1991, 67, 1047–1058. [Google Scholar] [CrossRef]
- Mineur, Y.S.; Huynh, L.X.; Crusio, W.E. Social Behavior Deficits in the Fmr1 Mutant Mouse. Behav. Brain Res. 2006, 168, 172–175. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.Y.; Rex, C.S.; Babayan, A.H.; Kramár, E.A.; Lynch, G.; Gall, C.M.; Lauterborn, J.C. Physiological Activation of Synaptic Rac>PAK (p-21 Activated Kinase) Signaling Is Defective in a Mouse Model of Fragile X Syndrome. J. Neurosci. 2010, 30, 10977–10984. [Google Scholar] [CrossRef] [PubMed]
- Spencer, C.M.; Graham, D.F.; Yuva-Paylor, L.A.; Nelson, D.L.; Paylor, R. Social Behavior in Fmr1 Knockout Mice Carrying a Human FMR1 Transgene. Behav. Neurosci. 2008, 122, 710–715. [Google Scholar] [CrossRef] [PubMed]
- Huber, K.M.; Gallagher, S.M.; Warren, S.T.; Bear, M.F. Altered Synaptic Plasticity in a Mouse Model of Fragile X Mental Retardation. Proc. Natl. Acad. Sci. USA 2002, 99, 7746–7750. [Google Scholar] [CrossRef]
- Dölen, G.; Osterweil, E.; Rao, B.S.S.; Smith, G.B.; Auerbach, B.D.; Chattarji, S.; Bear, M.F. Correction of Fragile X Syndrome in Mice. Neuron 2007, 56, 955–962. [Google Scholar] [CrossRef]
- LaSalle, J.M.; Reiter, L.T.; Chamberlain, S.J. Epigenetic Regulation of UBE3A and Roles in Human Neurodevelopmental Disorders. Epigenomics 2015, 7, 1213–1228. [Google Scholar] [CrossRef]
- Smith, S.E.P.; Zhou, Y.D.; Zhang, G.; Jin, Z.; Stoppel, D.C.; Anderson, M.P. Increased Gene Dosage of Ube3a Results in Autism Traits and Decreased Glutamate Synaptic Transmission in Mice. Sci. Transl. Med. 2011, 3, 103ra97. [Google Scholar] [CrossRef]
- Scott-Van Zeeland, A.A.; Abrahams, B.S.; Alvarez-Retuerto, A.I.; Sonnenblick, L.I.; Rudie, J.D.; Ghahremani, D.; Mumford, J.A.; Poldrack, R.A.; Dapretto, M.; Geschwind, D.H.; et al. Altered Functional Connectivity in Frontal Lobe Circuits Is Associated with Variation in the Autism Risk Gene CNTNAP2. Sci. Transl. Med. 2010, 2, 56ra80. [Google Scholar] [CrossRef]
- Moy, S.S.; Riddick, N.V.; Nikolova, V.D.; Teng, B.L.; Agster, K.L.; Nonneman, R.J.; Young, N.B.; Baker, L.K.; Nadler, J.J.; Bodfish, J.W. Repetitive Behavior Profile and Supersensitivity to Amphetamine in the C58/J Mouse Model of Autism. Behav. Brain Res. 2014, 259, 200–214. [Google Scholar] [CrossRef]
- Ergaz, Z.; Weinstein-Fudim, L.; Ornoy, A. Genetic and Non-Genetic Animal Models for Autism Spectrum Disorders (ASD). Reprod. Toxicol. 2016, 64, 116–140. [Google Scholar] [CrossRef]
- Samaco, R.C.; Mcgraw, C.M.; Ward, C.S.; Sun, Y.; Neul, J.L.; Zoghbi, H.Y. Female Mecp2+/− Mice Display Robust Behavioral Deficits on Two Different Genetic Backgrounds Providing a Framework for Pre-Clinical Studies. Hum. Mol. Genet. 2013, 22, 96–109. [Google Scholar] [CrossRef]
- Chao, H.T.; Chen, H.; Samaco, R.C.; Xue, M.; Chahrour, M.; Yoo, J.; Neul, J.L.; Gong, S.; Lu, H.C.; Heintz, N.; et al. Dysfunction in GABA Signalling Mediates Autism-like Stereotypies and Rett Syndrome Phenotypes. Nature 2010, 468, 263–269. [Google Scholar] [CrossRef]
- Sztainberg, Y.; Chen, H.M.; Swann, J.W.; Hao, S.; Tang, B.; Wu, Z.; Tang, J.; Wan, Y.W.; Liu, Z.; Rigo, F.; et al. Reversal of Phenotypes in MECP2 Duplication Mice Using Genetic Rescue or Antisense Oligonucleotides. Nature 2015, 528, 123–126. [Google Scholar] [CrossRef]
- Wenderski, W.; Wang, L.; Krokhotin, A.; Walsh, J.J.; Li, H.; Shoji, H.; Ghosh, S.; George, R.D.; Miller, E.L.; Elias, L.; et al. Loss of the Neural-Specific BAF Subunit ACTL6B Relieves Repression of Early Response Genes and Causes Recessive Autism. Proc. Natl. Acad. Sci. USA 2020, 117, 10055–10066. [Google Scholar] [CrossRef]
- Sragovich, S.; Malishkevich, A.; Piontkewitz, Y.; Giladi, E.; Touloumi, O.; Lagoudaki, R.; Grigoriadis, N.; Gozes, I. The Autism/Neuroprotection-Linked ADNP/NAP Regulate the Excitatory Glutamatergic Synapse. Transl. Psychiatry 2019, 9, 2. [Google Scholar] [CrossRef]
- Vulih-Shultzman, I.; Pinhasov, A.; Mandel, S.; Grigoriadis, N.; Touloumi, O.; Pittel, Z.; Gozes, I. Activity-Dependent Neuroprotective Protein Snippet NAP Reduces Tau Hyperphosphorylation and Enhances Learning in a Novel Transgenic Mouse Model. J. Pharmacol. Exp. Ther. 2007, 323, 438–449. [Google Scholar] [CrossRef]
- Amram, N.; Hacohen-Kleiman, G.; Sragovich, S.; Malishkevich, A.; Katz, J.; Touloumi, O.; Lagoudaki, R.; Grigoriadis, N.C.; Giladi, E.; Yeheskel, A.; et al. Sexual Divergence in Microtubule Function: The Novel Intranasal Microtubule Targeting SKIP Normalizes Axonal Transport and Enhances Memory. Mol. Psychiatry 2016, 21, 1467–1476. [Google Scholar] [CrossRef]
- Dere, E.; Dahm, L.; Lu, D.; Hammerschmidt, K.; Ju, A.; Tantra, M.; Kästner, A.; Chowdhury, K.; Ehrenreich, H. Heterozygous Ambra1 Deficiency in Mice: A Genetic Trait with Autism-like Behavior Restricted to the Female Gender. Front. Behav. Neurosci. 2014, 8, 181. [Google Scholar] [CrossRef]
- Carbonell, A.U.; Cho, C.H.; Tindi, J.O.; Counts, P.A.; Bates, J.C.; Erdjument-Bromage, H.; Cvejic, S.; Iaboni, A.; Kvint, I.; Rosensaft, J.; et al. Haploinsufficiency in the ANKS1B Gene Encoding AIDA-1 Leads to a Neurodevelopmental Syndrome. Nat. Commun. 2019, 10, 3529. [Google Scholar] [CrossRef]
- Nakamura, T.; Arima-Yoshida, F.; Sakaue, F.; Nasu-Nishimura, Y.; Takeda, Y.; Matsuura, K.; Akshoomoff, N.; Mattson, S.N.; Grossfeld, P.D.; Manabe, T.; et al. PX-RICS-Deficient Mice Mimic Autism Spectrum Disorder in Jacobsen Syndrome through Impaired GABAA Receptor Trafficking. Nat. Commun. 2016, 7, 10861. [Google Scholar] [CrossRef]
- Lu, D.H.; Liao, H.M.; Chen, C.H.; Tu, H.J.; Liou, H.C.; Gau, S.S.F.; Fu, W.M. Impairment of Social Behaviors in Arhgef10 Knockout Mice. Mol. Autism 2018, 9, 11. [Google Scholar] [CrossRef]
- Celen, C.; Chuang, J.C.; Luo, X.; Nijem, N.; Walker, A.K.; Chen, F.; Zhang, S.; Chung, A.S.; Nguyen, L.H.; Nassour, I.; et al. Arid1b Haploinsufficient Mice Reveal Neuropsychiatric Phenotypes and Reversible Causes of Growth Impairment. eLife 2017, 6, e25730. [Google Scholar] [CrossRef]
- Shibutani, M.; Horii, T.; Shoji, H.; Morita, S.; Kimura, M.; Terawaki, N.; Miyakawa, T.; Hatada, I. Arid1b Haploinsufficiency Causes Abnormal Brain Gene Expression and Autism-Related Behaviors in Mice. Int. J. Mol. Sci. 2017, 18, 1872. [Google Scholar] [CrossRef]
- Brinkmeier, M.L.; Geister, K.A.; Jones, M.; Waqas, M.; Maillard, I.; Camper, S.A. The Histone Methyltransferase Gene Absent, Small, or Homeotic Discs-1 Like Is Required for Normal Hox Gene Expression and Fertility in Mice. Biol. Reprod. 2015, 93, 121. [Google Scholar] [CrossRef]
- Zhu, Τ.; Liang, C.; Li, D.; Tian, M.; Liu, S.; Gao, G.; Guan, J.-S. Histone Methyltransferase Ash1L Mediates Activity-Dependent Repression of Neurexin-1α. Sci. Rep. 2016, 6, 26597. [Google Scholar] [CrossRef] [PubMed]
- Kerr, D.J.; Marsillo, A.; Guariglia, S.R.; Budylin, T.; Sadek, R.; Menkes, S.; Chauhan, A.; Wen, G.Y.; McCloskey, D.P.; Wieraszko, A.; et al. Aberrant Hippocampal Atp8a1 Levels Are Associated with Altered Synaptic Strength, Electrical Activity, and Autistic-like Behavior. Biochim. Biophys. Acta 2016, 1862, 1755–1765. [Google Scholar] [CrossRef]
- Oaks, A.W.; Zamarbide, M.; Tambunan, D.E.; Santini, E.; Di Costanzo, S.; Pond, H.L.; Johnson, M.W.; Lin, J.; Gonzalez, D.M.; Boehler, J.F.; et al. Cc2d1a Loss of Function Disrupts Functional and Morphological Development in Forebrain Neurons Leading to Cognitive and Social Deficits. Cereb. Cortex 2017, 27, 1670–1685. [Google Scholar] [CrossRef]
- Wersinger, S.R.; Ginns, E.I.; O’Carroll, A.M.; Lolait, S.J.; Young, W.S. Vasopressin V1b Receptor Knockout Reduces Aggressive Behavior in Male Mice. Mol. Psychiatry 2002, 7, 975–984. [Google Scholar] [CrossRef] [PubMed]
- Scattoni, M.L.; McFarlane, H.G.; Zhodzishsky, V.; Caldwell, H.K.; Young, W.S.; Ricceri, L.; Crawley, J.N. Reduced Ultrasonic Vocalizations in Vasopressin 1b Knockout Mice. Behav. Brain Res. 2008, 187, 371–378. [Google Scholar] [CrossRef] [PubMed]
- Ohashi, R.; Takao, K.; Miyakawa, T.; Shiina, N. Comprehensive Behavioral Analysis of RNG105 (Caprin1) Heterozygous Mice: Reduced Social Interaction and Attenuated Response to Novelty. Sci. Rep. 2016, 6, 20775. [Google Scholar] [CrossRef] [PubMed]
- Nagarajan, P.; Onami, T.M.; Rajagopalan, S.; Kania, S.; Donnell, R.; Venkatachalam, S. Role of Chromodomain Helicase DNA-Binding Protein 2 in DNA Damage Response Signaling and Tumorigenesis. Oncogene 2009, 28, 1053–1062. [Google Scholar] [CrossRef][Green Version]
- Kim, Y.J.; Khoshkhoo, S.; Frankowski, J.C.; Zhu, B.; Abbasi, S.; Lee, S.; Wu, Y.E.; Hunt, R.F. Chd2 Is Necessary for Neural Circuit Development and Long-Term Memory. Neuron 2018, 100, 1180–1193.e6. [Google Scholar] [CrossRef]
- Durak, O.; Gao, F.; Kaeser-Woo, Y.J.; Rueda, R.; Martorell, A.J.; Nott, A.; Liu, C.Y.; Watson, L.A.; Tsai, L.H. Chd8 Mediates Cortical Neurogenesis via Transcriptional Regulation of Cell Cycle and Wnt Signaling. Nat. Neurosci. 2016, 19, 1477–1488. [Google Scholar] [CrossRef]
- Gompers, A.L.; Su-Feher, L.; Ellegood, J.; Copping, N.A.; Riyadh, M.A.; Stradleigh, T.W.; Pride, M.C.; Schaffler, M.D.; Wade, A.A.; Catta-Preta, R.; et al. Germline Chd8 Haploinsufficiency Alters Brain Development in Mouse. Nat. Neurosci. 2017, 20, 1062–1073. [Google Scholar] [CrossRef]
- Jung, H.; Park, H.; Choi, Y.; Kang, H.; Lee, E.; Kweon, H.; Roh, J.D.; Ellegood, J.; Choi, W.; Kang, J.; et al. Sexually Dimorphic Behavior, Neuronal Activity, and Gene Expression in Chd8-Mutant Mice. Nat. Neurosci. 2018, 21, 1218–1228. [Google Scholar] [CrossRef]
- Miyata, S.; Taniguchi, M.; Koyama, Y.; Shimizu, S.; Tanaka, T.; Yasuno, F.; Yamamoto, A.; Iida, H.; Kudo, T.; Katayama, T.; et al. Association between Chronic Stress-Induced Structural Abnormalities in Ranvier Nodes and Reduced Oligodendrocyte Activity in Major Depression. Sci. Rep. 2016, 6, 23084. [Google Scholar] [CrossRef]
- Platt, R.J.; Zhou, Y.; Slaymaker, I.M.; Shetty, A.S.; Weisbach, N.R.; Kim, J.A.; Sharma, J.; Desai, M.; Sood, S.; Kempton, H.R.; et al. Chd8 Mutation Leads to Autistic-like Behaviors and Impaired Striatal Circuits. Cell Rep. 2017, 19, 335–350. [Google Scholar] [CrossRef]
- Lu, H.-C.; Tan, Q.; Rousseaux, M.W.C.; Wang, W.; Kim, J.-Y.; Richman, R.; Wan, Y.-W.; Yeh, S.-Y.; Patel, J.M.; Liu, X.; et al. Disruption of the ATXN1–CIC Complex Causes a Spectrum of Neurobehavioral Phenotypes in Mice and Humans. Nat. Genet. 2017, 49, 527–536. [Google Scholar] [CrossRef]
- Peñagarikano, O.; Lázaro, M.T.; Lu, X.H.; Gordon, A.; Dong, H.; Lam, H.A.; Peles, E.; Maidment, N.T.; Murphy, N.P.; Yang, X.W.; et al. Exogenous and Evoked Oxytocin Restores Social Behavior in the Cntnap2 Mouse Model of Autism. Sci. Transl. Med. 2015, 7, 271ra8. [Google Scholar] [CrossRef]
- Peñagarikano, O.; Abrahams, B.S.; Herman, E.I.; Winden, K.D.; Gdalyahu, A.; Dong, H.; Sonnenblick, L.I.; Gruver, R.; Almajano, J.; Bragin, A.; et al. Absence of CNTNAP2 Leads to Epilepsy, Neuronal Migration Abnormalities, and Core Autism-Related Deficits. Cell 2011, 147, 235–246. [Google Scholar] [CrossRef]
- Schaafsma, S.M.; Gagnidze, K.; Reyes, A.; Norstedt, N.; Månsson, K.; Francis, K.; Pfaff, D.W. Sex-Specific Gene-Environment Interactions Underlying ASD-like Behaviors. Proc. Natl. Acad. Sci. USA 2017, 114, 1383–1388. [Google Scholar] [CrossRef]
- Boitnott, A.; Garcia-Forn, M.; Ung, D.C.; Niblo, K.; Mendonca, D.; Park, Y.; Flores, M.; Maxwell, S.; Ellegood, J.; Qiu, L.R.; et al. Developmental and Behavioral Phenotypes in a Mouse Model of DDX3X Syndrome. Biol. Psychiatry 2021, 90, 742–755. [Google Scholar] [CrossRef]
- Ma, J.; Zhang, L.Q.; He, Z.X.; He, X.X.; Wang, Y.J.; Jian, Y.L.; Wang, X.; Zhang, B.B.; Su, C.; Lu, J.; et al. Autism Candidate Gene DIP2A Regulates Spine Morphogenesis via Acetylation of Cortactin. PLoS Biol. 2019, 17, e3000461. [Google Scholar] [CrossRef]
- Cheh, M.A.; Millonig, J.H.; Roselli, L.M.; Ming, X.; Jacobsen, E.; Kamdar, S.; Wagner, G.C. En2 Knockout Mice Display Neurobehavioral and Neurochemical Alterations Relevant to Autism Spectrum Disorder. Brain Res. 2006, 1116, 166–176. [Google Scholar] [CrossRef]
- Moy, S.S.; Nadler, J.J.; Young, N.B.; Nonneman, R.J.; Grossman, A.W.; Murphy, D.L.; D’Ercole, A.J.; Crawley, J.N.; Magnuson, T.R.; Lauder, J.M. Social Approach in Genetically Engineered Mouse Lines Relevant to Autism. Genes Brain Behav. 2009, 8, 129–142. [Google Scholar] [CrossRef]
- Scearce-Levie, K.; Roberson, E.D.; Gerstein, H.; Cholfin, J.A.; Mandiyan, V.S.; Shah, N.M.; Rubenstein, J.L.R.; Mucke, L. Abnormal Social Behaviors in Mice Lacking Fgf17. Genes Brain Behav. 2008, 7, 344–354. [Google Scholar] [CrossRef]
- Ronesi, J.A.; Collins, K.A.; Hays, S.A.; Tsai, N.P.; Guo, W.; Birnbaum, S.G.; Hu, J.H.; Worley, P.F.; Gibson, J.R.; Huber, K.M. Disrupted Homer Scaffolds Mediate Abnormal MGluR5 Function in a Mouse Model of Fragile X Syndrome. Nat. Neurosci. 2012, 15, 431–440. [Google Scholar] [CrossRef]
- Bhattacharya, A.; Kaphzan, H.; Alvarez-Dieppa, A.C.; Murphy, J.P.; Pierre, P.; Klann, E. Genetic Removal of P70 S6 Kinase 1 Corrects Molecular, Synaptic, and Behavioral Phenotypes in Fragile X Syndrome Mice. Neuron 2012, 76, 325–337. [Google Scholar] [CrossRef]
- Spencer, C.M.; Alekseyenko, O.; Serysheva, E.; Yuva-Paylor, L.A.; Paylor, R. Altered Anxiety-Related and Social Behaviors in the Fmr1 Knockout Mouse Model of Fragile X Syndrome. Genes Brain Behav. 2005, 4, 420–430. [Google Scholar] [CrossRef]
- Shu, W.; Cho, J.Y.; Jiang, Y.; Zhang, M.; Weisz, D.; Elder, G.A.; Schmeidler, J.; De Gasperi, R.; Gama Sosa, M.A.; Rabidou, D.; et al. Altered Ultrasonic Vocalization in Mice with a Disruption in the Foxp2 Gene. Proc. Natl. Acad. Sci. USA 2005, 102, 9643–9648. [Google Scholar] [CrossRef]
- Enard, W.; Gehre, S.; Hammerschmidt, K.; Hölter, S.M.; Blass, T.; Somel, M.; Brückner, M.K.; Schreiweis, C.; Winter, C.; Sohr, R.; et al. A Humanized Version of Foxp2 Affects Cortico-Basal Ganglia Circuits in Mice. Cell 2009, 137, 961–971. [Google Scholar] [CrossRef]
- Fujita, E.; Tanabe, Y.; Shiota, A.; Ueda, M.; Suwa, K.; Momoi, M.Y.; Momoi, T. Ultrasonic Vocalization Impairment of Foxp2 (R552H) Knockin Mice Related to Speech-Language Disorder and Abnormality of Purkinje Cells. Proc. Natl. Acad. Sci. USA 2008, 105, 3117–3122. [Google Scholar] [CrossRef]
- DeLorey, T.M.; Sahbaie, P.; Hashemi, E.; Li, W.W.; Salehi, A.; Clark, D.J. Somatosensory and Sensorimotor Consequences Associated with the Heterozygous Disruption of the Autism Candidate Gene, Gabrb3. Behav. Brain Res. 2011, 216, 36–45. [Google Scholar] [CrossRef]
- Li, S.; Kumar, P.; Joshee, S.; Kirschstein, T.; Subburaju, S.; Khalili, J.S.; Kloepper, J.; Du, C.; Elkhal, A.; Szabó, G.; et al. Endothelial Cell-Derived GABA Signaling Modulates Neuronal Migration and Postnatal Behavior. Cell Res. 2018, 28, 221–248. [Google Scholar] [CrossRef]
- Orefice, L.L.L.; Zimmerman, A.L.L.; Chirila, A.M.M.; Sleboda, S.J.J.; Head, J.P.P.; Ginty, D.D.D. Peripheral Mechanosensory Neuron Dysfunction Underlies Tactile and Behavioral Deficits in Mouse Models of ASDs. Cell 2016, 166, 299–313. [Google Scholar] [CrossRef]
- DeLorey, T.M.; Sahbaie, P.; Hashemi, E.; Homanics, G.E.; Clark, J.D. Gabrb3 Gene Deficient Mice Exhibit Impaired Social and Exploratory Behaviors, Deficits in Non-Selective Attention and Hypoplasia of Cerebellar Vermal Lobules: A Potential Model of Autism Spectrum Disorder. Behav. Brain Res. 2008, 187, 207–220. [Google Scholar] [CrossRef]
- Carter, M.D.; Shah, C.R.; Muller, C.L.; Crawley, J.N.; Carneiro, A.M.D.; Veenstra-VanderWeele, J. Absence of Preference for Social Novelty and Increased Grooming in Integrin B3 Knockout Mice: Initial Studies and Future Directions. Autism Res. 2011, 4, 57–67. [Google Scholar] [CrossRef]
- Wickramasekara, R.N.; Robertson, B.; Hulen, J.; Hallgren, J.; Stessman, H.A.F. Differential Effects by Sex with Kmt5b Loss. Autism Res. 2021, 14, 1554–1571. [Google Scholar] [CrossRef] [PubMed]
- Gemelli, T.; Berton, O.; Nelson, E.D.; Perrotti, L.I.; Jaenisch, R.; Monteggia, L.M. Postnatal Loss of Methyl-CpG Binding Protein 2 in the Forebrain Is Sufficient to Mediate Behavioral Aspects of Rett Syndrome in Mice. Biol. Psychiatry 2006, 59, 468–476. [Google Scholar] [CrossRef] [PubMed]
- Guy, J.; Hendrich, B.; Holmes, M.; Martin, J.E.; Bird, A. A Mouse Mecp2-Null Mutation Causes Neurological Symptoms That Mimic Rett Syndrome. Nat. Genet. 2001, 27, 322–326. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.Z.; Akbarian, S.; Tudor, M.; Jaenisch, R. Deficiency of Methyl-CpG Binding Protein-2 in CNS Neurons Results in a Rett-like Phenotype in Mice. Nat. Genet. 2001, 27, 327–331. [Google Scholar] [CrossRef] [PubMed]
- Nott, A.; Cheng, J.; Gao, F.; Lin, Y.T.; Gjoneska, E.; Ko, T.; Minhas, P.; Zamudio, A.V.; Meng, J.; Zhang, F.; et al. Histone Deacetylase 3 Associates with MeCP2 to Regulate FOXO and Social Behavior. Nat. Neurosci. 2016, 19, 1497–1505. [Google Scholar] [CrossRef]
- Martins, G.J.; Plachez, C.; Powell, E.M. Loss of Embryonic MET Signaling Alters Profiles of Hippocampal Interneurons. Dev. Neurosci. 2007, 29, 143–158. [Google Scholar] [CrossRef]
- Cheng, Y.; Wang, Z.M.; Tan, W.; Wang, X.; Li, Y.; Bai, B.; Li, Y.; Zhang, S.F.; Yan, H.L.; Chen, Z.L.; et al. Partial Loss of Psychiatric Risk Gene Mir137 in Mice Causes Repetitive Behavior and Impairs Sociability and Learning via Increased Pde10a. Nat. Neurosci. 2018, 21, 1689–1703. [Google Scholar] [CrossRef]
- Singh, K.; Loreth, D.; Pöttker, B.; Hefti, K.; Innos, J.; Schwald, K.; Hengstler, H.; Menzel, L.; Sommer, C.J.; Radyushkin, K.; et al. Neuronal Growth and Behavioral Alterations in Mice Deficient for the Psychiatric Disease-Associated Negr1 Gene. Front. Mol. Neurosci. 2018, 11. [Google Scholar] [CrossRef]
- Runge, K.; Mathieu, R.; Bugeon, S.; Lafi, S.; Beurrier, C.; Sahu, S.; Schaller, F.; Loubat, A.; Herault, L.; Gaillard, S.; et al. Disruption of NEUROD2 Causes a Neurodevelopmental Syndrome with Autistic Features via Cell-Autonomous Defects in Forebrain Glutamatergic Neurons. Mol. Psychiatry 2021, 26, 6125–6148. [Google Scholar] [CrossRef]
- Gilbert, J.; O’Connor, M.; Templet, S.; Moghaddam, M.; Di Via Ioschpe, A.; Sinclair, A.; Zhu, L.Q.; Xu, W.; Man, H.Y. NEXMIF/KIDLIA Knock-out Mouse Demonstrates Autism-Like Behaviors, Memory Deficits, and Impairments in Synapse Formation and Function. J. Neurosci. 2020, 40, 237–254. [Google Scholar] [CrossRef]
- Blundell, J.; Tabuchi, K.; Bolliger, M.F.; Blaiss, C.A.; Brose, N.; Liu, X.; Südhof, T.C.; Powell, C.M. Increased Anxiety-like Behavior in Mice Lacking the Inhibitory Synapse Cell Adhesion Molecule Neuroligin 2. Genes Brain Behav. 2009, 8, 114–126. [Google Scholar] [CrossRef]
- Blundell, J.; Blaiss, C.A.; Etherton, M.R.; Espinosa, F.; Tabuchi, K.; Walz, C.; Bolliger, M.F.; Südhof, T.C.; Powell, C.M. Neuroligin-1 Deletion Results in Impaired Spatial Memory and Increased Repetitive Behavior. J. Neurosci. 2010, 30, 2115–2129. [Google Scholar] [CrossRef]
- Jamain, S.; Radyushkin, K.; Hammerschmidt, K.; Granon, S.; Boretius, S.; Varoqueaux, F.; Ramanantsoa, N.; Gallego, J.; Ronnenberg, A.; Winter, D.; et al. Reduced Social Interaction and Ultrasonic Communication in a Mouse Model of Monogenic Heritable Autism. Proc. Natl. Acad. Sci. USA 2008, 105, 1710–1715. [Google Scholar] [CrossRef]
- Radyushkin, K.; Hammerschmidt, K.; Boretius, S.; Varoqueaux, F.; El-Kordi, A.; Ronnenberg, A.; Winter, D.; Frahm, J.; Fischer, J.; Brose, N.; et al. Neuroligin-3-Deficient Mice: Model of a Monogenic Heritable Form of Autism with an Olfactory Deficit. Genes Brain Behav. 2009, 8, 416–425. [Google Scholar] [CrossRef]
- Pobbe, R.L.H.; Pearson, B.L.; Defensor, E.B.; Bolivar, V.J.; Young, W.S.; Lee, H.J.; Blanchard, D.C.; Blanchard, R.J. Oxytocin Receptor Knockout Mice Display Deficits in the Expression of Autism-Related Behaviors. Horm. Behav. 2012, 61, 436–444. [Google Scholar] [CrossRef]
- Sala, M.; Braida, D.; Lentini, D.; Busnelli, M.; Bulgheroni, E.; Capurro, V.; Finardi, A.; Donzelli, A.; Pattini, L.; Rubino, T.; et al. Pharmacologic Rescue of Impaired Cognitive Flexibility, Social Deficits, Increased Aggression, and Seizure Susceptibility in Oxytocin Receptor Null Mice: A Neurobehavioral Model of Autism. Biol. Psychiatry 2011, 69, 875–882. [Google Scholar] [CrossRef]
- Macbeth, A.H.; Stepp, J.E.; Lee, H.J.; Young, W.S.; Caldwell, H.K. Normal Maternal Behavior, but Increased Pup Mortality, in Conditional Oxytocin Receptor Knockout Females. Behav. Neurosci. 2010, 124, 677–685. [Google Scholar] [CrossRef]
- Hayashi, S.; Inoue, Y.; Hattori, S.; Kaneko, M.; Shioi, G.; Miyakawa, T.; Takeichi, M. Loss of X-Linked Protocadherin-19 Differentially Affects the Behavior of Heterozygous Female and Hemizygous Male Mice. Sci. Rep. 2017, 7, 5801. [Google Scholar] [CrossRef]
- Matsumura, K.; Seiriki, K.; Okada, S.; Nagase, M.; Ayabe, S.; Yamada, I.; Furuse, T.; Shibuya, H.; Yasuda, Y.; Yamamori, H.; et al. Pathogenic POGZ Mutation Causes Impaired Cortical Development and Reversible Autism-like Phenotypes. Nat. Commun. 2020, 11, 859. [Google Scholar] [CrossRef]
- Vogt, D.; Cho, K.K.A.; Lee, A.T.; Sohal, V.S.; Rubenstein, J.L.R. The Parvalbumin/Somatostatin Ratio Is Increased in Pten Mutant Mice and by Human PTEN ASD Alleles. Cell Rep. 2015, 11, 944–956. [Google Scholar] [CrossRef]
- Cupolillo, D.; Hoxha, E.; Faralli, A.; De Luca, A.; Rossi, F.; Tempia, F.; Carulli, D. Autistic-Like Traits and Cerebellar Dysfunction in Purkinje Cell PTEN Knock-Out Mice. Neuropsychopharmacology 2016, 41, 1457–1466. [Google Scholar] [CrossRef]
- Cabral-Costa, J.V.; Andreotti, D.Z.; Mello, N.P.; Scavone, C.; Camandola, S.; Kawamoto, E.M. Intermittent Fasting Uncovers and Rescues Cognitive Phenotypes in PTEN Neuronal Haploinsufficient Mice. Sci. Rep. 2018, 8, 8595. [Google Scholar] [CrossRef]
- Williams, M.R.; De-Spenza, T.; Li, M.; Gulledge, A.T.; Luikart, B.W. Hyperactivity of Newborn Pten Knock-out Neurons Results from Increased Excitatory Synaptic Drive. J. Neurosci. 2015, 35, 943–959. [Google Scholar] [CrossRef]
- Zhang, W.; Ma, L.; Yang, M.; Shao, Q.; Xu, J.; Lu, Z.; Zhao, Z.; Chen, R.; Chai, Y.; Chen, J.F. Cerebral Organoid and Mouse Models Reveal a RAB39b-PI3K-MTOR Pathway-Dependent Dysregulation of Cortical Development Leading to Macrocephaly/Autism Phenotypes. Genes Dev. 2020, 34, 580–597. [Google Scholar] [CrossRef]
- Nyarenchi, O.M.; Scherer, A.; Wilson, S.; Fulkerson, D.H. Cloacal Exstrophy with Extensive Chiari II Malformation: Case Report and Review of the Literature. Child’s Nerv. Syst. 2014, 30, 337–343. [Google Scholar] [CrossRef]
- Mullen, B.R.; Khialeeva, E.; Hoffman, D.B.; Ghiani, C.A.; Carpenter, E.M. Decreased Reelin Expression and Organophosphate Pesticide Exposure Alters Mouse Behaviour and Brain Morphology. ASN Neuro 2013, 5, AN20120060. [Google Scholar] [CrossRef]
- Coba, M.P.; Ramaker, M.J.; Ho, E.V.; Thompson, S.L.; Komiyama, N.H.; Grant, S.G.N.; Knowles, J.A.; Dulawa, S.C. Dlgap1 Knockout Mice Exhibit Alterations of the Postsynaptic Density and Selective Reductions in Sociability. Sci. Rep. 2018, 8, 2281. [Google Scholar] [CrossRef]
- Planells-Cases, R.; Caprini, M.; Zhang, J.; Rockenstein, E.M.; Rivera, R.R.; Murre, C.; Masliah, E.; Montal, M. Neuronal Death and Perinatal Lethality in Voltage-Gated Sodium Channel AII-Deficient Mice. Biophys. J. 2000, 78, 2878–2891. [Google Scholar] [CrossRef]
- Tatsukawa, T.; Raveau, M.; Ogiwara, I.; Hattori, S.; Miyamoto, H.; Mazaki, E.; Itohara, S.; Miyakawa, T.; Montal, M.; Yamakawa, K. Scn2a Haploinsufficient Mice Display a Spectrum of Phenotypes Affecting Anxiety, Sociability, Memory Flexibility and Ampakine CX516 Rescues Their Hyperactivity. Mol. Autism 2019, 10, 15. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Shi, Y.; Du, X.; Wang, J.; Zhang, Y.; Shan, S.; Yuan, Y.; Wang, R.; Zhou, C.; Liu, Y.; et al. SENP1 in the Retrosplenial Agranular Cortex Regulates Core Autistic-like Symptoms in Mice. Cell Rep. 2021, 37, 109939. [Google Scholar] [CrossRef] [PubMed]
- Deliu, E.; Arecco, N.; Morandell, J.; Dotter, C.P.; Contreras, X.; Girardot, C.; Käsper, E.-L.; Kozlova, A.; Kishi, K.; Chiaradia, I.; et al. Haploinsufficiency of the Intellectual Disability Gene SETD5 Disturbs Developmental Gene Expression and Cognition. Nat. Neurosci. 2018, 21, 1717–1727. [Google Scholar] [CrossRef] [PubMed]
- Chung, C.; Ha, S.; Kang, H.; Lee, J.; Um, S.M.; Yan, H.; Yoo, Y.E.; Yoo, T.; Jung, H.; Lee, D.; et al. Early Correction of N-Methyl-D-Aspartate Receptor Function Improves Autistic-like Social Behaviors in Adult Shank2−/− Mice. Biol. Psychiatry 2019, 85, 534–543. [Google Scholar] [CrossRef] [PubMed]
- Ha, S.; Lee, D.; Cho, Y.S.; Chung, C.; Yoo, Y.E.; Kim, J.; Lee, J.; Kim, W.; Kim, H.; Bae, Y.; et al. Cerebellar Shank2 Regulates Excitatory Synapse Density, Motor Coordination, and Specific Repetitive and Anxiety-Like Behaviors. J. Neurosci. 2016, 36, 12129–12143. [Google Scholar] [CrossRef]
- Lim, C.S.; Kim, H.; Yu, N.K.; Kang, S.J.; Kim, T.H.; Ko, H.G.; Lee, J.H.; Yang, J.E.; Ryu, H.H.; Park, T.; et al. Enhancing Inhibitory Synaptic Function Reverses Spatial Memory Deficits in Shank2 Mutant Mice. Neuropharmacology 2017, 112, 104–112. [Google Scholar] [CrossRef]
- Speed, H.E.; Kouser, M.; Xuan, Z.; Reimers, J.M.; Ochoa, C.F.; Gupta, N.; Liu, S.; Powell, C.M. Autism-Associated Insertion Mutation (InsG) of Shank3 Exon 21 Causes Impaired Synaptic Transmission and Behavioral Deficits. J. Neurosci. 2015, 35, 9648–9665. [Google Scholar] [CrossRef]
- Richter, M.; Murtaza, N.; Scharrenberg, R.; White, S.H.; Johanns, O.; Walker, S.; Yuen, R.K.C.; Schwanke, B.; Bedürftig, B.; Henis, M.; et al. Altered TAOK2 Activity Causes Autism-Related Neurodevelopmental and Cognitive Abnormalities through RhoA Signaling. Mol. Psychiatry 2019, 24, 1329–1350. [Google Scholar] [CrossRef]
- Fazel Darbandi, S.; Robinson Schwartz, S.E.; Qi, Q.; Catta-Preta, R.; Pai, E.L.L.; Mandell, J.D.; Everitt, A.; Rubin, A.; Krasnoff, R.A.; Katzman, S.; et al. Neonatal Tbr1 Dosage Controls Cortical Layer 6 Connectivity. Neuron 2018, 100, 831–845.e7. [Google Scholar] [CrossRef]
- Huang, T.N.; Yen, T.L.; Qiu, L.R.; Chuang, H.C.; Lerch, J.P.; Hsueh, Y.P. Haploinsufficiency of Autism Causative Gene Tbr1 Impairs Olfactory Discrimination and Neuronal Activation of the Olfactory System in Mice. Mol. Autism 2019, 10, 5. [Google Scholar] [CrossRef]
- Nakatani, J.; Tamada, K.; Hatanaka, F.; Ise, S.; Ohta, H.; Inoue, K.; Tomonaga, S.; Watanabe, Y.; Chung, Y.J.; Banerjee, R.; et al. Abnormal Behavior in a Chromosome- Engineered Mouse Model for Human 15q11-13 Duplication Seen in Autism. Cell 2009, 137, 1235–1246. [Google Scholar] [CrossRef]
- Shemesh, Y.; Forkosh, O.; Mahn, M.; Anpilov, S.; Sztainberg, Y.; Manashirov, S.; Shlapobersky, T.; Elliott, E.; Tabouy, L.; Ezra, G.; et al. Ucn3 and CRF-R2 in the Medial Amygdala Regulate Complex Social Dynamics. Nat. Neurosci. 2016, 19, 1489–1496. [Google Scholar] [CrossRef]
- Johnson, J.L.; Stoica, L.; Liu, Y.; Zhu, P.J.; Bhattacharya, A.; Buffington, S.A.; Huq, R.; Eissa, N.T.; Larsson, O.; Porse, B.T.; et al. Inhibition of Upf2-Dependent Nonsense-Mediated Decay Leads to Behavioral and Neurophysiological Abnormalities by Activating the Immune Response. Neuron 2019, 104, 665–679.e8. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Shum, E.Y.; Jones, S.H.; Lou, C.-H.; Dumdie, J.; Kim, H.; Roberts, A.J.; Jolly, L.A.; Espinoza, J.L.; Skarbrevik, D.M.; et al. A Upf3b-Mutant Mouse Model with Behavioral and Neurogenesis Defects. Mol. Psychiatry 2018, 23, 1773–1786. [Google Scholar] [CrossRef] [PubMed]
- Ornoy, A. Valproic Acid in Pregnancy: How Much Are We Endangering the Embryo and Fetus? Reprod. Toxicol. 2009, 28, 1–10. [Google Scholar] [CrossRef]
- Kolozsi, E.; Mackenzie, R.N.; Roullet, F.I.; Decatanzaro, D.; Foster, J.A. Prenatal Exposure to Valproic Acid Leads to Reduced Expression of Synaptic Adhesion Molecule Neuroligin 3 in Mice. Neuroscience 2009, 163, 1201–1210. [Google Scholar] [CrossRef] [PubMed]
- Bambini-Junior, V.; Rodrigues, L.; Behr, G.A.; Moreira, J.C.F.; Riesgo, R.; Gottfried, C. Animal Model of Autism Induced by Prenatal Exposure to Valproate: Behavioral Changes and Liver Parameters. Brain Res. 2011, 1408, 8–16. [Google Scholar] [CrossRef]
- Massa, V.; Cabrera, R.M.; Menegola, E.; Giavini, E.; Finnell, R.H. Valproic Acid-Induced Skeletal Malformations: Associated Gene Expression Cascades. Pharmacogenet. Genom. 2005, 15, 787–800. [Google Scholar] [CrossRef]
- Htway, S.M.; Sein, M.T.; Nohara, K.; Win-Shwe, T.T. Effects of Developmental Arsenic Exposure on the Social Behavior and Related Gene Expression in C3H Adult Male Mice. Int. J. Environ. Res. Public Health 2019, 16, 174. [Google Scholar] [CrossRef]
- Yu, C.; Tai, F.; Song, Z.; Wu, R.; Zhang, X.; He, F. Pubertal Exposure to Bisphenol A Disrupts Behavior in Adult C57BL/6J Mice. Environ. Toxicol. Pharmacol. 2011, 31, 88–99. [Google Scholar] [CrossRef]
- Lan, A.; Kalimian, M.; Amram, B.; Kofman, O. Prenatal Chlorpyrifos Leads to Autism-like Deficits in C57Bl6/J Mice. Environ. Health 2017, 16, 43. [Google Scholar] [CrossRef]
- Hanks, A.N.; Dlugolenski, K.; Hughes, Z.A.; Seymour, P.A.; Majchrzak, M.J. Pharmacological Disruption of Mouse Social Approach Behavior: Relevance to Negative Symptoms of Schizophrenia. Behav. Brain Res. 2013, 252, 405–414. [Google Scholar] [CrossRef]
- Gao, X.M.; Elmer, G.I.; Adams-Huet, B.; Tamminga, C.A. Social Memory in Mice: Disruption with an NMDA Antagonist and Attenuation with Antipsychotic Drugs. Pharmacol. Biochem. Behav. 2009, 92, 236–242. [Google Scholar] [CrossRef]
- Qiao, H.; Noda, Y.; Kamei, H.; Nagai, T.; Furukawa, H.; Miura, H.; Kayukawa, Y.; Ohta, T.; Nabeshima, T. Clozapine, but Not Haloperidol, Reverses Social Behavior Deficit in Mice during Withdrawal from Chronic Phencyclidine Treatment. Neuroreport 2001, 12, 11–15. [Google Scholar] [CrossRef]
- Norregaard, L.; Gether, U. The Monoamine Neurotransmitter Transporters: Structure, Conformational Changes and Molecular Gating. Curr. Opin. Drug Discov. Devel. 2001, 4, 591–601. [Google Scholar]
- Zhang, H.; Li, S.; Wang, M.; Vukusic, B.; Pristupa, Z.B.; Liu, F. Regulation of Dopamine Transporter Activity by Carboxypeptidase E. Mol. Brain 2009, 2, 10. [Google Scholar] [CrossRef]
- Gainetdinov, R.R.; Caron, M.G. Genetics of Childhood Disorders: XXIV. ADHD, Part 8: Hyperdopaminergic Mice as an Animal Model of ADHD. J. Am. Acad. Child Adolesc. Psychiatry 2001, 40, 380–382. [Google Scholar] [CrossRef]
- Sontag, T.A.; Tucha, O.; Walitza, S.; Lange, K.W. Animal Models of Attention Deficit/Hyperactivity Disorder (ADHD): A Critical Review. ADHD Atten. Deficit Hyperact. Disord. 2010, 2, 1–20. [Google Scholar] [CrossRef]
- Jones, S.R.; Gainetdinov, R.R.; Wightman, R.M.; Caron, M.G. Mechanisms of Amphetamine Action Revealed in Mice Lacking the Dopamine Transporter. J. Neurosci. 1998, 18, 1979–1986. [Google Scholar] [CrossRef]
- Gainetdinov, R.R.; Jones, S.R.; Fumagalli, F.; Wightman, R.M.; Caron, M.G. Re-Evaluation of the Role of the Dopamine Transporter in Dopamine System Homeostasis1Published on the World Wide Web on 27 January 1998.1. Brain Res. Rev. 1998, 26, 148–153. [Google Scholar] [CrossRef]
- Jaber, M.; Dumartin, B.; Sagné, C.; Haycock, J.W.; Roubert, C.; Giros, B.; Bloch, B.; Caron, M.G. Differential Regulation of Tyrosine Hydroxylase in the Basal Ganglia of Mice Lacking the Dopamine Transporter. Eur. J. Neurosci. 1999, 11, 3499–3511. [Google Scholar] [CrossRef]
- Gainetdinov, R.R.; Wetsel, W.C.; Jones, S.R.; Levin, E.D.; Jaber, M.; Caron, M.G. Role of Serotonin in the Paradoxical Calming Effect of Psychostimulants on Hyperactivity. Science 1999, 283, 397–401. [Google Scholar] [CrossRef]
- Giros, B.; Jaber, M.; Jones, S.R.; Wightman, R.M.; Caron, M.G. Hyperlocomotion and Indifference to Cocaine and Amphetamine in Mice Lacking the Dopamine Transporter. Nature 1996, 379, 606–612. [Google Scholar] [CrossRef] [PubMed]
- Bruno, K.J.; Freet, C.S.; Twining, R.C.; Egami, K.; Grigson, P.S.; Hess, E.J. Abnormal Latent Inhibition and Impulsivity in Coloboma Mice, a Model of ADHD. Neurobiol. Dis. 2007, 25, 206–216. [Google Scholar] [CrossRef] [PubMed]
- Hess, E.J.; Collins, K.A.; Wilson, M.C. Mouse Model of Hyperkinesis Implicates SNAP-25 in Behavioral Regulation. J. Neurosci. 1996, 16, 3104–3111. [Google Scholar] [CrossRef] [PubMed]
- Heyser, C.J.; Wilson, M.C.; Gold, L.H. Coloboma Hyperactive Mutant Exhibits Delayed Neurobehavioral Developmental Milestones. Dev. Brain Res. 1995, 89, 264–269. [Google Scholar] [CrossRef]
- Wilson, M.C. Coloboma Mouse Mutant as an Animal Model of Hyperkinesis and Attention Deficit Hyperactivity Disorder. In Proceedings of the Neuroscience and Biobehavioral Reviews, Pergamon, Turkey, 1 January 2000; Volume 24, pp. 51–57. [Google Scholar]
- Steffensen, S.C.; Wilson, M.C.; Henriksen, S.J. Coloboma Contiguous Gene Deletion EncompassingSnap Alters Hippocampal Plasticity. Synapse 1996, 22, 281–289. [Google Scholar] [CrossRef]
- Lipp, H.-P.; Wahlsten, D. Absence of the Corpus Callosum. In Genetically Defined Animal Models of Neurobehavioral Dysfunctions; Birkhäuser Boston: Boston, MA, USA, 1992; pp. 217–252. [Google Scholar]
- Weiss, R.E.; Refetoff, S. Resistance to Thyroid Hormone. Rev. Endocr. Metab. Disord. 2000, 1, 97–108. [Google Scholar] [CrossRef]
- McDonald, M.P.; Wong, R.; Goldstein, G.; Weintraub, B.; Cheng, S.Y.; Crawley, J.N. Hyperactivity and Learning Deficits in Transgenic Mice Bearing a Human Mutant Thyroid Hormone B1 Receptor Gene. Learn. Mem. 1998, 5, 289–301. [Google Scholar] [CrossRef]
- Siesser, W.B.; Zhao, J.; Miller, L.R.; Cheng, S.Y.; McDonald, M.P. Transgenic Mice Expressing a Human Mutant B1 Thyroid Receptor Are Hyperactive, Impulsive, and Inattentive. Genes Brain Behav. 2006, 5, 282–297. [Google Scholar] [CrossRef]
- Thompson, C.C. Thyroid Hormone Action in Neural Development. Cereb. Cortex 2000, 10, 939–945. [Google Scholar] [CrossRef]
- Nakajo, S.; Tsukada, K.; Omata, K.; Nakamura, Y.; Nakaya, K. A New Brain-Specific 14-KDa Protein Is a Phosphoprotein. Its Complete Amino Acid Sequence and Evidence for Phosphorylation. Eur. J. Biochem. 1993, 217, 1057–1063. [Google Scholar] [CrossRef]
- Totterdell, S.; Hanger, D.; Meredith, G.E. The Ultrastructural Distribution of Alpha-Synuclein-like Protein in Normal Mouse Brain. Brain Res. 2004, 1004, 61–72. [Google Scholar] [CrossRef]
- Totterdell, S.; Meredith, G.E. Localization of Alpha-Synuclein to Identified Fibers and Synapses in the Normal Mouse Brain. Neuroscience 2005, 135, 907–913. [Google Scholar] [CrossRef]
- Chartier-Harlin, M.C.; Kachergus, J.; Roumier, C.; Mouroux, V.; Douay, X.; Lincoln, S.; Levecque, C.; Larvor, L.; Andrieux, J.; Hulihan, M.; et al. α-Synuclein Locus Duplication as a Cause of Familial Parkinson’s Disease. Lancet 2004, 364, 1167–1169. [Google Scholar] [CrossRef]
- Zarranz, J.J.; Alegre, J.; Gómez-Esteban, J.C.; Lezcano, E.; Ros, R.; Ampuero, I.; Vidal, L.; Hoenicka, J.; Rodriguez, O.; Atarés, B.; et al. The New Mutation, E46K, of α-Synuclein Causes Parkinson and Lewy Body Dementia. Ann. Neurol. 2004, 55, 164–173. [Google Scholar] [CrossRef]
- Decressac, M.; Mattsson, B.; Lundblad, M.; Weikop, P.; Björklund, A. Progressive Neurodegenerative and Behavioural Changes Induced by AAV-Mediated Overexpression of α-Synuclein in Midbrain Dopamine Neurons. Neurobiol. Dis. 2012, 45, 939–953. [Google Scholar] [CrossRef]
- Matsui, H.; Kenmochi, N.; Namikawa, K. Age- and α-Synuclein-Dependent Degeneration of Dopamine and Noradrenaline Neurons in the Annual Killifish Nothobranchius Furzeri. Cell Rep. 2019, 26, 1727–1733.e6. [Google Scholar] [CrossRef]
- Senior, S.L.; Ninkina, N.; Deacon, R.; Bannerman, D.; Buchman, V.L.; Cragg, S.J.; Wade-Martins, R. Increased Striatal Dopamine Release and Hyperdopaminergic-like Behaviour in Mice Lacking Both Alpha-Synuclein and Gamma-Synuclein. Eur. J. Neurosci. 2008, 27, 947–957. [Google Scholar] [CrossRef]
- Faraone, S.V.; Doyle, A.E. The Nature and Heritability of Attention-Deficit/Hyperactivity Disorder. Child Adolesc. Psychiatr. Clin. N. Am. 2001, 10, 299–316. [Google Scholar] [CrossRef]
- Faraone, S.V.; Biederman, J.; Weiffenbach, B.; Keith, T.; Chu, M.P.; Weaver, A.; Spencer, T.J.; Wilens, T.E.; Frazier, J.; Cleves, M.; et al. Brief Reports Dopamine D 4 Gene 7-Repeat Allele and Attention Deficit Hyperactivity Disorder. Am. J. Psychiatry 1999, 156, 768. [Google Scholar] [CrossRef]
- Grady, D.L.; Chi, H.-C.; Ding, Y.-C.; Smith, M.; Wang, E.; Schuck, S.; Flodman, P.; Spence, M.A.; Swanson, J.M.; Moyzis, R.K. High Prevalence of Rare Dopamine Receptor D4 Alleles in Children Diagnosed with Attention-Deficit Hyperactivity Disorder. Mol. Psychiatry 2003, 8, 536–545. [Google Scholar] [CrossRef]
- Swanson, J.M.; Sunohara, G.A.; Kennedy, J.L.; Regino, R.; Fineberg, E.; Wigal, T.; Lerner, M.; Williams, L.; LaHoste, G.J.; Wigal, S. Association of the Dopamine Receptor D4 (DRD4) Gene with a Refined Phenotype of Attention Deficit Hyperactivity Disorder (ADHD): A Family-Based Approach. Mol. Psychiatry 1998, 3, 38–41. [Google Scholar] [CrossRef] [PubMed]
- Avale, M.E.; Falzone, T.L.; Gelman, D.M.; Low, M.J.; Grandy, D.K.; Rubinstein, M. The Dopamine D4 Receptor Is Essential for Hyperactivity and Impaired Behavioral Inhibition in a Mouse Model of Attention Deficit/Hyperactivity Disorder. Mol. Psychiatry 2004, 9, 718–726. [Google Scholar] [CrossRef]
- Safe, S.H. Polychlorinated Biphenyls (PCBs): Environmental Impact, Biochemical and Toxic Responses, and Implications for Risk Assessment. Crit. Rev. Toxicol. 1994, 24, 87–149. [Google Scholar] [CrossRef] [PubMed]
- Shain, W.; Bush, B.; Seegal, R. Neurotoxicity of Polychlorinated Biphenyls: Structure-Activity Relationship of Individual Congeners. Toxicol. Appl. Pharmacol. 1991, 111, 33–42. [Google Scholar] [CrossRef]
- Silbergeld, E.K.; Goldberg, A.M. Lead-Induced Behavioral Dysfunction: An Animal Model of Hyperactivity. Exp. Neurol. 1974, 42, 146–157. [Google Scholar] [CrossRef]
- Silbergeld, E.K.; Goldberg, A.M. Pharmacological and Neurochemical Investigations of Lead-Induced Hyperactivity. Neuropharmacology 1975, 14, 431–444. [Google Scholar] [CrossRef]
- Zhu, J.; Fan, F.; McCarthy, D.M.; Zhang, L.; Cannon, E.N.; Spencer, T.J.; Biederman, J.; Bhide, P.G. A Prenatal Nicotine Exposure Mouse Model of Methylphenidate Responsive ADHD-Associated Cognitive Phenotypes. Int. J. Dev. Neurosci. 2017, 58, 26–34. [Google Scholar] [CrossRef]
- Buck, J.M.; Sanders, K.N.; Wageman, C.R.; Knopik, V.S.; Stitzel, J.A.; O’Neill, H.C. Developmental Nicotine Exposure Precipitates Multigenerational Maternal Transmission of Nicotine Preference and ADHD-like Behavioral, Rhythmometric, Neuropharmacological, and Epigenetic Anomalies in Adolescent Mice. Neuropharmacology 2019, 149, 66–82. [Google Scholar] [CrossRef]
- McCarthy, D.M.; Morgan, T.J.; Lowe, S.E.; Williamson, M.J.; Spencer, T.J.; Biederman, J.; Bhide, P.G. Nicotine Exposure of Male Mice Produces Behavioral Impairment in Multiple Generations of Descendants. PLoS Biol. 2018, 16, e2006497. [Google Scholar] [CrossRef]
- Son, G.H.; Chung, S.; Geum, D.; Kang, S.S.; Choi, W.S.; Kim, K.; Choi, S. Hyperactivity and Alteration of the Midbrain Dopaminergic System in Maternally Stressed Male Mice Offspring. Biochem. Biophys. Res. Commun. 2007, 352, 823–829. [Google Scholar] [CrossRef]
- Laucht, M.; Esser, G.; Baving, L.; Gerhold, M.; Hoesch, I.; Ihle, W.; Steigleider, P.; Stock, B.; Stoehr, R.M.; Weindrich, D.; et al. Behavioral Sequelae of Perinatal Insults and Early Family Adversity at 8 Years of Age. J. Am. Acad. Child Adolesc. Psychiatry 2000, 39, 1229–1237. [Google Scholar] [CrossRef]
- Mcintosh, D.E.; Mulkins, R.S.; Dean, R.S. Utilization of Maternal Perinatal Risk Indicators in the Differential Diagnosis of Adhd and Uadd Children. Int. J. Neurosci. 1995, 81, 35–46. [Google Scholar] [CrossRef]
- Nasevicius, A.; Ekker, S.C. Effective Targeted Gene ‘Knockdown’ in Zebrafish. Nat. Genet. 2000, 26, 216–220. [Google Scholar] [CrossRef]
- Bill, B.R.; Petzold, A.M.; Clark, K.J.; Schimmenti, L.A.; Ekker, S.C. A Primer for Morpholino Use in Zebrafish. Zebrafish 2009, 6, 69–77. [Google Scholar] [CrossRef]
- Eisen, J.S.; Smith, J.C. Controlling Morpholino Experiments: Don’t Stop Making Antisense. Development 2008, 135, 1735–1743. [Google Scholar] [CrossRef]
- Stainier, D.Y.R.; Raz, E.; Lawson, N.D.; Ekker, S.C.; Burdine, R.D.; Eisen, J.S.; Ingham, P.W.; Schulte-Merker, S.; Yelon, D.; Weinstein, B.M.; et al. Guidelines for Morpholino Use in Zebrafish. PLoS Genet. 2017, 13, e1007000. [Google Scholar] [CrossRef]
- Doyon, Y.; McCammon, J.M.; Miller, J.C.; Faraji, F.; Ngo, C.; Katibah, G.E.; Amora, R.; Hocking, T.D.; Zhang, L.; Rebar, E.J.; et al. Heritable Targeted Gene Disruption in Zebrafish Using Designed Zinc-Finger Nucleases. Nat. Biotechnol. 2008, 26, 702–708. [Google Scholar] [CrossRef]
- Huang, P.; Xiao, A.; Zhou, M.; Zhu, Z.; Lin, S.; Zhang, B. Heritable Gene Targeting in Zebrafish Using Customized TALENs. Nat. Biotechnol. 2011, 29, 699–700. [Google Scholar] [CrossRef]
- Wang, H.; Yang, H.; Shivalila, C.S.; Dawlaty, M.M.; Cheng, A.W.; Zhang, F.; Jaenisch, R. One-Step Generation of Mice Carrying Mutations in Multiple Genes by CRISPR/Cas-Mediated Genome Engineering. Cell 2013, 153, 910–918. [Google Scholar] [CrossRef]
- Varshney, G.K.; Burgess, S.M. Mutagenesis and Phenotyping Resources in Zebrafish for Studying Development and Human Disease. Brief. Funct. Genom. 2014, 13, 82–94. [Google Scholar] [CrossRef]
- Vejnar, C.E.; Moreno-Mateos, M.A.; Cifuentes, D.; Bazzini, A.A.; Giraldez, A.J. Optimized CRISPR–Cas9 System for Genome Editing in Zebrafish. Cold Spring Harb. Protoc. 2016, 2016, pdb.prot086850. [Google Scholar] [CrossRef]
- Hwang, W.Y.; Fu, Y.; Reyon, D.; Maeder, M.L.; Tsai, S.Q.; Sander, J.D.; Peterson, R.T.; Yeh, J.R.J.; Joung, J.K. Efficient Genome Editing in Zebrafish Using a CRISPR-Cas System. Nat. Biotechnol. 2013, 31, 227–229. [Google Scholar] [CrossRef]
- Moreno-Mateos, M.A.; Vejnar, C.E.; Beaudoin, J.-D.; Fernandez, J.P.; Mis, E.K.; Khokha, M.K.; Giraldez, A.J. CRISPRscan: Designing Highly Efficient SgRNAs for CRISPR-Cas9 Targeting in Vivo. Nat. Methods 2015, 12, 982–988. [Google Scholar] [CrossRef]
- Liu, X.; Hu, G.; Ye, J.; Ye, B.; Shen, N.; Tao, Y.; Zhang, X.; Fan, Y.; Liu, H.; Zhang, Z.; et al. De Novo ARID1B Mutations Cause Growth Delay Associated with Aberrant Wnt/β–Catenin Signaling. Hum. Mutat. 2020, 41, 1012–1024. [Google Scholar] [CrossRef] [PubMed]
- Ishibashi, M.; Manning, E.; Shoubridge, C.; Krecsmarik, M.; Hawkins, T.A.; Giacomotto, J.; Zhao, T.; Mueller, T.; Bader, P.I.; Cheung, S.W.; et al. Copy Number Variants in Patients with Intellectual Disability Affect the Regulation of ARX Transcription Factor Gene. Hum. Genet. 2015, 134, 1163–1182. [Google Scholar] [CrossRef] [PubMed]
- Oksenberg, N.; Stevison, L.; Wall, J.D.; Ahituv, N. Function and Regulation of AUTS2, a Gene Implicated in Autism and Human Evolution. PLoS Genet. 2013, 9, e1003221. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, K.V.; Hennessey, J.A.; Barnett, A.S.; Yin, X.; Stadt, H.A.; Foster, E.; Shah, R.A.; Yazawa, M.; Dolmetsch, R.E.; Kirby, M.L.; et al. Calcium Influx through L-Type CaV1.2 Ca2+ Channels Regulates Mandibular Development. J. Clin. Investig. 2013, 123, 1638–1646. [Google Scholar] [CrossRef]
- Patowary, A.; Won, S.Y.; Oh, S.J.; Nesbitt, R.R.; Archer, M.; Nickerson, D.; Raskind, W.H.; Bernier, R.; Lee, J.E.; Brkanac, Z. Family-Based Exome Sequencing and Case-Control Analysis Implicate CEP41 as an ASD Gene. Transl. Psychiatry 2019, 9, 4. [Google Scholar] [CrossRef]
- Suls, A.; Jaehn, J.A.; Kecskés, A.; Weber, Y.; Weckhuysen, S.; Craiu, D.C.; Siekierska, A.; Djémié, T.; Afrikanova, T.; Gormley, P.; et al. De Novo Loss-of-Function Mutations in CHD2 Cause a Fever-Sensitive Myoclonic Epileptic Encephalopathy Sharing Features with Dravet Syndrome. Am. J. Hum. Genet. 2013, 93, 967–975. [Google Scholar] [CrossRef]
- Bernier, R.; Golzio, C.; Xiong, B.; Stessman, H.A.; Coe, B.P.; Penn, O.; Witherspoon, K.; Gerdts, J.; Baker, C.; Vulto-Van Silfhout, A.T.; et al. Disruptive CHD8 Mutations Define a Subtype of Autism Early in Development. Cell 2014, 158, 263–276. [Google Scholar] [CrossRef]
- Sugathan, A.; Biagioli, M.; Golzio, C.; Erdin, S.; Blumenthal, I.; Manavalan, P.; Ragavendran, A.; Brand, H.; Lucente, D.; Miles, J.; et al. CHD8 Regulates Neurodevelopmental Pathways Associated with Autism Spectrum Disorder in Neural Progenitors. Proc. Natl. Acad. Sci. USA 2014, 111, E4468–E4477. [Google Scholar] [CrossRef]
- Hoffman, E.J.; Turner, K.J.; Fernandez, J.M.; Cifuentes, D.; Ghosh, M.; Ijaz, S.; Jain, R.A.; Kubo, F.; Bill, B.R.; Baier, H.; et al. Estrogens Suppress a Behavioral Phenotype in Zebrafish Mutants of the Autism Risk Gene, CNTNAP2. Neuron 2016, 89, 725–733. [Google Scholar] [CrossRef]
- Turner, T.N.; Sharma, K.; Oh, E.C.; Liu, Y.P.; Collins, R.L.; Sosa, M.X.; Auer, D.R.; Brand, H.; Sanders, S.J.; Moreno-De-Luca, D.; et al. Loss of δ-Catenin Function in Severe Autism. Nature 2015, 520, 51–56. [Google Scholar] [CrossRef]
- Kim, O.H.; Cho, H.J.; Han, E.; Hong, T.I.; Ariyasiri, K.; Choi, J.H.; Hwang, K.S.; Jeong, Y.M.; Yang, S.Y.; Yu, K.; et al. Zebrafish Knockout of Down Syndrome Gene, DYRK1A, Shows Social Impairments Relevant to Autism. Mol. Autism 2017, 8, 1–14. [Google Scholar] [CrossRef]
- Sicca, F.; Ambrosini, E.; Marchese, M.; Sforna, L.; Servettini, I.; Valvo, G.; Brignone, M.S.; Lanciotti, A.; Moro, F.; Grottesi, A.; et al. Gain-of-Function Defects of Astrocytic Kir4.1 Channels in Children with Autism Spectrum Disorders and Epilepsy. Sci. Rep. 2016, 6, 34325. [Google Scholar] [CrossRef]
- Bögershausen, N.; Tsai, I.C.; Pohl, E.; Kiper, P.O.S.; Beleggia, F.; Ferda Percin, E.; Keupp, K.; Matchan, A.; Milz, E.; Alanay, Y.; et al. RAP1-Mediated MEK/ERK Pathway Defects in Kabuki Syndrome. J. Clin. Investig. 2015, 125, 3585–3599. [Google Scholar] [CrossRef]
- Van Laarhoven, P.M.; Neitzel, L.R.; Quintana, A.M.; Geiger, E.A.; Zackai, E.H.; Clouthier, D.E.; Artinger, K.B.; Ming, J.E.; Shaikh, T.H. Kabuki Syndrome Genes KMT2D and KDM6A: Functional Analyses Demonstrate Critical Roles in Craniofacial, Heart and Brain Development. Hum. Mol. Genet. 2015, 24, 4443–4453. [Google Scholar] [CrossRef]
- Leong, W.Y.; Lim, Z.H.; Korzh, V.; Pietri, T.; Goh, E.L.K. Methyl-CpG Binding Protein 2 (Mecp2) Regulates Sensory Function through Sema5b and Robo2. Front. Cell. Neurosci. 2015, 9, 481. [Google Scholar] [CrossRef]
- Van Der Vaart, M.; Svoboda, O.; Weijts, B.G.; Espín-Palazón, R.; Sapp, V.; Pietri, T.; Bagnat, M.; Muotri, A.R.; Traver, D. Mecp2 Regulates Tnfa during Zebrafish Embryonic Development and Acute Inflammation. DMM Dis. Model. Mech. 2017, 10, 1439–1451. [Google Scholar] [CrossRef]
- Elsen, G.E.; Choi, L.Y.; Prince, V.E.; Ho, R.K. The Autism Susceptibility Gene Met Regulates Zebrafish Cerebellar Development and Facial Motor Neuron Migration. Dev. Biol. 2009, 335, 78–92. [Google Scholar] [CrossRef]
- Blanchet, P.; Bebin, M.; Bruet, S.; Cooper, G.M.; Thompson, M.L.; Duban-Bedu, B.; Gerard, B.; Piton, A.; Suckno, S.; Deshpande, C.; et al. MYT1L Mutations Cause Intellectual Disability and Variable Obesity by Dysregulating Gene Expression and Development of the Neuroendocrine Hypothalamus. PLoS Genet. 2017, 13, e1006957. [Google Scholar] [CrossRef]
- Miller, A.C.; Voelker, L.H.; Shah, A.N.; Moens, C.B. Neurobeachin Is Required Postsynaptically for Electrical and Chemical Synapse Formation. Curr. Biol. 2015, 25, 16–28. [Google Scholar] [CrossRef]
- Ruzzo, E.K.; Pérez-Cano, L.; Jung, J.Y.; Wang, L.K.; Kashef-Haghighi, D.; Hartl, C.; Singh, C.; Xu, J.; Hoekstra, J.N.; Leventhal, O.; et al. Inherited and De Novo Genetic Risk for Autism Impacts Shared Networks. Cell 2019, 178, 850–866.e26. [Google Scholar] [CrossRef]
- Ribeiro, D.; Nunes, A.R.; Gliksberg, M.; Anbalagan, S.; Levkowitz, G.; Oliveira, R.F. Oxytocin Receptor Signalling Modulates Novelty Recognition but Not Social Preference in Zebrafish. J. Neuroendocrinol. 2020, 32, 12834. [Google Scholar] [CrossRef]
- Vecchia, E.D.; Di Donato, V.; Young, A.M.J.; Del Bene, F.; Norton, W.H.J. Reelin Signaling Controls the Preference for Social Novelty in Zebrafish. Front. Behav. Neurosci. 2019, 13, 214. [Google Scholar] [CrossRef]
- Plaster, N.; Sonntag, C.; Schilling, T.F.; Hammerschmidt, M. REREa/Atrophin-2 Interacts with Histone Deacetylase and Fgf8 Signaling to Regulate Multiple Processes of Zebrafish Development. Dev. Dyn. 2007, 236, 1891–1904. [Google Scholar] [CrossRef]
- Liu, C.-X.; Li, C.-Y.; Hu, C.-C.; Wang, Y.; Lin, J.; Jiang, Y.-H.; Li, Q.; Xu, X. CRISPR/Cas9-Induced Shank3b Mutant Zebrafish Display Autism-like Behaviors. Mol. Autism 2018, 9, 23. [Google Scholar] [CrossRef]
- Kozol, R.A.; James, D.M.; Varela, I.; Sumathipala, S.H.; Züchner, S.; Dallman, J.E. Restoring Shank3 in the Rostral Brainstem of Shank3ab−/− Zebrafish Autism Models Rescues Sensory Deficits. Commun. Biol. 2021, 4, 1411. [Google Scholar] [CrossRef]
- Kozol, R.A.; Cukier, H.N.; Zou, B.; Mayo, V.; De Rubeis, S.; Cai, G.; Griswold, A.J.; Whitehead, P.L.; Haines, J.L.; Gilbert, J.R.; et al. Two Knockdown Models of the Autism Genes SYNGAP1 and SHANK3 in Zebrafish Produce Similar Behavioral Phenotypes Associated with Embryonic Disruptions of Brain Morphogenesis. Hum. Mol. Genet. 2015, 24, 4006–4023. [Google Scholar] [CrossRef]
- Anderson, J.L.; Mulligan, T.S.; Shen, M.C.; Wang, H.; Scahill, C.M.; Tan, F.J.; Du, S.J.; Busch-Nentwich, E.M.; Farber, S.A. MRNA Processing in Mutant Zebrafish Lines Generated by Chemical and CRISPR-Mediated Mutagenesis Produces Unexpected Transcripts That Escape Nonsense-Mediated Decay. PLoS Genet. 2017, 13, e1007105. [Google Scholar] [CrossRef]
- Rea, V.; Van Raay, T.J. Using Zebrafish to Model Autism Spectrum Disorder: A Comparison of ASD Risk Genes Between Zebrafish and Their Mammalian Counterparts. Front. Mol. Neurosci. 2020, 13, 207. [Google Scholar] [CrossRef] [PubMed]
- Smith, V.; Brown, N. Prenatal Valproate Exposure and Risk of Autism Spectrum Disorders and Childhood Autism. Arch. Dis. Child. Educ. Pract. Ed. 2014, 99, 198. [Google Scholar] [PubMed]
- Bromley, R.L.; Mawer, G.E.; Briggs, M.; Cheyne, C.; Clayton-Smith, J.; García-Fiñana, M.; Kneen, R.; Lucas, S.B.; Shallcross, R.; Baker, G.A. The Prevalence of Neurodevelopmental Disorders in Children Prenatally Exposed to Antiepileptic Drugs. J. Neurol. Neurosurg. Psychiatry 2013, 84, 637–643. [Google Scholar] [CrossRef] [PubMed]
- Wood, A. Prenatal Exposure to Sodium Valproate Is Associated with Increased Risk of Childhood Autism and Autistic Spectrum Disorder. Evid.-Based Nurs. 2014, 17, 84. [Google Scholar] [CrossRef]
- Sailer, L.; Duclot, F.; Wang, Z.; Kabbaj, M. Consequences of Prenatal Exposure to Valproic Acid in the Socially Monogamous Prairie Voles. Sci. Rep. 2019, 9, 2453. [Google Scholar] [CrossRef]
- Lee, Y.; Kim, Y.H.; Yun, J.S.; Lee, C.J. Valproic Acid Decreases the Proliferation of Telencephalic Cells in Zebrafish Larvae. Neurotoxicol. Teratol. 2013, 39, 91–99. [Google Scholar] [CrossRef]
- Baronio, D.; Puttonen, H.A.J.; Sundvik, M.; Semenova, S.; Lehtonen, E.; Panula, P. Embryonic Exposure to Valproic Acid Affects the Histaminergic System and the Social Behaviour of Adult Zebrafish (Danio rerio). Br. J. Pharmacol. 2018, 175, 797–809. [Google Scholar] [CrossRef]
- Zimmermann, F.F.; Gaspary, K.V.; Siebel, A.M.; Bonan, C.D. Oxytocin Reversed MK-801-Induced Social Interaction and Aggression Deficits in Zebrafish. Behav. Brain Res. 2016, 311, 368–374. [Google Scholar] [CrossRef]
- Seibt, K.J.; Piato, A.L.; da Luz Oliveira, R.; Capiotti, K.M.; Vianna, M.R.; Bonan, C.D. Antipsychotic Drugs Reverse MK-801-Induced Cognitive and Social Interaction Deficits in Zebrafish (Danio rerio). Behav. Brain Res. 2011, 224, 135–139. [Google Scholar] [CrossRef]
- Miller, N.; Greene, K.; Dydinski, A.; Gerlai, R. Effects of Nicotine and Alcohol on Zebrafish (Danio rerio) Shoaling. Behav. Brain Res. 2013, 240, 192–196. [Google Scholar] [CrossRef]
- Scerbina, T.; Chatterjee, D.; Gerlai, R. Dopamine Receptor Antagonism Disrupts Social Preference in Zebrafish: A Strain Comparison Study. Amino Acids 2012, 43, 2059–2072. [Google Scholar] [CrossRef]
- Wang, Y.; Zhong, H.; Wang, C.; Gao, D.; Zhou, Y.; Zuo, Z. Maternal Exposure to the Water Soluble Fraction of Crude Oil, Lead and Their Mixture Induces Autism-like Behavioral Deficits in Zebrafish (Danio rerio) Larvae. Ecotoxicol. Environ. Saf. 2016, 134, 23–30. [Google Scholar] [CrossRef]
- Eddins, D.; Cerutti, D.; Williams, P.; Linney, E.; Levin, E.D. Zebrafish Provide a Sensitive Model of Persisting Neurobehavioral Effects of Developmental Chlorpyrifos Exposure: Comparison with Nicotine and Pilocarpine Effects and Relationship to Dopamine Deficits. Neurotoxicol. Teratol. 2010, 32, 99–108. [Google Scholar] [CrossRef]
- Regan, S.L.; Williams, M.T.; Vorhees, C.V. Latrophilin-3 Disruption: Effects on Brain and Behavior. Neurosci. Biobehav. Rev. 2021, 127, 619–629. [Google Scholar] [CrossRef]
- Chiu, C.N.; Rihel, J.; Lee, D.A.; Singh, C.; Mosser, E.A.; Chen, S.; Sapin, V.; Pham, U.; Engle, J.; Niles, B.J.; et al. A Zebrafish Genetic Screen Identifies Neuromedin U as a Regulator of Sleep/Wake States. Neuron 2016, 89, 842–856. [Google Scholar] [CrossRef]
- Huang, J.; Zhong, Z.; Wang, M.; Chen, X.; Tan, Y.; Zhang, S.; He, W.; He, X.; Huang, G.; Lu, H.; et al. Circadian Modulation of Dopamine Levels and Dopaminergic Neuron Development Contributes to Attention Deficiency and Hyperactive Behavior. J. Neurosci. 2015, 35, 2572–2587. [Google Scholar] [CrossRef]
- de Calbiac, H.; Dabacan, A.; Marsan, E.; Tostivint, H.; Devienne, G.; Ishida, S.; Leguern, E.; Baulac, S.; Muresan, R.C.; Kabashi, E.; et al. Depdc5 Knockdown Causes MTOR-Dependent Motor Hyperactivity in Zebrafish. Ann. Clin. Transl. Neurol. 2018, 5, 510–523. [Google Scholar] [CrossRef]
- Yang, L.; Chang, S.; Lu, Q.; Zhang, Y.; Wu, Z.; Sun, X.; Cao, Q.; Qian, Y.; Jia, T.; Xu, B.; et al. A New Locus Regulating MICALL2 Expression Was Identified for Association with Executive Inhibition in Children with Attention Deficit Hyperactivity Disorder. Mol. Psychiatry 2018, 23, 1014–1020. [Google Scholar] [CrossRef]
- Wan, J.; Yourshaw, M.; Mamsa, H.; Rudnik-Schöneborn, S.; Menezes, M.P.; Hong, J.E.; Leong, D.W.; Senderek, J.; Salman, M.S.; Chitayat, D.; et al. Mutations in the RNA Exosome Component Gene EXOSC3 Cause Pontocerebellar Hypoplasia and Spinal Motor Neuron Degeneration. Nat. Genet. 2012, 44, 704–708. [Google Scholar] [CrossRef]
- Fan, Y.; Yin, W.; Hu, B.; Kline, A.D.; Zhang, V.W.; Liang, D.; Sun, Y.; Wang, L.; Tang, S.; Powis, Z.; et al. De Novo Mutations of CCNK Cause a Syndromic Neurodevelopmental Disorder with Distinctive Facial Dysmorphism. Am. J. Hum. Genet. 2018, 103, 448–455. [Google Scholar] [CrossRef]
- O’Rawe, J.A.; Wu, Y.; Dörfel, M.J.; Rope, A.F.; Au, P.Y.B.; Parboosingh, J.S.; Moon, S.; Kousi, M.; Kosma, K.; Smith, C.S.; et al. TAF1 Variants Are Associated with Dysmorphic Features, Intellectual Disability, and Neurological Manifestations. Am. J. Hum. Genet. 2015, 97, 922–932. [Google Scholar] [CrossRef]
- Stankiewicz, P.; Khan, T.N.; Szafranski, P.; Slattery, L.; Streff, H.; Vetrini, F.; Bernstein, J.A.; Brown, C.W.; Rosenfeld, J.A.; Rednam, S.; et al. Haploinsufficiency of the Chromatin Remodeler BPTF Causes Syndromic Developmental and Speech Delay, Postnatal Microcephaly, and Dysmorphic Features. Am. J. Hum. Genet. 2017, 101, 503–515. [Google Scholar] [CrossRef]
- Pietri, T.; Roman, A.C.; Guyon, N.; Romano, S.A.; Washbourne, P.; Moens, C.B.; de Polavieja, G.G.; Sumbre, G. The First Mecp2-Null Zebrafish Model Shows Altered Motor Behaviors. Front. Neural Circuits 2013, 7, 118. [Google Scholar] [CrossRef]
- Nozawa, K.; Lin, Y.; Kubodera, R.; Shimizu, Y.; Tanaka, H.; Ohshima, T. Zebrafish Mecp2 Is Required for Proper Axonal Elongation of Motor Neurons and Synapse Formation. Dev. Neurobiol. 2017, 77, 1101–1113. [Google Scholar] [CrossRef]
- Norton, W. Towards Developmental Models of Psychiatric Disorders in Zebrafish. Front. Neural Circuits 2013, 7, 79. [Google Scholar] [CrossRef]
- Walitza, S.; Renner, T.J.; Dempfle, A.; Konrad, K.; Wewetzer, C.; Halbach, A.; Herpertz-Dahlmann, B.; Remschmidt, H.; Smidt, J.; Linder, M.; et al. Transmission Disequilibrium of Polymorphic Variants in the Tryptophan Hydroxylase-2 Gene in Attention-Deficit/Hyperactivity Disorder. Mol. Psychiatry 2005, 10, 1126–1132. [Google Scholar] [CrossRef]
- Li, J.; Wang, Y.; Zhou, R.; Zhang, H.; Yang, L.; Wang, B.; Faraone, S.V. Association between Polymorphisms in Serotonin Transporter Gene and Attention Deficit Hyperactivity Disorder in Chinese Han Subjects. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2007, 144B, 14–19. [Google Scholar] [CrossRef] [PubMed]
- Gizer, I.R.; Ficks, C.; Waldman, I.D. Candidate Gene Studies of ADHD: A Meta-Analytic Review. Hum. Genet. 2009, 126, 51–90. [Google Scholar] [CrossRef] [PubMed]
- Ebstein, R.P.; Novick, O.; Umansky, R.; Priel, B.; Osher, Y.; Blaine, D.; Bennett, E.R.; Nemanov, L.; Katz, M.; Belmaker, R.H. Dopamine D4 Receptor (D4DR) Exon III Polymorphism Associated with the Human Personality Trait of Novelty Seeking. Nat. Genet. 1996, 12, 78–80. [Google Scholar] [CrossRef] [PubMed]
- Hawi, Z.; Dring, M.; Kirley, A.; Foley, D.; Kent, L.; Craddock, N.; Asherson, P.; Curran, S.; Gould, A.; Richards, S.; et al. Serotonergic System and Attention Deficit Hyperactivity Disorder (ADHD): A Potential Susceptibility Locus at the 5-HT1B Receptor Gene in 273 Nuclear Families from a Multi-Centre Sample. Mol. Psychiatry 2002, 7, 718–725. [Google Scholar] [CrossRef] [PubMed]
- Nowicki, M.; Tran, S.; Chatterjee, D.; Gerlai, R. Inhibition of Phosphorylated Tyrosine Hydroxylase Attenuates Ethanol-Induced Hyperactivity in Adult Zebrafish (Danio rerio). Pharmacol. Biochem. Behav. 2015, 138, 32–39. [Google Scholar] [CrossRef]
- Fernandes, Y.; Tran, S.; Abraham, E.; Gerlai, R. Embryonic Alcohol Exposure Impairs Associative Learning Performance in Adult Zebrafish. Behav. Brain Res. 2014, 265, 181–187. [Google Scholar] [CrossRef]
- de Abreu, M.S.; Genario, R.; Giacomini, A.C.V.V.; Demin, K.A.; Lakstygal, A.M.; Amstislavskaya, T.G.; Fontana, B.D.; Parker, M.O.; Kalueff, A.V. Zebrafish as a Model of Neurodevelopmental Disorders. Neuroscience 2020, 445, 3–11. [Google Scholar] [CrossRef]
- Carvan, M.J.; Loucks, E.; Weber, D.N.; Williams, F.E. Ethanol Effects on the Developing Zebrafish: Neurobehavior and Skeletal Morphogenesis. Neurotoxicol. Teratol. 2004, 26, 757–768. [Google Scholar] [CrossRef]
- Luchiari, A.C.; Salajan, D.C.; Gerlai, R. Acute and Chronic Alcohol Administration: Effects on Performance of Zebrafish in a Latent Learning Task. Behav. Brain Res. 2015, 282, 76–83. [Google Scholar] [CrossRef]
- Cleal, M.; Fontana, B.D.; Parker, M.O. The Cognitive and Behavioral Effects of D-Amphetamine and Nicotine Sensitization in Adult Zebrafish. Psychopharmacology 2021, 238, 2191. [Google Scholar] [CrossRef]
- Mech, A.M.; Merteroglu, M.; Sealy, I.M.; Teh, M.T.; White, R.J.; Havelange, W.; Brennan, C.H.; Busch-Nentwich, E.M. Behavioral and Gene Regulatory Responses to Developmental Drug Exposures in Zebrafish. Front. Psychiatry 2022, 12, 2383. [Google Scholar] [CrossRef]
- Lawrence, T. Body Length, Activity Level, and Avoidance Learning in Zebrafish Exposed to Nicotine as Embryos. Master’s Thesis, Department of Psychology, Western Kentucky University, Bowling Green, KY, USA, 2001. [Google Scholar]
- Fann, B. Effects of Nicotine on Contextual Fear Conditioning in Adult Zebrafish (Danio rerio). Bachelor’s Thesis, University of Lynchburg, Lynchburg, VA, USA, 2020. [Google Scholar]
- Zhang, J.; Peterson, S.M.; Weber, G.J.; Zhu, X.; Zheng, W.; Freeman, J.L. Decreased Axonal Density and Altered Expression Profiles of Axonal Guidance Genes Underlying Lead (Pb) Neurodevelopmental Toxicity at Early Embryonic Stages in the Zebrafish. Neurotoxicol. Teratol. 2011, 33, 715–720. [Google Scholar] [CrossRef]
- Lovato, A.K.; Creton, R.; Colwill, R.M. Effects of Embryonic Exposure to Polychlorinated Biphenyls (PCBs) on Larval Zebrafish Behavior. Neurotoxicol. Teratol. 2016, 53, 1–10. [Google Scholar] [CrossRef]
- Levin, E.D.; Sledge, D.; Roach, S.; Petro, A.; Donerly, S.; Linney, E. Persistent Behavioral Impairment Caused by Embryonic Methylphenidate Exposure in Zebrafish. Neurotoxicol. Teratol. 2011, 33, 668–673. [Google Scholar] [CrossRef]
- Spulber, S.; Kilian, P.; Wan Ibrahim, W.N.; Onishchenko, N.; Ulhaq, M.; Norrgren, L.; Negri, S.; Di Tuccio, M.; Ceccatelli, S. PFOS Induces Behavioral Alterations, Including Spontaneous Hyperactivity That Is Corrected by Dexamfetamine in Zebrafish Larvae. PLoS ONE 2014, 9, e94227. [Google Scholar] [CrossRef] [PubMed]

















| ASD Phenotypes | Behaviours |
|---|---|
| Social communication/Interaction | Usually having little or inconstant eye contact |
| Lack of sharing interest, emotion, or pleasure when performing recreational activities | |
| Difficulty in responding or being slow to respond to signs for attention | |
| Especially talkative about a favourite topic | |
| Displaying facial expressions, movements, and gestures not related to a discussed topic | |
| Change in tone of voice (can even be poetic or robot-like) | |
| Problems with understanding other people | |
| Difficulty adjusting behaviours to social situations | |
| Restrictive/Repetitive behaviours | Repeating certain behaviours or phrases (echolalia) |
| Having an unusual and prolonged interest in numbers, details, or specific facts | |
| Exhibiting particularly focused interests, such as interests in objects in motion | |
| More or less sensitive than a neurotypical person to sensory input (light, sound, clothing, or temperature) | |
| Aptitudes/Potentials | Can learn things in surprising ways and remember specific details and information for long periods |
| Excellence in mathematics, science, music, or art disciplines at school |
| ADHD Phenotypes | Symptoms |
|---|---|
| Inattention | Overlook or miss details and make careless mistakes in every aspect of life |
| Difficulty sustaining attention in conversations, lectures, or lengthy reading | |
| Distracted when spoken to directly | |
| Lose focus and get easily side-tracked | |
| Difficulty in organizing, managing time, and meeting deadlines | |
| Avoid tasks requiring important mental effort | |
| Often lose personal objects (pencils, books, keys, wallet, phone) | |
| Easily forget to perform simple daily tasks (homework, appointment) | |
| Hyperactivity-Impulsivity | Fidget and squirm while seated |
| Stand up brusquely in situations when staying seated is expected | |
| Run, dash around, or climb at inappropriate times | |
| Incapacity to play or conduct an activity quietly | |
| Excessive talking and always in motion | |
| Incapacity to wait one’s turn | |
| Often interrupt or intrude others | |
| Very active in conversations and finish other people’s phrases or answer without being asked |
| ASD | ADHD | |
|---|---|---|
| Definition | A range of neurodevelopmental conditions that are accompanied by repetitive behaviours and causes difficulty with social skills, communication, and thinking. | A neurodevelopmental disorder characterised by impulsively and difficulty in concentration, attention, and staying still. |
| Similarities | Poor social skills Difficulty in making eye contact Deficits in attention Difficulty in managing one’s emotions Speech/language delays Treatments involve medication and behavioural therapy | |
| Differences | Less frequent | Very common |
| Social communication skills are impaired | Executive skills are impaired | |
| Repetitive body movements and preference for routine | High activity level and impulsivity: always moving, talkative, interrupts others | |
| Restricted interest | Distractibility | |
| Difficulty in nonverbal communication (difficulty in understanding facial expressions) | Difficulty in memory, forgetful | |
| Other conditions sharing the same symptomatology | Speech delays, hearing problems, or other developmental delays Restricted interests Hyperlexia Psychological disorders such as obsessive compulsive disorder, avoidance personality disorder Lead poisoning Genetic disorders such as tardive dyskinesia, Angelman syndrome, Rett syndrome | Mood disorders such as depression and anxiety Alcohol and substance abuse Dyslexia Conduct and oppositional defiant disorder Bipolar disorder Seizure and sleep disorders Tourette’s syndrome |
| Mouse (Mus musculus) | Zebrafish (Danio rerio) | |
|---|---|---|
| Graphical representation |  |  |
| Lifespan | 1–2 years | 2–5 years |
| Habitat | Diverse environments | Freshwater streams and rivers |
| Sexual maturity | Male: 8 weeks; Female: 6 weeks | 10–12 weeks (juveniles are hermaphroditic) |
| Gestation | 19–21 days (6–8 pups, 5–10 times/year) | Less than 24 h (200–300 eggs/week) |
| Advantages in neuroscience research | Can be used to investigate complex behaviours High genetic similarity to humans | Can be used to investigate complex behaviours Excellent and rapid reproductive rate Ease of neural analysis due to their transparent body in early life High genetic similarity to humans |
| Limitations in neuroscience research | Expensive to maintain Ethical limitations Long experimental cycle | Average flexibility, predictivity and translational value |
| Drugs | Behaviours Observed | References |
|---|---|---|
| Arsenic | Poor sociability and poor social novelty preference | [209] |
| Bisphenol A | Altered female exploratory and anxiety behaviour, increased levels of affiliation to female stimulus mice and decreased levels of affiliation to male stimulus mice | [210] |
| Chlorpyrifos | Reduced preference towards an unfamiliar conspecific in the social preference test and reduced social conditioned place preference | [211] |
| d-Amphetamine | Reduction in sociability with no stimulation of locomotor activity | [212] |
| GABA-A | Reduction in sociability | [212] |
| Ketamine | Social deficits | [213] |
| Phencyclidine (PCP) | Reduction in sociability | [214] |
| Valproic acid | Decreased social interaction, increased repetitive behaviours, lower sensitivity to pain, increased anxiety, reduced locomotor activity Increased fear memories | [206,207] |
| Genes | Modification Technique | Phenotypes | References |
|---|---|---|---|
| AT-rich interaction domain 1B (arid1b) | MO | Reduction in body length and alteration of chondrogenic/osteogenic genes expression | [268] |
| Aristaless related homeobox (arx) | MO | Alterations in neurons and brain development | [269] |
| Activator of transcription and developmental regulator AUTS2 (auts2) | MO | Microcephaly, small head and body zebrafish, reduced locomotor activity | [270] |
| Calcium voltage-gated channel subunit alpha1 C (cacna1c) | MO | Cardiac alterations | [271] |
| Centrosomal protein 41 (cep41) | MO | Neuronal defects and deficits in social behaviour | [272] |
| Chromodomain helicase DNA binding protein 2 (chd2) | MO | Microcephaly, abnormalities in body shape and motor impairments | [273] |
| Chromodomain helicase DNA binding protein 8 (chd8) | CRISPR/Cas9, MO | Macrocephaly, decreased gastro-intestinal motility | [274,275] |
| Contactin associated protein 2 (cntnap2) | ZFN | Decreased forebrain GABAergic neurons at 4 dpf, microcephaly and motor impairments | [276] |
| Catenin delta 2 (ctnnd2) | MO | Reduced body length and various notochord alterations | [277] |
| Dual specificity tyrosine phosphorylation regulated kinase 1A (dyrk1a) | TALEN | Increased brain apoptosis, microcephaly, decreased anxiety and decreased freezing times, deficits in social behaviours | [278] |
| Potassium inwardly rectifying channel subfamily J member 10 (kcnj10) | MO | Motor and developmental alterations | [279] |
| Lysine demethylase 6A (kdm6a) | MO | Reduction in body length and notochord alterations | [280,281] |
| Methyl-CpG binding protein 2 (mecp2) | ENU, MO | Neuronal and immune response alterations | [282,283] |
| MET proto-oncogene, receptor tyrosine kinase (met) | MO | Increased mortality and neuronal defects | [284] |
| Myelin transcription factor 1 like (myt1l) | MO | Loss of oxytocin expression in the preoptic neuroendocrine area | [285] |
| Neurobeachin (nbea) | ENU, TALEN | Abnormal response to startle stimuli | [286] |
| Nuclear receptor subfamily 3 group C member 2 (nr3c2) | CRISPR/Cas9 | Alteration in social behaviour and sleep | [287] |
| Oxytocin/neurophysin I prepropeptide (oxt) | TALEN | Altered oxytocin signalling and memory alterations | [288] |
| Reelin (reln) | TALEN | Altered social behaviour and disrupted serotonin signalling pathway | [289] |
| Arginine-glutamic acid dipeptide repeats (rere) | ENU | Deficits in vision and hearing, altered startle response to stimuli | [290] |
| SH3 and multiple ankyrin repeat domains 3 (shank3) | CRISPR/Cas9, MO | Abnormal mid-hindbrain boundary, increased apoptosis in CNS, decreased GABAergic neurons, impaired social preference, hypoactivity, seizure-like behaviours | [291,292] |
| Synaptic Ras GTPase activating protein 1 (syngap1) | MO | Microcephaly, developmental delay, high mortality, increased apoptosis in CNS, motor impairment | [293] |
| Genes | Names | SFARI Gene Score | ADHD Association |
|---|---|---|---|
| arid1b | AT rich interactive domain 1B | High confidence, Syndromic (1S) | Yes |
| chd8 | Chromatin helicase DNA-binding protein 8 | High confidence, Syndromic (1S) | Yes |
| dync1h1 | Dynein cytoplasmic 1 heavy chain 1 | High confidence (1) | No |
| fmr1 | Fragile X syndrome mental retardation 1 | High confidence, Syndromic (1S) | Yes |
| mecp2 | Methyl CpG binding protein 2 | High confidence, Syndromic (1S) | Yes |
| mef2c | Myocyte enhancer factor 2c | High confidence, Syndromic (1S) | Yes |
| pten | Phosphatase and tensin homolog | High confidence, Syndromic (1S) | Yes |
| scn1a/scn2a | Sodium channel voltage-gated, type I-like, alpha subunit/type II-like, alpha subunit | High confidence, Syndromic (1S) | Yes |
| cntnap2 | Contactin associated protein-like 2 | Syndromic (S) | Yes |
| dyrk1a | Dual-specificity tyrosine-(Y)-phosphorylation regulated kinase 1A | High confidence, Syndromic (1S) | No |
| grin2b | Glutamate receptor ionotropic N-methyl D-aspartate 2B | High confidence (1) | Yes |
| nrxn1 | Neurexin 1 | High confidence (1) | Yes |
| shank3 | SH3 and multiple ankyrin repeat domains | High confidence, Syndromic (1S) | No |
| syngap1 | Synaptic Ras GTPase activating protein 1 | High confidence, Syndromic (1S) | Yes |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dougnon, G.; Matsui, H. Modelling Autism Spectrum Disorder (ASD) and Attention-Deficit/Hyperactivity Disorder (ADHD) Using Mice and Zebrafish. Int. J. Mol. Sci. 2022, 23, 7550. https://doi.org/10.3390/ijms23147550
Dougnon G, Matsui H. Modelling Autism Spectrum Disorder (ASD) and Attention-Deficit/Hyperactivity Disorder (ADHD) Using Mice and Zebrafish. International Journal of Molecular Sciences. 2022; 23(14):7550. https://doi.org/10.3390/ijms23147550
Chicago/Turabian StyleDougnon, Godfried, and Hideaki Matsui. 2022. "Modelling Autism Spectrum Disorder (ASD) and Attention-Deficit/Hyperactivity Disorder (ADHD) Using Mice and Zebrafish" International Journal of Molecular Sciences 23, no. 14: 7550. https://doi.org/10.3390/ijms23147550
APA StyleDougnon, G., & Matsui, H. (2022). Modelling Autism Spectrum Disorder (ASD) and Attention-Deficit/Hyperactivity Disorder (ADHD) Using Mice and Zebrafish. International Journal of Molecular Sciences, 23(14), 7550. https://doi.org/10.3390/ijms23147550