
Mitochondrial a Kinase Anchor Proteins in Cardiovascular Health and Disease: A Review Article on Behalf of the Working Group on Cellular and Molecular Biology of the Heart of the Italian Society of Cardiology

 ,
, 
Abstract
:1. Introduction
2. Mitochondrial AKAPs
2.1. D-AKAP1
2.1.1. D-AKAP1 in Cardiovascular System and Metabolism
2.1.2. D-AKAP1 and Myocardial Infarction
2.1.3. D-AKAP1 and Cardiac Hypertrophy Induced by Pressure Overload
2.2. D-AKAP2
2.2.1. D-AKAP2 and Cardiovascular Function
2.2.2. D-AKAP2 and Myocardial Infarction
2.2.3. D-AKAP2 and Cardiac Arrhythmias
2.3. ACBD3/PAP7/GCP60
2.4. OPA1
2.5. WAVE-1
2.6. RAB32
2.7. SKIP
3. Mitochondrial AKAPs as Novel Therapeutic Targets
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wang, J.; Gareri, C.; Rockman, H.A. G-Protein-Coupled Receptors in Heart Disease. Circ. Res. 2018, 123, 716–735. [Google Scholar] [CrossRef] [PubMed]
- Rockman, H.A.; Koch, W.J.; Lefkowitz, R.J. Seven-transmembrane-spanning receptors and heart function. Nature 2002, 415, 206–212. [Google Scholar] [CrossRef]
- Perrino, C.; Rockman, H.A. Reversal of cardiac remodeling by modulation of adrenergic receptors: A new frontier in heart failure. Curr. Opin. Cardiol. 2007, 22, 443–449. [Google Scholar] [CrossRef] [PubMed]
- Ippolito, M.; Benovic, J.L. Biased agonism at beta-adrenergic receptors. Cell Signal. 2021, 80, 109905. [Google Scholar] [CrossRef] [PubMed]
- Bond, R.A.; Garcia-Rojas, E.Y.L.; Hegde, A.; Walker, J.K.L. Therapeutic Potential of Targeting ß-Arrestin. Front. Pharmacol. 2019, 10, 124. [Google Scholar] [CrossRef] [PubMed]
- Lezoualc’H, F.; Fazal, L.; Laudette, M.; Conte, C. Cyclic AMP Sensor EPAC Proteins and Their Role in Cardiovascular Function and Disease. Circ. Res. 2016, 118, 881–897. [Google Scholar] [CrossRef]
- Boularan, C.; Gales, C. Cardiac cAMP: Production, hydrolysis, modulation and detection. Front. Pharmacol. 2015, 6, 203. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Chen, J.; Fontes, S.K.; Bautista, E.N.; Cheng, Z. Physiological and pathological roles of protein kinase A in the heart. Cardiovasc. Res. 2022, 118, 386–398. [Google Scholar] [CrossRef]
- Colombe, A.S.; Pidoux, G. Cardiac cAMP-PKA Signaling Compartmentalization in Myocardial Infarction. Cells 2021, 10, 922. [Google Scholar] [CrossRef]
- Ercu, M.; Klussmann, E. Roles of A-Kinase Anchoring Proteins and Phosphodiesterases in the Cardiovascular System. J. Cardiovasc. Dev. Dis. 2018, 5, 14. [Google Scholar] [CrossRef] [Green Version]
- Dodge-Kafka, K.L.; Langeberg, L.; Scott, J.D. Compartmentation of cyclic nucleotide signaling in the heart: The role of A-kinase anchoring proteins. Circ. Res. 2006, 98, 993–1001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marin, W. A-kinase anchoring protein 1 (AKAP1) and its role in some cardiovascular diseases. J. Mol. Cell. Cardiol. 2020, 138, 99–109. [Google Scholar] [CrossRef]
- Aye, T.T.; Soni, S.; van Veen, T.A.; van der Heyden, M.A.; Cappadona, S.; Varro, A.; de Weger, R.A.; de Jonge, N.; Vos, M.A.; Heck, A.J.; et al. Reorganized PKA-AKAP associations in the failing human heart. J. Mol. Cell. Cardiol. 2012, 52, 511–518. [Google Scholar] [CrossRef] [PubMed]
- Perino, A.; Ghigo, A.; Scott, J.D.; Hirsch, E. Anchoring Proteins as Regulators of Signaling Pathways. Circ. Res. 2012, 111, 482–492. [Google Scholar] [CrossRef] [Green Version]
- Murabito, A.; Cnudde, S.; Hirsch, E.; Ghigo, A. Potential therapeutic applications of AKAP disrupting peptides. Clin. Sci. 2020, 134, 3259–3282. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Merrill, R.A.; Strack, S. A-Kinase Anchoring Protein 1: Emerging Roles in Regulating Mitochondrial Form and Function in Health and Disease. Cells 2020, 9, 298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forte, M.; Schirone, L.; Ameri, P.; Basso, C.; Catalucci, D.; Modica, J.; Chimenti, C.; Crotti, L.; Frati, G.; Rubattu, S.; et al. The role of mitochondrial dynamics in cardiovascular diseases. Br. J. Pharmacol. 2021, 178, 2060–2076. [Google Scholar] [CrossRef] [PubMed]
- Schaper, J.; Froede, R.; Hein, S.; Buck, A.; Hashizume, H.; Speiser, B.; Friedl, A.; Bleese, N. Impairment of the myocardial ultrastructure and changes of the cytoskeleton in dilated cardiomyopathy. Circulation 1991, 83, 504–514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carlucci, A.; Lignitto, L.; Feliciello, A. Control of mitochondria dynamics and oxidative metabolism by cAMP, AKAPs and the proteasome. Trends Cell Biol. 2008, 18, 604–613. [Google Scholar] [CrossRef]
- Sung, J.Y.; Engmann, O.; Teylan, M.A.; Nairn, A.C.; Greengard, P.; Kim, Y. WAVE1 controls neuronal activity-induced mitochondrial distribution in dendritic spines. Proc. Natl. Acad. Sci. USA 2008, 105, 3112–3116. [Google Scholar] [CrossRef] [Green Version]
- Sherpa, R.T.; Fiore, C.; Moshal, K.S.; Wadsworth, A.; Rudokas, M.W.; Agarwal, S.R.; Harvey, R.D. Mitochondrial A-kinase anchoring proteins in cardiac ventricular myocytes. Physiol. Rep. 2021, 9, e15015. [Google Scholar] [CrossRef] [PubMed]
- Di Benedetto, G.; Lefkimmiatis, K.; Pozzan, T. The basics of mitochondrial cAMP signalling: Where, when, why. Cell Calcium 2021, 93, 102320. [Google Scholar] [CrossRef] [PubMed]
- Ould Amer, Y.; Hebert-Chatelain, E. Mitochondrial cAMP-PKA signaling: What do we really know? Biochim. Biophys. Acta Bioenerg. 2018, 1859, 868–877. [Google Scholar] [CrossRef] [PubMed]
- Merrill, R.A.; Strack, S. Mitochondria: A kinase anchoring protein 1, a signaling platform for mitochondrial form and function. Int. J. Biochem. Cell Biol. 2014, 48, 92–96. [Google Scholar] [CrossRef] [Green Version]
- Schiattarella, G.G.; Cattaneo, F.; Carrizzo, A.; Paolillo, R.; Boccella, N.; Ambrosio, M.; Damato, A.; Pironti, G.; Franzone, A.; Russo, G.; et al. Akap1 Regulates Vascular Function and Endothelial Cells Behavior. Hypertension 2018, 71, 507–517. [Google Scholar] [CrossRef]
- Schiattarella, G.G.; Cattaneo, F.; Pironti, G.; Magliulo, F.; Carotenuto, G.; Pirozzi, M.; Polishchuk, R.; Borzacchiello, D.; Paolillo, R.; Oliveti, M.; et al. Akap1 Deficiency Promotes Mitochondrial Aberrations and Exacerbates Cardiac Injury Following Permanent Coronary Ligation via Enhanced Mitophagy and Apoptosis. PLoS ONE 2016, 11, e0154076. [Google Scholar] [CrossRef] [Green Version]
- Schiattarella, G.G.; Boccella, N.; Paolillo, R.; Cattaneo, F.; Trimarco, V.; Franzone, A.; D’Apice, S.; Giugliano, G.; Rinaldi, L.; Borzacchiello, D.; et al. Loss of Akap1 Exacerbates Pressure Overload-Induced Cardiac Hypertrophy and Heart Failure. Front. Physiol. 2018, 9, 558. [Google Scholar] [CrossRef] [Green Version]
- Patel, H.H.; Hamuro, L.L.; Chun, B.J.; Kawaraguchi, Y.; Quick, A.; Rebolledo, B.; Pennypacker, J.; Thurston, J.; Rodriguez-Pinto, N.; Self, C.; et al. Disruption of Protein Kinase A Localization Using a Trans-activator of Transcription (TAT)-conjugated A-kinase-anchoring Peptide Reduces Cardiac Function. J. Biol. Chem. 2010, 285, 27632–27640. [Google Scholar] [CrossRef] [Green Version]
- Maric, D.; Paterek, A.; Delaunay, M.; López, I.P.; Arambasic, M.; Diviani, D. A-Kinase Anchoring Protein 2 Promotes Protection against Myocardial Infarction. Cells 2021, 10, 2861. [Google Scholar] [CrossRef]
- Tingley, W.G.; Pawlikowska, L.; Zaroff, J.G.; Kim, T.; Nguyen, T.; Young, S.G.; Vranizan, K.; Kwok, P.-Y.; Whooley, M.A.; Conklin, B.R. Gene-trapped mouse embryonic stem cell-derived cardiac myocytes and human genetics implicate AKAP10 in heart rhythm regulation. Proc. Natl. Acad. Sci. USA 2007, 104, 8461–8466. [Google Scholar] [CrossRef] [Green Version]
- Yue, X.; Qian, Y.; Gim, B.; Lee, I. Acyl-CoA-Binding Domain-Containing 3 (ACBD3; PAP7; GCP60): A Multi-Functional Membrane Domain Organizer. Int. J. Mol. Sci. 2019, 20, 2028. [Google Scholar] [CrossRef] [Green Version]
- Wai, T.; García-Prieto, J.; Baker, M.J.; Merkwirth, C.; Benit, P.; Rustin, P.; Rupérez, F.J.; Barbas, C.; Ibañez, B.; Langer, T. Imbalanced OPA1 processing and mitochondrial fragmentation cause heart failure in mice. Science 2015, 350, aad0116. [Google Scholar] [CrossRef] [PubMed]
- Piquereau, J.; Caffin, F.; Novotova, M.; Prola, A.; Garnier, A.; Mateo, P.; Fortin, D.; Huynh, L.H.; Nicolas, V.; Alavi, M.V.; et al. Down-regulation of OPA1 alters mouse mitochondrial morphology, PTP function, and cardiac adaptation to pressure overload. Cardiovasc. Res. 2012, 94, 408–417. [Google Scholar] [CrossRef] [Green Version]
- Cheng, A.; Arumugam, T.V.; Liu, D.; Khatri, R.G.; Mustafa, K.; Kwak, S.; Ling, H.-P.; Gonzales, C.; Xin, O.; Jo, D.-G.; et al. Pancortin-2 interacts with WAVE1 and Bcl-xL in a mitochondria-associated protein complex that mediates ischemic neuronal death. J. Neurosci. 2007, 27, 1519–1528. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Sung, J.Y.; Ceglia, I.; Lee, K.-W.; Ahn, J.-H.; Halford, J.M.; Kim, A.M.; Kwak, S.P.; Park, J.B.; Ryu, S.H.; et al. Phosphorylation of WAVE1 regulates actin polymerization and dendritic spine morphology. Nature 2006, 442, 814–817. [Google Scholar] [CrossRef] [PubMed]
- Danial, N.N.; Gramm, C.F.; Scorrano, L.; Zhang, C.-Y.; Krauss, S.; Ranger, A.M.; Datta, S.; Greenberg, M.E.; Licklider, L.J.; Lowell, B.B.; et al. BAD and glucokinase reside in a mitochondrial complex that integrates glycolysis and apoptosis. Nature 2003, 424, 952–956. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Okada, T.; Assmann, A.; Soto, J.; Liew, C.W.; Bugger, H. Insulin signaling regulates mitochondrial function in pancreatic beta-cells. PLoS ONE 2009, 4, e7983. [Google Scholar] [CrossRef] [Green Version]
- Drizyte-Miller, K.; Chen, J.; Cao, H.; Schott, M.B.; McNiven, M.A. The small GTPase Rab32 resides on lysosomes to regulate mTORC1 signaling. J. Cell Sci. 2020, 133, jcs236661. [Google Scholar] [CrossRef]
- Rybnicek, J.; Samtleben, S.; Herrera-Cruz, M.S.; Simmen, T. Expression of a T39N mutant Rab32 protein arrests mitochondria movement within neurites of differentiated SH-SY5Y cells. Small GTPases 2020, 11, 289–292. [Google Scholar] [CrossRef]
- Chen, P.L.; Huang, K.T.; Cheng, C.Y.; Li, J.C.; Chan, H.Y.; Lin, T.Y.; Su, M.P.; Yang, W.Y.; Chang, H.C.; Wang, H.D.; et al. Vesicular transport mediates the uptake of cytoplasmic proteins into mitochondria in Drosophila melanogaster. Nat. Commun. 2020, 11, 2592. [Google Scholar] [CrossRef]
- Aguilar, A.; Weber, J.; Boscher, J.; Freund, M.; Ziessel, C.; Eckly, A.; Magnenat, S.; Bourdon, C.; Hechler, B.; Mangin, P.H.; et al. Combined deficiency of RAB32 and RAB38 in the mouse mimics Hermansky-Pudlak syndrome and critically impairs thrombosis. Blood Adv. 2019, 3, 2368–2380. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Harashima, S.-I.; Liu, Y.; Usui, R.; Inagaki, N. Sphingosine kinase 1-interacting protein is a novel regulator of glucose-stimulated insulin secretion. Sci. Rep. 2017, 7, 779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, Z.-Q.; Zhang, J.; Huang, Y.; Hoover, H.E.; Vessey, D.A.; Karliner, J.S. A sphingosine kinase 1 mutation sensitizes the myocardium to ischemia/reperfusion injury. Cardiovasc. Res. 2007, 76, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Cardone, L.; De Cristofaro, T.; Affaitati, A.; Garbi, C.; Ginsberg, M.D.; Saviano, M.; Varrone, S.; Rubin, C.S.; Gottesman, M.E.; Avvedimento, E.V.; et al. A-kinase anchor protein 84/121 are targeted to mitochondria and mitotic spindles by overlapping amino-terminal motifs. J. Mol. Biol. 2002, 320, 663–675. [Google Scholar] [CrossRef]
- Ginsberg, M.D.; Feliciello, A.; Jones, J.K.; Avvedimento, E.V.; Gottesman, M.E. PKA-dependent Binding of mRNA to the Mitochondrial AKAP121 Protein. J. Mol. Biol. 2003, 327, 885–897. [Google Scholar] [CrossRef]
- Feliciello, A.; Rubin, C.S.; Avvedimento, E.V.; Gottesman, M.E. Expression of A Kinase Anchor Protein 121 Is Regulated by Hormones in Thyroid and Testicular Germ Cells. J. Biol. Chem. 1998, 273, 23361–23366. [Google Scholar] [CrossRef] [Green Version]
- Perrino, C.; Schiattarella, G.G. (Zebra) fishing for relevant genes in heart regeneration. J. Cardiovasc. Med. 2010, 11, 631–632. [Google Scholar] [CrossRef]
- Carlucci, A.; Adornetto, A.; Scorziello, A.; Viggiano, D.; Foca, M.; Cuomo, O.; Annunziato, L.; Gottesman, M.; Feliciello, A. Proteolysis of AKAP121 regulates mitochondrial activity during cellular hypoxia and brain ischaemia. EMBO J. 2008, 27, 1073–1084. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.; Scimia, M.C.; Wilkinson, D.; Trelles, R.D.; Wood, M.R.; Bowtell, D.; Dillin, A.; Mercola, M.; Ze’ev, A.R. Fine-tuning of Drp1/Fis1 availability by AKAP121/Siah2 regulates mitochondrial adaptation to hypoxia. Mol. Cell 2011, 44, 532–544. [Google Scholar] [CrossRef] [Green Version]
- Livigni, A.; Scorziello, A.; Agnese, S.; Adornetto, A.; Carlucci, A.; Garbi, C.; Castaldo, I.; Annunziato, L.; Avvedimento, E.V.; Feliciello, A. Mitochondrial AKAP121 Links cAMP and src Signaling to Oxidative Metabolism. Mol. Biol. Cell 2006, 17, 263–271. [Google Scholar] [CrossRef] [Green Version]
- Cardone, L.; Carlucci, A.; Affaitati, A.; Livigni, A.; Decristofaro, T.; Garbi, C.; Varrone, S.; Ullrich, A.; Gottesman, M.E.; Avvedimento, E.V.; et al. Mitochondrial AKAP121 Binds and Targets Protein Tyrosine Phosphatase D1, a Novel Positive Regulator of src Signaling. Mol. Cell. Biol. 2004, 24, 4613–4626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Chen, Y.; Gucek, M.; Xu, H. The mitochondrial outer membrane protein MDI promotes local protein synthesis and mtDNA replication. EMBO J. 2016, 35, 1045–1057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gabrovsek, L.; Collins, K.B.; Aggarwal, S.; Saunders, L.M.; Lau, H.-T.; Suh, D.; Sancak, Y.; Trapnell, C.; Ong, S.-E.; Smith, F.D.; et al. A-kinase-anchoring protein 1 (dAKAP1)-based signaling complexes coordinate local protein synthesis at the mitochondrial surface. J. Biol. Chem. 2020, 295, 10749–10765. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, Z.-H.; Liu, Y.; Chen, Y.; Sun, N.; Gucek, M.; Zhang, F.; Xu, H. PINK1 Inhibits Local Protein Synthesis to Limit Transmission of Deleterious Mitochondrial DNA Mutations. Mol. Cell 2019, 73, 1127–1137.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newhall, K.J.; Criniti, A.R.; Cheah, C.S.; Smith, K.C.; Kafer, K.E.; Burkart, A.D.; McKnight, G.S. Dynamic Anchoring of PKA Is Essential during Oocyte Maturation. Curr. Biol. 2006, 16, 321–327. [Google Scholar] [CrossRef] [Green Version]
- Qi, B.; He, L.; Zhao, Y.; Zhang, L.; He, Y.; Li, J.; Li, C.; Zhang, B.; Huang, Q.; Xing, J.; et al. Akap1 deficiency exacerbates diabetic cardiomyopathy in mice by NDUFS1-mediated mitochondrial dysfunction and apoptosis. Diabetologia 2020, 63, 1072–1087. [Google Scholar] [CrossRef]
- Perrino, C.; Feliciello, A.; Schiattarella, G.G.; Esposito, G.; Guerriero, R.; Zaccaro, L.; Del Gatto, A.; Saviano, M.; Garbi, C.; Carangi, R.; et al. AKAP121 downregulation impairs protective cAMP signals, promotes mitochondrial dysfunction, and increases oxidative stress. Cardiovasc. Res. 2010, 88, 101–110. [Google Scholar] [CrossRef] [Green Version]
- Frey, N.; Olson, E. Cardiac Hypertrophy: The Good, the Bad, and the Ugly. Annu. Rev. Physiol. 2003, 65, 45–79. [Google Scholar] [CrossRef]
- Chien, K.R. Stress Pathways and Heart Failure. Cell 1999, 98, 555–558. [Google Scholar] [CrossRef] [Green Version]
- Perrino, C.; Prasad, S.V.N.; Mao, L.; Noma, T.; Yan, Z.; Kim, H.-S.; Smithies, O.; Rockman, H.A. Intermittent pressure overload triggers hypertrophy-independent cardiac dysfunction and vascular rarefaction. J. Clin. Investig. 2006, 116, 1547–1560. [Google Scholar] [CrossRef] [Green Version]
- Perrino, C.; Schroder, J.N.; Lima, B.; Villamizar, N.; Nienaber, J.J.; Milano, C.A.; Naga Prasad, S.V. Dynamic regulation of phosphoinositide 3-kinase-gamma activity and beta-adrenergic receptor trafficking in end-stage human heart failure. Circulation 2007, 116, 2571–2579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abrenica, B.; AlShaaban, M.; Czubryt, M.P. The A-kinase anchor protein AKAP121 is a negative regulator of cardiomyocyte hypertrophy. J. Mol. Cell. Cardiol. 2009, 46, 674–681. [Google Scholar] [CrossRef] [PubMed]
- Boccella, N.; Paolillo, R.; Coretti, L.; D’Apice, S.; Lama, A.; Giugliano, G.; Schiattarella, G.G.; Cuomo, M.; D’Aquino, I.; Cavaliere, G.; et al. Transverse aortic constriction induces gut barrier alterations, microbiota remodeling and systemic inflammation. Sci. Rep. 2021, 11, 7404. [Google Scholar] [CrossRef]
- Eggers, C.T.; Schafer, J.C.; Goldenring, J.R.; Taylor, S.S. D-AKAP2 Interacts with Rab4 and Rab11 through Its RGS Domains and Regulates Transferrin Receptor Recycling. J. Biol. Chem. 2009, 284, 32869–32880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, L.J.-S.; Durick, K.; Weiner, J.A.; Chun, J.; Taylor, S.S. D-AKAP2, a novel protein kinase A anchoring protein with a putative RGS domain. Proc. Natl. Acad. Sci. USA 1997, 94, 11184–11189. [Google Scholar] [CrossRef] [Green Version]
- Chung, J.; Wittig, J.G.; Ghamari, A.; Maeda, M.; Dailey, T.A.; Bergonia, H. Erythropoietin signaling regulates heme biosynthesis. eLife 2017, 6, e24767. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, T.; Yajima, K.; Hibino, T.; Kato, K.; Matsuo, H.; Segawa, T.; Watanabe, S.; Oguri, M.; Yokoi, K.; Nozawa, Y.; et al. Association of gene polymorphisms with myocardial infarction in individuals with different lipid profiles. Int. J. Mol. Med. 2007, 20, 581–590. [Google Scholar] [CrossRef] [Green Version]
- Fan, J.; Liu, J.; Culty, M.; Papadopoulos, V. Acyl-coenzyme A binding domain containing 3 (ACBD3; PAP7; GCP60): An emerging signaling molecule. Prog. Lipid Res. 2010, 49, 218–234. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Taufalele, P.V.; Cochran, J.D.; Robillard-Frayne, I.; Marx, J.M.; Soto, J.; Rauckhorst, A.J.; Tayyari, F.; Pewa, A.D.; Gray, L.R.; et al. Mitochondrial pyruvate carriers are required for myocardial stress adaptation. Nat. Metab. 2020, 2, 1248–1264. [Google Scholar] [CrossRef]
- Darshi, M.; Mendiola, V.L.; Mackey, M.R.; Murphy, A.N.; Koller, A.; Perkins, G.A.; Ellisman, M.H.; Taylor, S.S. ChChd3, an Inner Mitochondrial Membrane Protein, Is Essential for Maintaining Crista Integrity and Mitochondrial Function. J. Biol. Chem. 2011, 286, 2918–2932. [Google Scholar] [CrossRef] [Green Version]
- Hu, C.; Shu, L.; Huang, X.; Yu, J.; Li, L.; Gong, L.; Yang, M.; Wu, Z.; Gao, Z.; Zhao, Y.; et al. OPA1 and MICOS Regulate mitochondrial crista dynamics and formation. Cell Death Dis. 2020, 11, 940. [Google Scholar] [CrossRef]
- Pidoux, G.; Witczak, O.; Jarnaess, E.; Myrvold, L.; Urlaub, H.; Stokka, A.J.; Küntziger, T.; Taskén, K. Optic atrophy 1 is an A-kinase anchoring protein on lipid droplets that mediates adrenergic control of lipolysis. EMBO J. 2011, 30, 4371–4386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Wang, Y.; Xu, J.; Tian, F.; Hu, S.; Chen, Y.; Fu, Z. Melatonin attenuates myocardial ischemia-reperfusion injury via improving mitochondrial fusion/mitophagy and activating the AMPK-OPA1 signaling pathways. J. Pineal Res. 2019, 66, e12542. [Google Scholar] [CrossRef] [PubMed]
- Varanita, T.; Soriano, M.E.; Romanello, V.; Zaglia, T.; Quintana-Cabrera, R.; Semenzato, M.; Menabò, R.; Costa, V.; Civiletto, G.; Pesce, P.; et al. The Opa1-Dependent Mitochondrial Cristae Remodeling Pathway Controls Atrophic, Apoptotic, and Ischemic Tissue Damage. Cell Metab. 2015, 21, 834–844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nan, J.; Hu, H.; Sun, Y.; Zhu, L.; Wang, Y.; Zhong, Z.; Zhao, J.; Zhang, N.; Wang, Y.; Wang, Y.; et al. TNFR2 Stimulation Promotes Mitochondrial Fusion via Stat3- and NF-kB-Dependent Activation of OPA1 Expression. Circ. Res. 2017, 121, 392–410. [Google Scholar] [CrossRef]
- Chen, L.; Liu, T.; Tran, A.; Lu, X.; Tomilov, A.A.; Davies, V.; Cortopassi, G.; Chiamvimonvat, N.; Bers, D.M.; Votruba, M.; et al. OPA1 Mutation and Late-Onset Cardiomyopathy: Mitochondrial Dysfunction and mtDNA Instability. J. Am. Heart Assoc. 2012, 1, e003012. [Google Scholar] [CrossRef] [Green Version]
- Kang, R.; Tang, D.; Yu, Y.; Wang, Z.; Hu, T.; Wang, H.; Cao, L. WAVE1 regulates Bcl-2 localization and phosphorylation in leukemia cells. Leukemia 2010, 24, 177–186. [Google Scholar] [CrossRef] [Green Version]
- Rawe, V.Y.; Ramalho-Santos, J.; Payne, C.; Chemes, H.E.; Schatten, G. WAVE1, an A-kinase anchoring protein, during mammalian spermatogenesis. Hum. Reprod. 2004, 19, 2594–2604. [Google Scholar] [CrossRef]
- Dahl, J.P.; Wang-Dunlop, J.; Gonzales, C.; Goad, M.E.P.; Mark, R.J.; Kwak, S.P. Characterization of the WAVE1 Knock-Out Mouse: Implications for CNS Development. J. Neurosci. 2003, 23, 3343–3352. [Google Scholar] [CrossRef] [Green Version]
- Alto, N.M.; Soderling, J.; Scott, J.D. Rab32 is an A-kinase anchoring protein and participates in mitochondrial dynamics. J. Cell Biol. 2002, 158, 659–668. [Google Scholar] [CrossRef] [Green Version]
- Bui, M.; Gilady, S.Y.; Fitzsimmons, R.E.; Benson, M.D.; Lynes, E.M.; Gesson, K.; Alto, N.M.; Strack, S.; Scott, J.D.; Simmen, T. Rab32 Modulates Apoptosis Onset and Mitochondria-associated Membrane (MAM) Properties. J. Biol. Chem. 2010, 285, 31590–31602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ortiz-Sandoval, C.G.; Hughes, S.C.; Dacks, J.B.; Simmen, T. Interaction with the effector dynamin-related protein 1 (Drp1) is an ancient function of Rab32 subfamily proteins. Cell. Logist. 2014, 4, e986399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Means, C.K.; Lygren, B.; Langeberg, L.K.; Jain, A.; Dixon, R.E.; Vega, A.L.; Gold, M.G.; Petrosyan, S.; Taylor, S.S.; Murphy, A.N.; et al. An entirely specific type I A-kinase anchoring protein that can sequester two molecules of protein kinase A at mitochondria. Proc. Natl. Acad. Sci. USA 2011, 108, E1227–E1235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jozefczuk, E.; Guzik, T.; Siedlinski, M. Significance of sphingosine-1-phosphate in cardiovascular physiology and pathology. Pharmacol. Res. 2020, 156, 104793. [Google Scholar] [CrossRef]
- Lacaná, E.; Maceyka, M.; Milstien, S.; Spiegel, S. Cloning and Characterization of a Protein Kinase A Anchoring Protein (AKAP)-related Protein That Interacts with and Regulates Sphingosine Kinase 1 Activity. J. Biol. Chem. 2002, 277, 32947–32953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mukherjee, A.B.; Weinstein, M.E. Sequence of centromere separation of mitotic chromosomes during human cellular aging. Mech. Ageing Dev. 1998, 45, 59–64. [Google Scholar] [CrossRef]
- Ting, X.; Lv, W.; Liu, D.; Jing, Y.; Hu, F. Opa1 Reduces Hypoxia-Induced Cardiomyocyte Death by Improving Mitochondrial Quality Control. Front. Cell Dev. Biol. 2020, 8, 853. [Google Scholar]
- Tröger, J.; Moutty, M.C.; Skroblin, P.; Klussmann, E. A-kinase anchoring proteins as potential drug targets. Br. J. Pharmacol. 2012, 166, 420–433. [Google Scholar] [CrossRef]
- Smith, F.D.; Omar, M.H.; Nygren, P.J.; Soughayer, J.; Hoshi, N.; Lau, H.-T.; Snyder, C.G.; Branon, T.C.; Ghosh, D.; Langeberg, L.K.; et al. Single nucleotide polymorphisms alter kinase anchoring and the subcellular targeting of A-kinase anchoring proteins. Proc. Natl. Acad. Sci. USA 2018, 115, E11465–E11474. [Google Scholar] [CrossRef] [Green Version]
- Kennedy, E.J.; Scott, J.D. Selective Disruption of the AKAP Signaling Complexes. Methods Mol. Biol. 2015, 1294, 137–150. [Google Scholar]
- Hung, V.; Zou, P.; Rhee, H.-W.; Udeshi, N.D.; Cracan, V.; Svinkina, T.; Carr, S.A.; Mootha, V.K.; Ting, A.Y. Proteomic Mapping of the Human Mitochondrial Intermembrane Space in Live Cells via Ratiometric APEX Tagging. Mol. Cell 2014, 55, 332–341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rinaldi, L.; Sepe, M.; Donne, R.D.; Conte, K.; Arcella, A.; Borzacchiello, D.; Amente, S.; De Vita, F.; Porpora, M.; Garbi, C.; et al. Mitochondrial AKAP1 supports mTOR pathway and tumor growth. Cell Death Dis. 2017, 8, e2842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suryavanshi, S.V.; Jadhav, S.M.; McConnell, B.K. Polymorphisms/Mutations in A-Kinase Anchoring Proteins (AKAPs): Role in the Cardiovascular System. J. Cardiovasc. Dev. Dis. 2018, 5, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
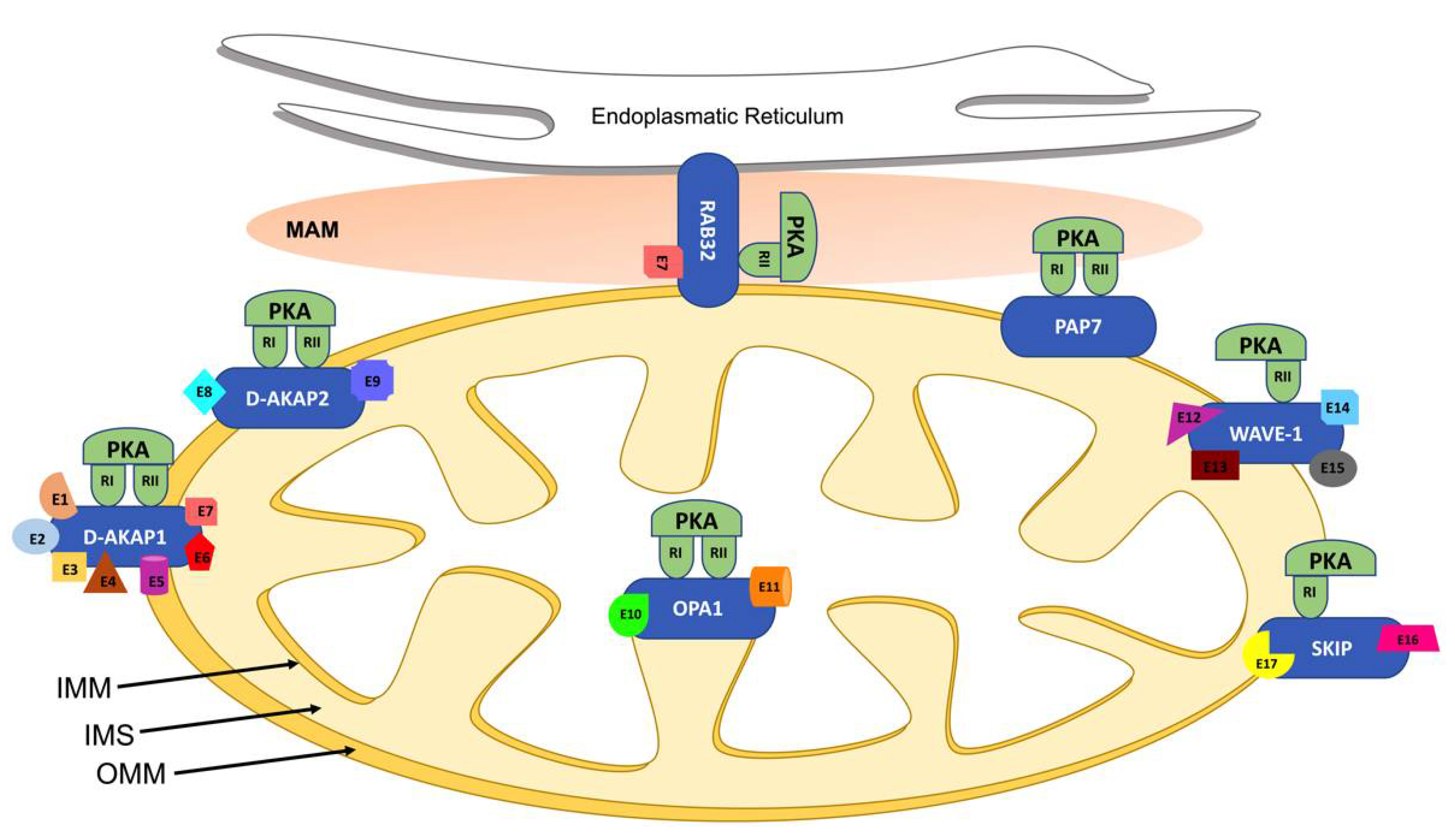
| Gene | Protein | Effectors | PKA Binding | Mitochondrial Localization | Mitochondrial Effects | Cardiovascular Functions | References |
|---|---|---|---|---|---|---|---|
| Akap1 | D-AKAP1 | PP1(E1), Src(E2), PDE4(E3), Calcineurin (E4), AGO2(E5), NCX3(E6), DRP1(E7) | RI, RII | OMM | Apoptosis, mitochondrial fusion, mitochondrial respiration | Modulates infarct size after coronary artery ligation and post-ischemic cardiac remodelling, cardiac hypertrophy and pressure-overload-induced cardiac dysfunction, endothelial function and angiogenesis | [25,26,27] |
| Akap10 | D-AKAP 2 | Rab 4(E8), Rab11(E9) | RI, RII | OMM | Mitochondrial dynamics, ROS production and apoptosis | Modulates cardiovascular integrity barrier and controller of pacemaker cells’ sensitivity to cholinergic stimulation | [28,29,30] |
| Acbd3 | PAP7 | RI, RII | OMM | Cholesterol transport | Unknown | [31] | |
| Opa1 | OPA1 | OMA1(E10), YME1L(E11) | RI, RII | IMM | Mitochondrial fusion and fission, stabilizing mitochondrial cristae, increasing mitochondrial respiratory efficiency | Modulates cardiac function and hypertrophy, metabolic shift of increased glucose uptake | [32,33] |
| Wasf | WAVE-1 | BAD(E12), Pancortin2(E13), GK(E14), BCL-XL(E15) | RII | OMM | Mitochondrial trafficking | Unknown | [34,35,36,37] |
| Rab32 | RAB32 | DRP1(E7) | RII | MAM | Endoplasmic reticulum Ca2+ handling, mitochondrial fusion, apoptosis | Unknown | [38,39,40,41] |
| Sphkap | SKIP | ChChdl3(E16), S1P(E17) | RI | OMM | Mitophagy | Modulates infarct size, apoptosis and cytochrome c release after myocardial ischemia-reperfusion injury | [42,43] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Paolillo, R.; D’Apice, S.; Schiattarella, G.G.; Ameri, P.; Borzacchiello, D.; Catalucci, D.; Chimenti, C.; Crotti, L.; Sciarretta, S.; Torella, D.; et al. Mitochondrial a Kinase Anchor Proteins in Cardiovascular Health and Disease: A Review Article on Behalf of the Working Group on Cellular and Molecular Biology of the Heart of the Italian Society of Cardiology. Int. J. Mol. Sci. 2022, 23, 7691. https://doi.org/10.3390/ijms23147691
Paolillo R, D’Apice S, Schiattarella GG, Ameri P, Borzacchiello D, Catalucci D, Chimenti C, Crotti L, Sciarretta S, Torella D, et al. Mitochondrial a Kinase Anchor Proteins in Cardiovascular Health and Disease: A Review Article on Behalf of the Working Group on Cellular and Molecular Biology of the Heart of the Italian Society of Cardiology. International Journal of Molecular Sciences. 2022; 23(14):7691. https://doi.org/10.3390/ijms23147691
Chicago/Turabian StylePaolillo, Roberta, Stefania D’Apice, Gabriele Giacomo Schiattarella, Pietro Ameri, Domenica Borzacchiello, Daniele Catalucci, Cristina Chimenti, Lia Crotti, Sebastiano Sciarretta, Daniele Torella, and et al. 2022. "Mitochondrial a Kinase Anchor Proteins in Cardiovascular Health and Disease: A Review Article on Behalf of the Working Group on Cellular and Molecular Biology of the Heart of the Italian Society of Cardiology" International Journal of Molecular Sciences 23, no. 14: 7691. https://doi.org/10.3390/ijms23147691
APA StylePaolillo, R., D’Apice, S., Schiattarella, G. G., Ameri, P., Borzacchiello, D., Catalucci, D., Chimenti, C., Crotti, L., Sciarretta, S., Torella, D., Feliciello, A., & Perrino, C. (2022). Mitochondrial a Kinase Anchor Proteins in Cardiovascular Health and Disease: A Review Article on Behalf of the Working Group on Cellular and Molecular Biology of the Heart of the Italian Society of Cardiology. International Journal of Molecular Sciences, 23(14), 7691. https://doi.org/10.3390/ijms23147691