
Ketamine plus Alcohol: What We Know and What We Can Expect about This

 , ,
, ,  and
and
Abstract
:1. Introduction
2. Epidemiological Features of Ethanol plus Ketamine Consumption
Fatal Outcomes
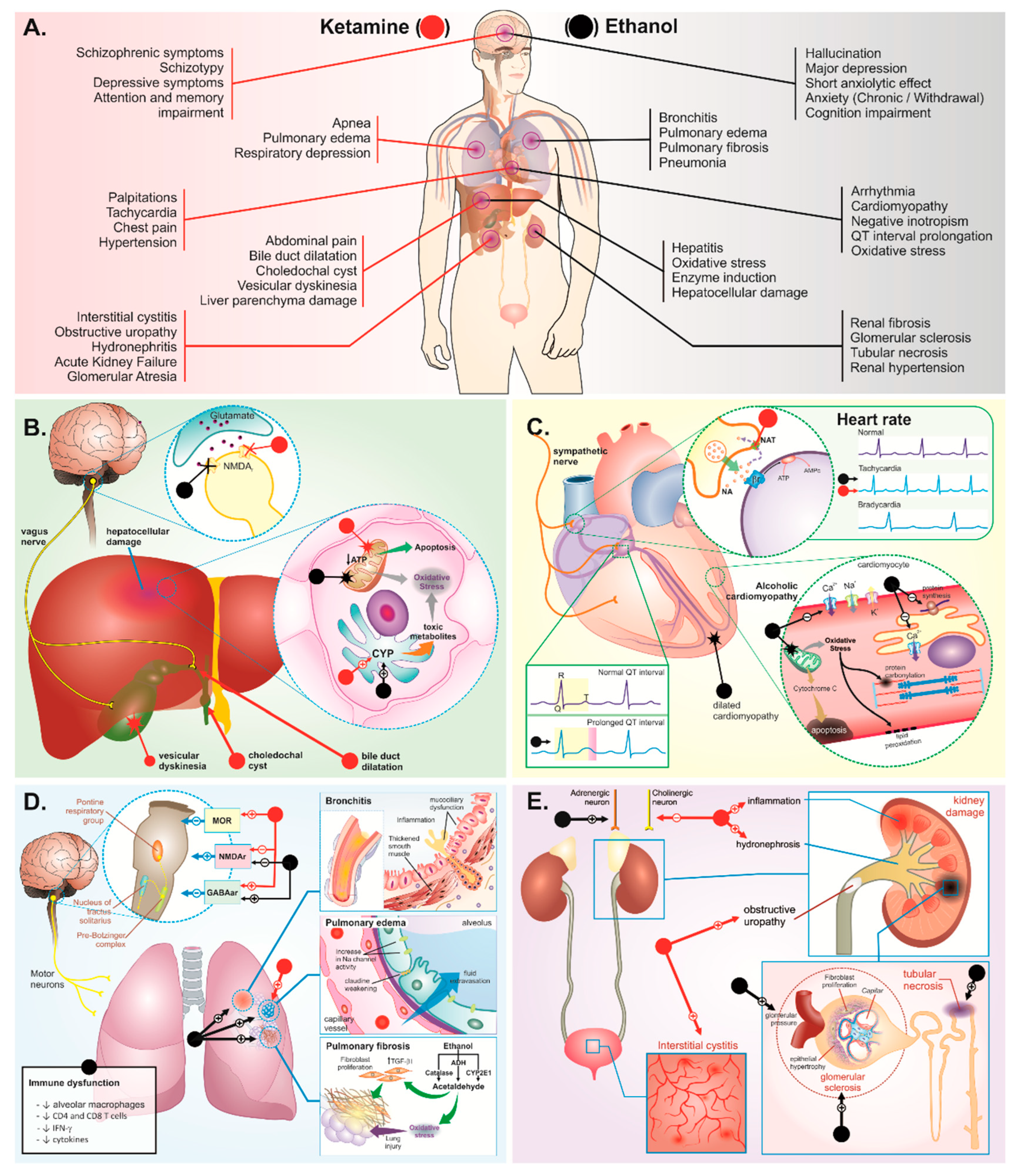
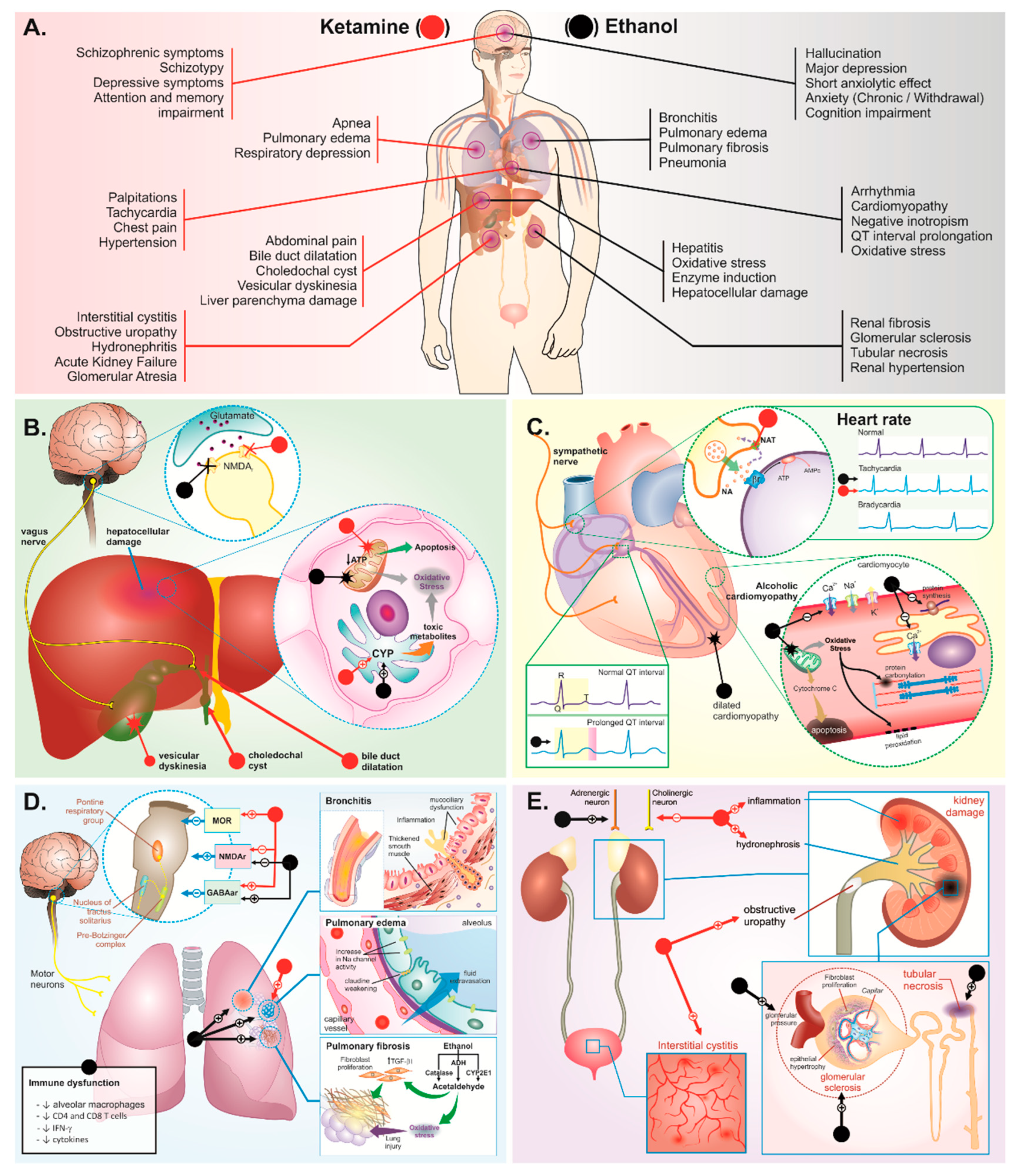
3. Body Systems Consequences of Ketamine plus Ethanol Abuse
3.1. Liver and Biliary System Damage
3.2. Cardiorespiratory System
3.3. Urinary System
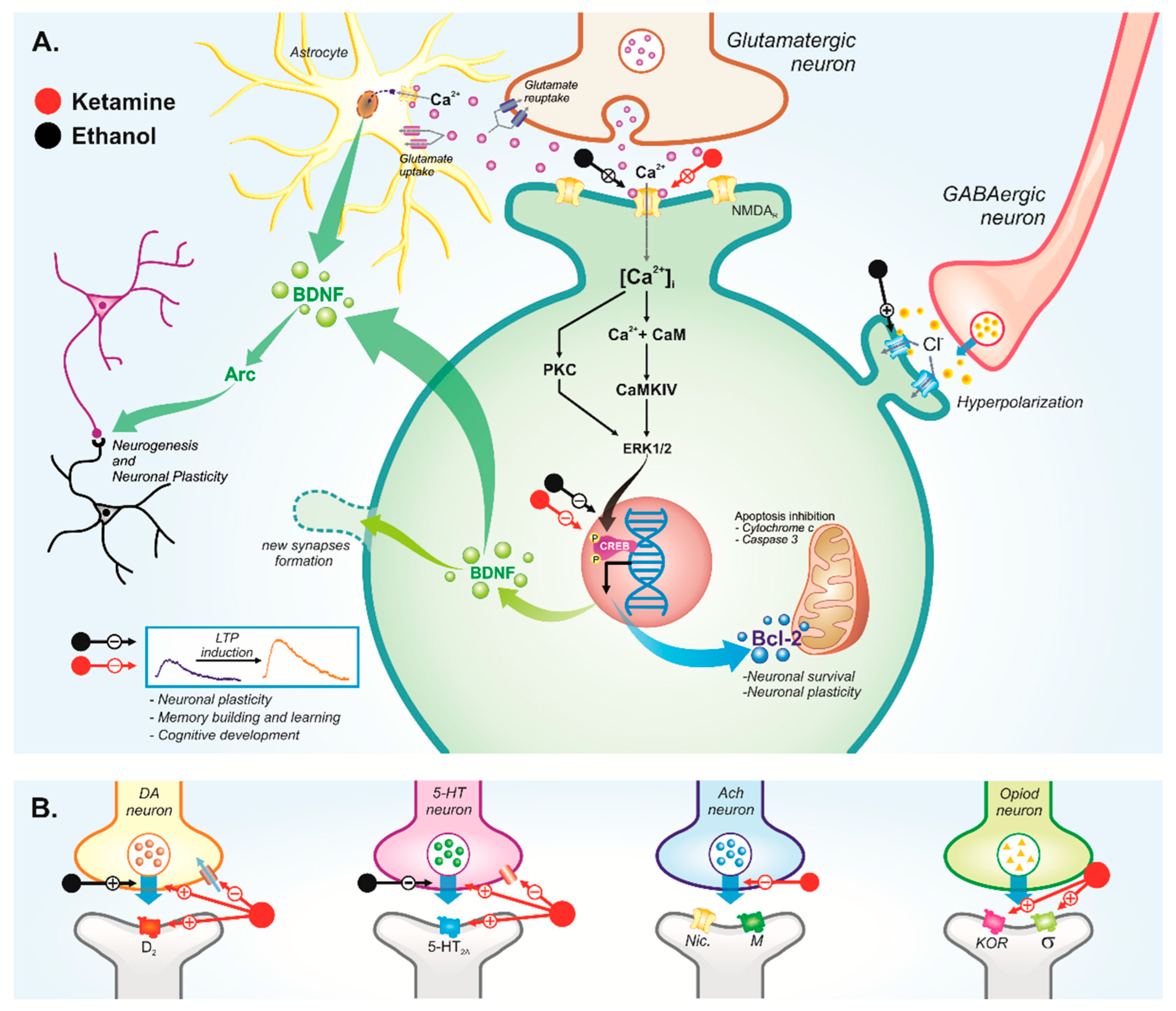
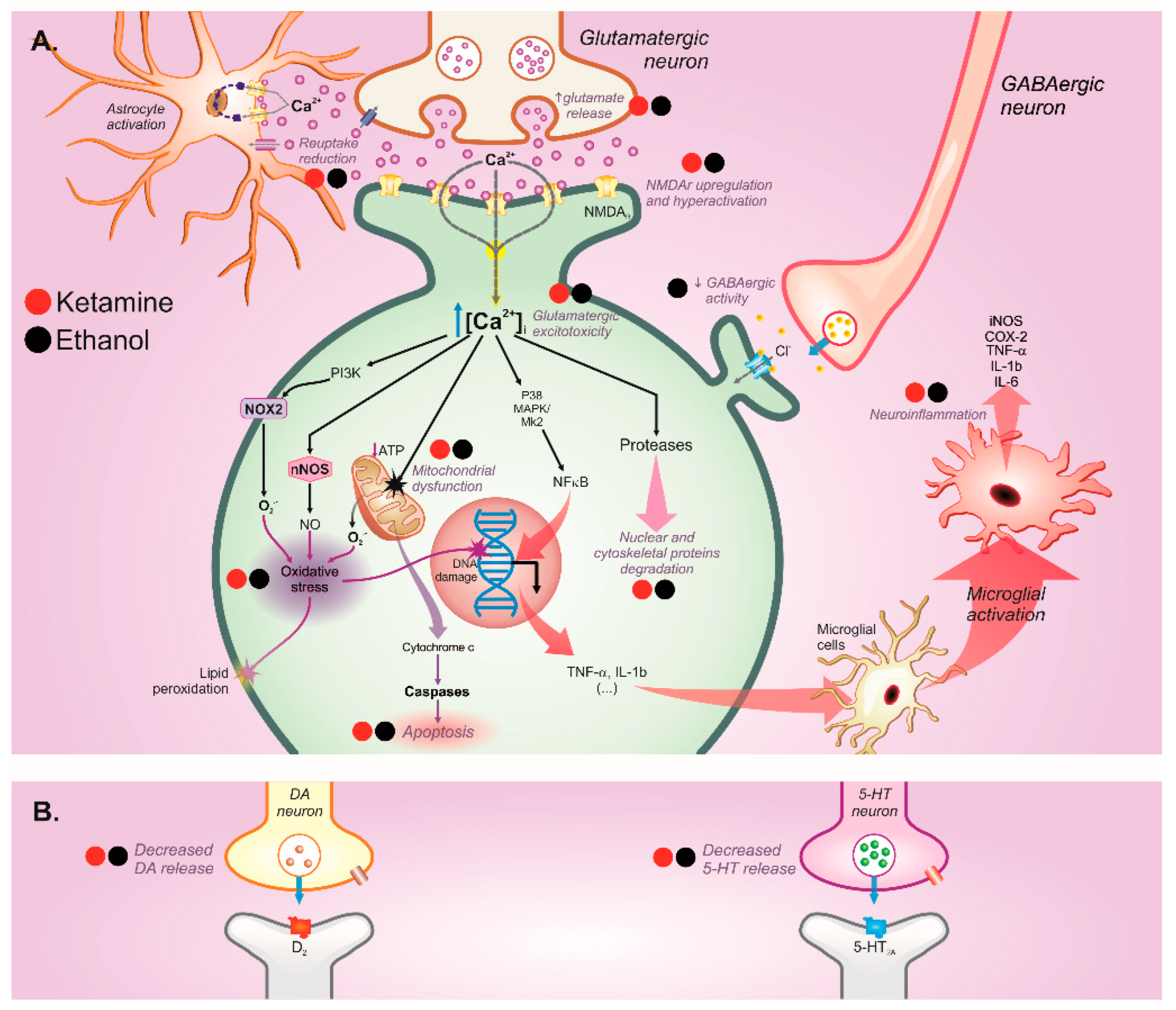
3.4. Central Nervous System
3.4.1. Behavioral Disorders
Psychosis
Depression
Anxiety
Cognitive Disorders
4. Potential Pharmacological Interactions between Ketamine and Ethanol Use
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Olsen, Y. What Is Addiction? History, Terminology, and Core Concepts. Med. Clin. 2022, 106, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Bombana, H.S.; Bogstrand, S.T.; Gjerde, H.; Jamt, R.E.G.; de Carvalho, H.B.; Andreuccetti, G.; de Oliveira Bernini, C.; Muñoz, D.R.; Leyton, V.; Greve, J.M.D. Use of alcohol and illicit drugs by trauma patients in Sao Paulo, Brazil. Injury 2022, 53, 30–36. [Google Scholar] [CrossRef] [PubMed]
- Wagner, V.; Flores-Aranda, J.; Villela Guilhon, A.C.; Knight, S.; Bertrand, K. How do Past, Present and Future Weigh into Trajectories of Precarity? The Time Perspectives of Young Psychoactive Substance Users Living in Situations of Social Precarity in Montreal. Qual. Health Res. 2022, 32, 195–209. [Google Scholar] [CrossRef] [PubMed]
- Dos Santos, A.R.; Perrelli, J.G.A.; Silva, T.T.D.M.; Lopes, M.V.D.O.; Frazão, I.D.S. Instruments related to drug use in adolescents: An integrated review. Texto Contexto Enfermagem. 2018, 27, e0370017. [Google Scholar]
- Silva, J.L.; Silva, R.L.; Silva, A.L.; Silva, G.C.B.e.; Cortez, A.C.L. Uso de Substâncias Psicoativas “Drogas”: Uma Revisão De Literatura/Psychoactive Substance Use “Drugs”: A Review. Northeast. Braz. Health J. 2013, 1, 2–8. [Google Scholar]
- Cohen, S.M.; Alexander, R.S.; Holt, S.R. The Spectrum of Alcohol Use: Epidemiology, Diagnosis, and Treatment. Med. Clin. N. Am. 2022, 106, 43–60. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Alcohol; World Health Organization: Geneva, Switzerland, 2018. [Google Scholar]
- Manzo-Avalos, S.; Saavedra-Molina, A. Cellular and mitochondrial effects of alcohol consumption. Int. J. Environ. Res. Public Health 2010, 7, 4281–4304. [Google Scholar] [CrossRef] [Green Version]
- Rehm, J.; Mathers, C.; Popova, S.; Thavorncharoensap, M.; Teerawattananon, Y.; Patra, J. Global burden of disease and injury and economic cost attributable to alcohol use and alcohol-use disorders. Lancet 2009, 373, 2223–2233. [Google Scholar] [CrossRef]
- Marco, C.A.; Sich, M.; Ganz, E.; Clark, A.N.J.; Graham, M. Penetrating trauma: Relationships to recreational drug and alcohol use. Am. J. Emerg. Med. 2022, 52, 8–12. [Google Scholar] [CrossRef]
- Vwaire Orhurhu, A.J.; Vashisht, R.; Claus, L.E.; Cohen Afiliações, S.P. Toxicidade da Cetamina Atividade de Educação Continuada. Available online: https://www.ncbi.nlm.nih.gov/books/NBK541087/?report=printable (accessed on 26 March 2022).
- Lee, T.S.H.; Liu, Y.H.; Huang, Y.J.; Tang, W.K.; Wang, Y.; Hu, S.; Lin, C.P.; Li, C.S.R.; Hung, C.C. Clinical and behavior characteristics of individuals who used ketamine. Sci. Rep. 2022, 12, 1–8. [Google Scholar] [CrossRef]
- UNODC—United Nations Office on Drug and Crime. World Drug Report; United Nations Office on Drug and Crime: Vienna, Asutria, 2015. [Google Scholar]
- Peoples, R.W.; White, G.; Lovinger, D.M.; Weight, F.F. Ethanol inhibition of N-methyl-D-aspartate-activated current in mouse hippocampal neurones: Whole-cell patch-clamp analysis. Br. J. Pharmacol. 1997, 122, 1035–1042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaczmarek, B.; Kowalski, K.; Bogudzińska, B.; Piotrowski, P. Ketamine as A Novel Drug for Depression Treatment. Psychiatr. Danubina 2021, 33, 468–474. [Google Scholar] [CrossRef] [PubMed]
- Roberto, M.; Kirson, D.; Khom, S. The role of the central amygdala in alcohol dependence. Cold Spring Harb. Perspect. Med. 2021, 11, 1–23. [Google Scholar] [CrossRef] [Green Version]
- Honey, C.R.; Miljkovic, Z.; Macdonald, J.F. Ketamine and Phencyclidine Cause a Voltage-Dependent Block of Responses To L-Aspartic Acid. Neurosci. Lett. 1985, 61, 135–139. [Google Scholar] [CrossRef]
- Macdonald, J.F.; Miljkovic, Z.; Pennefather, P. Use-Dependent Block of Excitatory Amino Acid Currents in Cultured Neurons by Ketamine. J. Neurophysiol. 1987, 58, 251–266. Available online: www.physiology.org/journal/jn (accessed on 28 March 2022). [CrossRef] [PubMed]
- Liu, Q.; Xu, T.Y.; Zhang, Z.B.; Leung, C.K.; You, D.Y.; Wang, S.W.; Yi, S.; Jing, Q.; Xie, R.F.; Li, H.J.; et al. Effects of co-administration of ketamine and ethanol on the dopamine system via the cortex-striatum circuitry. Life Sci. 2017, 179, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, L.M.P.; Cartágenes, S.C.; Barros, M.A.; Carvalheiro, T.C.V.S.; Castro, N.C.F.; Schamne, M.G.; Prediger, R.D.; Monteiro, M.C.; Fontes-Júnior, E.A.; Cunha, R.A.; et al. Repeated cycles of binge-like ethanol exposure induce immediate and delayed neurobehavioral changes and hippocampal dysfunction in adolescent female rats. Behav. Brain Res. 2018, 350, 99–108. [Google Scholar] [CrossRef]
- Fernandes, L.M.P.; Lopes, K.S.; Santana, L.N.S.; Fontes-Júnior, E.A.; Ribeiro, C.H.M.A.; Silva, M.C.F.; de Oliveira Paraense, R.S.; Crespo-López, M.E.; Gomes, A.R.Q.; Lima, R.R.; et al. Repeated cycles of binge-like ethanol intake in adolescent female rats induce motor function impairment and oxidative damage in motor cortex and liver, but not in blood. Oxid. Med. Cell. Longev. 2018, 2018, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, K.J.; Doshi, M.R.; Holzhausen, J.M.; Natavio, A.; Cadiz, M.; Winegardner, J.E. Treatment of Severe Alcohol Withdrawal. Ann. Pharmacother. 2016, 50, 389–401. [Google Scholar] [CrossRef]
- Siomek-Gorecka, A.; Dlugosz, A.; Czarnecki, D. The molecular basis of alcohol use disorder (Aud). genetics, epigenetics, and nutrition in aud: An amazing triangle. Int. J. Mol. Sci. 2021, 22, 4262. [Google Scholar] [CrossRef]
- Curran, H.V.; Morgan, C. Cognitive, dissociative and psychotogenic effects of ketamine in recreational users on the night of drug use and 3 days later. Addiction 2000, 95, 575–590. [Google Scholar] [CrossRef] [PubMed]
- De Carvalho Cartágenes, S.; Fernandes, L.M.P.; Carvalheiro, T.C.V.S.; de Sousa, T.M.; Gomes, A.R.Q.; Monteiro, M.C.; de Oliveira Paraense, R.S.; Crespo-López, M.E.; Lima, R.R.; Fontes-Júnior, E.A.; et al. “Special K” drug on adolescent rats: Oxidative damage and neurobehavioral impairments. Oxid. Med. Cell. Longev. 2019, 2019, 5452727. [Google Scholar] [CrossRef] [PubMed]
- Hase, A.; Erdmann, M.; Limbach Hasler, G. Analysis of recreational psychedelic substance use experiences classified by substance. Psychopharmacology 2022, 239, 643–659. [Google Scholar] [CrossRef] [PubMed]
- Pan American Health Organization—PAHO. Alcoohl, Key Facts. Available online: https://www.paho.org/en/topics/alcohol (accessed on 1 February 2022).
- National Institute on Alcohol Abuse e Alcoholism (NIAAA); Department of Health and Human Services; National Institutes of Health NIAAA Newsletter; National Institute on Alcohol Abuse and Alcoholism. Featuring Information from the National Institute on Alcohol Abuse and Alcoholism; Office of Research Translation and Communications, NIAAA, NIH, DHHS: Bethesda, MD, EUA, 2004.
- Lin, H.; Kim, J.G.; Park, S.W.; Noh, H.J.; Kim, J.M.; Yoon, C.Y.; Woo, N.S.; Kim, B.; Cho, S., II; Choi, B.H.; et al. Enhancement of 5-HT2A receptor function and blockade of Kv1.5 by MK801 and ketamine: Implications for PCP derivative-induced disease models. Exp. Mol. Med. 2018, 50, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molero, P.; Ramos-Quiroga, J.A.; Martin-Santos, R.; Calvo-Sánchez, E.; Gutiérrez-Rojas, L.; Meana, J.J. Antidepressant Efficacy and Tolerability of Ketamine and Esketamine: A Critical Review. CNS Drugs 2018, 32, 411–420. [Google Scholar] [CrossRef] [PubMed]
- Van Amsterdam, J.; van den Brink, W. Harm related to recreational ketamine use and its relevance for the clinical use of ketamine. A systematic review and comparison study. Expert Opin. Drug Saf. 2022, 21, 83–94. [Google Scholar] [CrossRef]
- Siegel, R.K. Phencyclidine and ketamine intoxication: A study of four populations of recreational users. Public Health Serv. Alcohol. 1978, 21, 119–147. [Google Scholar]
- Sassano-Higgins, S.; Baron, D.; Juarez, G.; Esmaili, N.; Gold, M. A review of ketamine abuse and diversion. Depress. Anxiety 2016, 33, 718–727. [Google Scholar] [CrossRef]
- Larabi, I.A.; Fabresse, N.; Etting, I.; Nadour, L.; Pfau, G.; Raphalen, J.H.; Philippe, P.; Edel, Y.; Alvarez, J.C. Prevalence of New Psychoactive Substances (NPS) and conventional drugs of abuse (DOA) in high risk populations from Paris (France) and its suburbs: A cross sectional study by hair testing (2012–2017). Drug Alcohol Depend. 2019, 204, 107508. [Google Scholar] [CrossRef]
- Johnston, L.D.; O’malley, P.M.; Miech, R.A.; Bachman, J.G.; Schulenberg, J.E. Sponsored by The National Institute on Drug Abuse at the National Institutes of Health. Monitoring the Future National Survey Results on Drug Use; Institute for Social Research, University of Michigan: Ann Arbor, MI, USA, 1975. [Google Scholar]
- Pavarin, R.M.; Marani, S.; Turino, E. Ketamine abusers referring to emergency departments in northern Italy: A cross-sectional study. Ann. Ist. Super. Sanita 2019, 55, 338–344. [Google Scholar]
- Wood, D.M.; Nicolaou, M.; Dargan, P.I. Epidemiology of Recreational Drug Toxicity in a Nightclub Environment. Subst. Use Misuse 2009, 44, 1495–1502. [Google Scholar] [CrossRef] [PubMed]
- Winstock, A.R.; Mitcheson, L.; Gillatt, D.A.; Cottrell, A.M. The prevalence and natural history of urinary symptoms among recreational ketamine users. BJU Int. 2012, 110, 1762–1766. [Google Scholar] [CrossRef] [PubMed]
- Morgan, C.J.A.; Muetzelfeldt, L.; Curran, H.V. Ketamine use, cognition and psychological wellbeing: A comparison of frequent, infrequent and ex-users with polydrug and non-using controls. Addiction 2009, 104, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Morgan, C.J.A.; Muetzelfeldt, L.; Curran, H.V. Consequences of chronic ketamine self-administration upon neurocognitive function and psychological wellbeing: A 1-year longitudinal study. Addiction 2010, 105, 121–133. [Google Scholar] [CrossRef] [PubMed]
- Galea, S.; Coffin, P.O.; Ahern, J.; Leon, A.C.; Vlahov, D.; Tardiff, K. Opiates, cocaine and alcohol combinations in accidental drug overdose deaths in New York City 1990–1998. Addiction 2003, 98, 739–747. [Google Scholar]
- Kerr, T.; Fairbairn, N.; Tyndall, M.; Marsh, D.; Li, K.; Montaner, J.; Wood, E. Predictors of non-fatal overdose among a cohort of polysubstance-using injection drug users. Drug Alcohol Depend. 2007, 87, 39–45. [Google Scholar] [CrossRef]
- Sergeev, B.; Karpets, A.; Sarang, A.; Tikhonov, M. Prevalence and Circumstances of Opiate Overdose Among Injection Drug Users in the Russian Federation. J. Urban Health Bull. N. Y. Acad. Med. 2003, 80, 212–219. [Google Scholar] [CrossRef] [Green Version]
- Darke, S.; Duflou, J.; Farrell, M.; Peacock, A.; Lappin, J. Characteristics and circumstances of death related to the self-administration of ketamine. Addiction 2021, 116, 339–345. [Google Scholar] [CrossRef]
- Schifano, F. Trapped in the “K-hole”: Overview of deaths associated with ketamine misuse in the UK. J. Clin. Psychopharmacol. 2008, 28, 114–116. [Google Scholar] [CrossRef] [Green Version]
- Kalsi, S.S.; Wood, D.M.; Dargan, P.I. The epidemiology and patterns of acute and chronic toxicity associated with recreational ketamine use. Emerg. Health Threat. J. 2011, 4, 7107. [Google Scholar] [CrossRef]
- Sen, K.L.R. A review of the nonmedical use of ketamine: Use, users and consequences. J. Psychoact. Drugs 2000, 32, 419–433. [Google Scholar]
- Ng, S.; Lee, H.; Chan, Y.; Lau, F. Dilated Common Bile Ducts in Ketamine Abusers. 2009. Available online: www.hkmj.org (accessed on 28 March 2022).
- Wong, S.; Lee, K.; Wong, J.; Ng, W.; Cheung, Y.; Lai, P. Dilated Common Bile Ducts Mimicking Choledochal Cysts in Ketamine Abusers. 2009. Available online: www.hkmj.org (accessed on 28 March 2022).
- Yu, W.L.; Cho, C.C.M.; Lung, P.F.C.; Hung, E.H.Y.; Hui, J.W.Y.; Chau, H.H.L.; Chan, A.W.; Ahuja, A.T. Ketamine-related cholangiopathy: A retrospective study on clinical and imaging findings. Abdom. Imaging 2014, 39, 1241–1246. [Google Scholar] [CrossRef] [PubMed]
- Lo, R.S.; Krishnamoorthy, R.; Freeman, J.G.; Austin, A.S. Cholestasis and biliary dilatation associated with chronic ketamine abuse: A case series. Item Type Article Cholestasis and biliary dilatation associated with chronic ketamine abuse: A case series. Singap. Med. J. 2011, 52, e52–e55. Available online: http://hdl.handle.net/20.500.12904/637 (accessed on 29 March 2022).
- Poon, T.L.; Wong, K.F.; Chan, M.Y.; Fung, K.W.; Chu, S.K.; Man, C.W.; You, M.K.; Leung, S.K. Upper gastrointestinal problems in inhalational ketamine abusers. J. Dig. Dis. 2010, 11, 106–110. [Google Scholar] [CrossRef]
- Selby, N.M.; Anderson, J.; Bungay, P.; Chesterton, L.J.; Kolhe, N.V. Obstructive nephropathy and kidney injury associated with ketamine abuse. NDT Plus 2008, 1, 310–312. [Google Scholar] [CrossRef] [Green Version]
- Wong, G.L.H.; Tam, Y.H.; Ng, C.F.; Chan, A.W.H.; Choi, P.C.L.; Chu, W.C.W.; Lai, P.B.; Chan, H.L.; Wong, V.W. Liver Injury Is Common Among Chronic Abusers of Ketamine. Clin. Gastroenterol. Hepatol. 2014, 12, 1759–1762.e1. [Google Scholar] [CrossRef]
- Lee, S.T.; Wu, T.T.; Yu, P.Y.; Chen, R.M. Apoptotic insults to human HepG2 cells induced by S-(+)-ketamine occurs through activation of a Bax-mitochondria-caspase protease pathway. Br. J. Anaesth. 2009, 102, 80–89. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.Y.; Xie, D.P.; Liu, J.Z. Microinjection of glutamate into dorsal motor nucleus of the vagus excites gallbladder motility through NMDA receptor—Nitric oxide—cGMP pathway. Neurogastroenterol. Motil. 2004, 16, 347–353. [Google Scholar] [CrossRef]
- Rocco, A.; Compare, D.; Angrisani, D.; Sanduzzi Zamparelli Nardone, G. Alcoholic disease: Liver and beyond. World J. Gastroenterol. 2014, 20, 14652–14659. [Google Scholar] [CrossRef]
- Seth, D.; Haber, P.S.; Syn, W.K.; Diehl, A.M.; Day, C.P. Pathogenesis of alcohol-induced liver disease: Classical concepts and recent advances. J. Gastroenterol. Hepatol. 2011, 26, 1089–1105. [Google Scholar] [CrossRef]
- Mello, T.; Ceni, E.; Surrenti, C.; Galli, A. Alcohol induced hepatic fibrosis: Role of acetaldehyde. Mol. Asp. Med. 2008, 29, 17–21. [Google Scholar] [CrossRef] [PubMed]
- Weiner, A.L.; Vieira, L.; Mckay, C.A.; Bayer, M.J. Selected Topics: Toxicology Ketamine Abusers Presenting to The Emergency Department: A Case Series. J. Emerg. Med. 2000, 18, 447–451. [Google Scholar] [CrossRef]
- Wai, M.S.; Jiang, Y.; Chan, W.M.; Tsui, T.Y.M.; Tang, H.C.; Lam, W.P.; Fan, M.; Yew, D.T. Long term ketamine and ketamine plus alcohol toxicity—What can we leran from animal models? Mini Rev. Med. Chem. 2013, 13, 273–279. [Google Scholar]
- Wei, Y.; Yang, J.; Song, W.; Guo, Q. Gourd-shaped bladder associated with ketamine abuse. Urol. Int. 2012, 89, 123–124. [Google Scholar] [CrossRef] [PubMed]
- Turkish, A.; Luo, J.J.; Lefkowitch, J.H. Ketamine abuse, biliary tract disease, and secondary sclerosing cholangitis. Hepatology 2013, 58, 825–827. [Google Scholar] [CrossRef] [PubMed]
- Smith, K.M.; Larive, L.L.; Romanelli, F. Club drugs: Methylenedioxymethamphetamine, flunitrazepam, ketamine hydrochloride, and γ-hydroxybutyrate. Am. J. Health-Syst. Pharm. 2022, 59, 1067–1076. Available online: www.clubdrugs.org (accessed on 16 February 2022). [CrossRef] [PubMed]
- Kohrs, R.; Durieux, M.E. Ketamine: Teaching an Old Drug New Tricks. Anesth. Analg. 1998, 87, 1186–1193. [Google Scholar]
- Yuan, H.; Du, S.; Deng, Y.; Xu, X.; Zhang, Q.; Wang, M.; Wang, P.; Su, Y.; Liang, X.; Sun, Y.; et al. Effects of microRNA-208a on inflammation and oxidative stress in ketamine-induced cardiotoxicity through Notch/NF-κB signal pathways by CHD9. Biosci. Rep. 2019, 39, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Mustroph, J.; Lebek, S.; Maier, L.S.; Neef, S. Mechanisms of cardiac ethanol toxicity and novel treatment options. Pharmacol. Ther. 2019, 197, 1–10. [Google Scholar] [CrossRef]
- Kinkead, R.; Bach, K.B.; Johnson, S.M.; Hodgeman, B.A.; Mitchell, G.S. Plasticity in respiratory motor control: Intermittent hypoxia and hypercapnia activate opposing serotonergic and noradrenergic modulatory systems. Comp. Biochem. Physiol. Part A 2001, 130, 207–218. [Google Scholar] [CrossRef]
- Imam, M.Z.; Kuo, A.; Ghassabian, S.; Smith, M.T. Progress in understanding mechanisms of opioid-induced gastrointestinal adverse effects and respiratory depression. Neuropharmacology 2018, 131, 238–255. [Google Scholar] [CrossRef] [PubMed]
- Waters, K.A.; Machaalani, R. Role of NMDA receptors in development of respiratory control. Respir. Physiol. Neurobiol. 2005, 149, 123–130. [Google Scholar] [CrossRef] [PubMed]
- Traphagen, N.; Tian, Z.; Allen-Gipson, D. Chronic ethanol exposure: Pathogenesis of pulmonary disease and dysfunction. Biomolecules 2015, 5, 2840–2853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chu, P.S.; Ma, W.K.; Yu, C.; Yiu, M.K.; Man, C.W. Destruction of the urinary tract by ketamine abuse: Hong Kong Local experience. SurgPract 2010, 14, 44–48. [Google Scholar] [CrossRef]
- Chu, P.S.K.; Ma, W.K.; Wong, S.C.W.; Chu, R.W.H.; Cheng, C.H.; Wong, S.; Tse, J.M.; Lau, F.L.; You, M.K.; Man, C.W. The destruction of the lower urinary tract by ketamine abuse: A new syndrome? BJU Int. 2008, 102, 1616–1622. [Google Scholar] [CrossRef]
- Shahani, R.; Streutker, C.; Dickson, B.; Stewart, R.J. Ketamine-Associated Ulcerative Cystitis: A New Clinical Entity. Urology 2007, 69, 810–812. [Google Scholar] [CrossRef]
- Rajandram, R.; Yap, N.Y.; Ong, T.A.; Mun, K.S.; Mohamad Wali, H.A.; Hasan, M.S.; Razack, A.H.A.; Ali Mohd, M. Oral ketamine induced pathological changes of the urinary tract in a rat model. Malays. J. Pathol. 2017, 39, 47. [Google Scholar]
- Frenkel, C. Urban BW. Molecular actions of racemic ketamine on human cns sodium channels. Br. J. Anaesth. 1992, 69, 292–297. [Google Scholar] [CrossRef]
- Han, J.; Kim, N.; Joo, H.; Kim, E. Ketamine blocks Ca 2-activated K channels in rabbit cerebral arterial smooth muscle cells. Am. J. Physiol. -Heart Circ. Physiol. 2003, 285, H1347–H1355. Available online: www.ajpheart.org (accessed on 16 February 2022). [CrossRef]
- Kawano, T.; Oshita, S.; Takahashi, A.; Tsutsumi, Y.; Tanaka, K.; Tomiyama, Y.; Kitahata, H.; Nakaya, Y. Molecular Mechanisms Underlying Ketamine-mediated Inhibition of Sarcolemmal Adenosine Triphosphate-sensitive Potassium Channels. Anesthesiology 2005, 102, 93–101. Available online: http://pubs.asahq.org/anesthesiology/article-pdf/102/1/93/357819/0000542-200501000-00017.pdf (accessed on 16 February 2022). [CrossRef]
- Chen, X.; Shu, S.; Bayliss, D.A. HCN1 channel subunits are a molecular substrate for hypnotic actions of ketamine. J. Neurosci. 2009, 29, 600–609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, E.; Chang, H.Y.; Chang, S.Y.; Sun, G.H.; Yu, D.S.; Cha, T.L. Involvement of purinergic neurotransmission in ketamine induced bladder dysfunction. J. Urol. 2011, 186, 1134–1141. [Google Scholar] [CrossRef] [PubMed]
- Yeung, L.Y.; Rudd, J.A.; Lam, W.P.; Mak, Y.T.; Yew, D.T. Mice are prone to kidney pathology after prolonged ketamine addiction. Toxicol. Lett. 2009, 191, 275–278. [Google Scholar] [CrossRef] [PubMed]
- Robinson, D.; Hanna-Mitchell, A.; Rantell, A.; Thiagamoorthy, G.; Cardozo, L. Are we justified in suggesting change to caffeine, alcohol, and carbonated drink intake in lower urinary tract disease? Report from the ICI-RS 2015. Neurourol. Urodyn. 2017, 36, 876–881. [Google Scholar] [CrossRef]
- Li, D.; Xu, J.; Liu, F.; Wang, X.; Yang, H.; Li, X. Alcohol Drinking and the Risk of Chronic Kidney Damage: A Meta-Analysis of 15 Prospective Cohort Studies. Alcohol. Clin. Exp. Res. 2019, 43, 1360–1372. [Google Scholar] [CrossRef]
- Cheungpasitporn, W.; Thongprayoon, C.; Kittanamongkolchai, W.; Brabec, B.A.; O’corragain, O.A.; Edmonds, P.J.; Erickson, S.B. High alcohol consumption and the risk of renal damage: A systematic review and meta-analysis. QJM 2015, 108, 539–548. [Google Scholar] [CrossRef] [Green Version]
- Leal, S.; Ricardo Jorge, D.O.; Joana, B.; Maria, S.S.; Isabel, S.S. Heavy Alcohol Consumption Effects on Blood Pressure and on Kidney Structure Persist after Long-Term Withdrawal. Kidney Blood Press. Res. 2017, 42, 664–675. [Google Scholar] [CrossRef]
- Brzóska, M.M.; Moniuszko-Jakoniuk, J.; Piłat-Marcinkiewicz, B.; Sawicki, B. Liver and kidney function and histology in rats exposed to cadmium and ethanol. Alcohol Alcohol. 2003, 38, 2–10. [Google Scholar] [CrossRef] [Green Version]
- Husain, K.; Ferder, L.; Ansari, R.A.; Lalla, J. Chronic ethanol ingestion induces aortic inflammation/oxidative endothelial injury and hypertension in rats. Hum. Exp. Toxicol. 2011, 30, 930–939. [Google Scholar] [CrossRef] [Green Version]
- Tirapelli, L.F.; Martins-Oliveira, A.; Batalhão, M.E.; Tirapelli, D.P.; Carnio, E.C.; Tanus-Santos, J.E.; Queiroz, R.H.; Padovan, C.M.; Tirapelli, C.R. Ethanol consumption increases the expression of endothelial nitric oxide synthase, inducible nitric oxide synthase and metalloproteinases in the rat kidney. J. Pharm. Pharmacol. 2012, 64, 68–76. [Google Scholar] [CrossRef]
- Dinu, D.; Nechifor, M.T.; Movileanu, L. Ethanol-induced alterations of the antioxidant defense system in rat kidney. J. Biochem. Mol. Toxicol. 2006, 19, 386–395. [Google Scholar] [CrossRef] [PubMed]
- Hill, G.S.; Heudes, D.; Jacquot, C.; Gauthier, É.; Bariéty, J. Morphometric evidence for impairment of renal autoregulation in advanced essential hypertension. Kidney Int. 2006, 69, 823–831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ojeda, M.L.; Barrero, M.J.; Nogales, F.; Murillo, M.L.; Carreras, O. Oxidative effects of chronic ethanol consumption on the functions of heart and kidney: Folic acid supplementation. Alcohol Alcohol. 2012, 47, 404–412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdulla, M.H.; Johns, E.J. Nitric oxide impacts on angiotensin AT2 receptor modulation of high-pressure baroreflex control of renal sympathetic nerve activity in anaesthetized rats. Acta Physiol. 2014, 210, 832–844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morgan, C.J.A.; Curran, H.V. Ketamine use: A review. Addiction 2012, 107, 27–38. [Google Scholar] [CrossRef]
- Hansen, G.; Jensen, B. The Psychotropic Effect of Ketamine. J. Psychoact. Drugs 1988, 20, 419–425. [Google Scholar] [CrossRef]
- Fernandes, L.M.P.; Bezerra, F.R.; Monteiro, M.C.; Silva, M.L.; de Oliveira, F.R.; Lima, R.R.; Fontes-Júnior, E.A.; Maia, C.S.F. Thiamine deficiency, oxidative metabolic pathways and ethanol-induced neurotoxicity: How poor nutrition contributes to the alcoholic syndrome, as Marchiafava-Bignami disease. Eur. J. Clin. Nutr. Nat. Publ. Group 2017, 71, 580–5866. [Google Scholar] [CrossRef]
- Fernandes, L.M.P.; de Andrade, E.F.; Monteiro, M.C.; Cartágenes, S.C.; Lima, R.R.; Prediger, R.D.; Maia, C.S.F. Ethanol: Neurotoxicity and Brain Disorders. In Addictive Substances and Neurological Disease: Alcohol, Tobacco, Caffeine, and Drugs of Abuse in Everyday Lifestyles; Elsevier: Amsterdam, The Netherlands, 2017; pp. 201–215. [Google Scholar]
- Morgan, C.J.A.; Curran, H.V. Acute and chronic effects of ketamine upon human memory: A review. Psychopharmacology 2006, 188, 408–424. [Google Scholar] [CrossRef]
- Stefanovic, A.; Brandner, B.; Klaassen, E.; Cregg, R.; Nagaratnam, M.; Bromley, L.M.; Das, R.K.; Rossell, S.L.; Morgan, C.J.; Curran, H.V. Acute and chronic effects of ketamine on semantic priming: Modeling schizophrenia. J. Clin. Psychopharmacol. 2009, 29, 124–133. [Google Scholar] [CrossRef]
- Edward Roberts, R. Abnormalities in white matter microstructure associated with chronic ketamine. Neuropsychopharmacology 2014, 39, 329–338. [Google Scholar] [CrossRef] [Green Version]
- Blagrove, M.; Morgan, C.J.A.; Curran, H.V.; Bromley, L.; Brandner, B. The incidence of unpleasant dreams after sub-anaesthetic ketamine. Psychopharmacology 2009, 203, 109–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muetzelfeldt, L.; Kamboj, S.K.; Rees, H.; Taylor, J.; Morgan, C.J.A.; Curran, H.V. Journey through the K-hole: Phenomenological aspects of ketamine use. Drug Alcohol Depend. 2008, 95, 219–229. [Google Scholar] [CrossRef] [PubMed]
- Morgan, C.J.A.; Duffin, S.; Hunt, S.; Monaghan, L.; Mason, O.; Curran, H.V. Neurocognitive function and schizophrenia-proneness in individuals dependent on ketamine, on high potency cannabis (skunk) or on cocaine. Pharmacopsychiatry 2012, 45, 269–274. [Google Scholar] [CrossRef]
- Morgan, C.J.A.; Mofeez, A.; Brandner, B.; Bromley, L.; Curran, H.V. Acute Effects of Ketamine on Memory Systems and Psychotic Symptoms in Healthy Volunteers. Neuropsychopharmacology 2004, 29, 208–218. [Google Scholar] [CrossRef] [PubMed]
- Morgan, C.J.A.; Riccelli, M.; Maitland, C.H.; Curran, H.V. Long-term effects of ketamine: Evidence for a persisting impairment of source memory in recreational users. Drug Alcohol Depend. 2004, 75, 301–308. [Google Scholar] [CrossRef]
- Morgan, C.J.A.; Monaghan, L.; Curran, H.V. Beyond the K-hole: A 3-year longitudinal investigation of the cognitive and subjective effects of ketamine in recreational users who have substantially reduced their use of the drug. Addiction 2004, 99, 1450–1461. [Google Scholar] [CrossRef]
- Mason, O.J.; Morgan, C.J.M.; Stefanovic, A.; Curran, H.V. The Psychotomimetic States Inventory (PSI): Measuring psychotic-type experiences from ketamine and cannabis. Schizophr. Res. 2008, 103, 138–142. [Google Scholar] [CrossRef]
- Freeman, T.P.; Morgan, C.J.A.; Klaassen, E.; Das, R.K.; Stefanovic, A.; Brandner, B.; Curran, H.V. Superstitious conditioning as a model of delusion formation following chronic but not acute ketamine in humans. Psychopharmacology 2009, 206, 563–573. [Google Scholar] [CrossRef]
- Adell, A. Brain NMDA receptors in schizophrenia and depression. Biomolecules 2020, 10, 947. [Google Scholar] [CrossRef]
- Shelley, A.M.; Silipo, G.; Javitt, D.C. Diminished responsiveness of ERPs in schizophrenic subjects to changes in auditory stimulation parameters: Implications for theories of cortical dysfunction. Schizophr. Res. 1999, 37, 65–79. [Google Scholar] [CrossRef]
- Stone, J.M.; Erlandsson, K.; Arstad, E.; Squassante, L.; Teneggi, V.; Bressan, R.A.; Krystal, J.H.; Ell, P.J.; Pilowsky, L.S. Relationship between ketamine-induced psychotic symptoms and NMDA receptor occupancy—A [123I]CNS-1261 SPET study. Psychopharmacology 2008, 197, 401–408. [Google Scholar] [CrossRef]
- Hakami, T.; Jones, N.C.; Tolmacheva, E.A.; Gaudias, J.; Chaumont, J.; Salzberg, M.; O’Brien, T.J.; Pinault, D. NMDA receptor hypofunction leads to generalized and persistent aberrant γ oscillations independent of hyperlocomotion and the state of consciousness. PLoS ONE 2009, 4, e6755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, K.H.; Williams, L.; Haig, A.; Gordon, E. “Gamma (40 Hz) phase synchronicity” and symptom dimensions in schizophrenia. Cogn. Neuropsychiatry 2003, 8, 57–71. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, E.M.; Timi, P.; Hong, L.E.; O’Donnell, P. Reverse translation of clinical electrophysiological biomarkers in behaving rodents under acute and chronic NMDA receptor antagonism. Neuropsychopharmacology 2015, 40, 719–727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdallah, C.G.; de Feyter, H.M.; Averill, L.A.; Jiang, L.; Averill, C.L.; Chowdhury, G.M.I.; Purohit, P.; de Graaf, R.A.; Esterlis, I.; Juchem, C.; et al. The effects of ketamine on prefrontal glutamate neurotransmission in healthy and depressed subjects. Neuropsychopharmacology 2018, 43, 2154–2160. [Google Scholar] [CrossRef]
- Callicott, J.H.; Bertolino, A.; Mattay, V.S.; Langheim, F.J.; Duyn, J.; Coppola, R.; Goldberg, T.E.; Weinberger, D.R. Physiological dysfunction of the dorso lateral prefrontal cortex in schizophrenia revisited. Cereb. Cortex. 2000, 10, 1078–1092. [Google Scholar] [CrossRef]
- Dienel, S.J.; Enwright, J.F.; Hoftman, G.D.; Lewis, D.A. Markers of glutamate and GABA neurotransmission in the prefrontal cortex of schizophrenia subjects: Disease effects differ across anatomical levels of resolution. Schizophr. Res. 2020, 217, 86–94. [Google Scholar] [CrossRef]
- Lathi, A.C.; Holcomb, H.H.; Medoff, D.R.; Tamminga, C.A. Ketamine activates psychosis and alters limbic blood flow in schizophrenia. Neuroreport 1995, 6, 869–872. [Google Scholar]
- Kondziella, D.; Brenner, E.; Eyjolfsson, E.M.; Markinhuhta, K.R.; Carlsson, M.L.; Sonnewald, U. Glial-neuronal interactions are impaired in the schizophrenia model of repeated MK801 exposure. Neuropsychopharmacology 2006, 31, 1880–1887. [Google Scholar] [CrossRef]
- Vollenweider, F.X.; Vontobel, P.; Éye, I.; Hell, D.; Leenders, K.L. Effects of (S)-ketamine on Striatal Dopamine: A [11 C]raclopride PET Study of a Model Psychosis in Humans. 2002. Available online: www.elsevier.com/locate/jpsychires (accessed on 16 February 2022).
- Martin, P.; CMHS. Systemic PCP treatment elevates brain extracellular 5-HT: A microdialysis study in awake rats. Neuroreport 1998, 9, 2985–2988. [Google Scholar] [CrossRef]
- Nelson, C.L.; Burk, J.A.; Bruno, J.P.; Sarter, M. Effects of acute and repeated systemic administration of ketamine on prefrontal acetylcholine release and sustained attention performance in rats. Psychopharmacology 2002, 161, 168–179. [Google Scholar] [CrossRef] [PubMed]
- Pal, D.; Hambrecht-Wiedbusch, V.S.; Silverstein, B.H.; Mashour, G.A. Electroencephalographic coherence and cortical acetylcholine during ketamine-induced unconsciousness. Br. J. Anaesth. 2015, 114, 979–989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seeman, P.; Ko, F.; Tallerico, T. Dopamine receptor contribution to the action of PCP, LSD and ketamine psychotomimetics. Mol. Psychiatry 2005, 10, 877–883. [Google Scholar] [CrossRef] [PubMed]
- Kapur, S.; Seeman, P. NMDA receptor antagonists ketamine and PCP have direct effects on the dopamine D2 and serotonin 5-HT2 receptors—Implications for models of schizophrenia. Mol. Psychiatry 2002, 7, 837–844. [Google Scholar] [CrossRef] [Green Version]
- Kinoshita, H.; Nishitani, N.; Nagai, Y.; Andoh, C.; Asaoka, N.; Kawai, H.; Shibui, N.; Nagayasu, K.; Shirakawa, H.; Nakagawa, T.; et al. Ketamine-Induced Prefontal Serotonin Realease Is Mediated by Cholinergic Neurons in the Penduculopontine Tegmental Nucleus. Int. J. Neuropsychopharmacol. 2018, 21, 305–310. [Google Scholar] [CrossRef] [Green Version]
- Frohlich, J.; van Horn, J.D. Reviewing the ketamine model for schizophrenia. J. Psychopharmacol. 2014, 28, 287–302. [Google Scholar] [CrossRef]
- Shibuya, H.; Mori, H.; Toru, M. Sigma Receptors in Schizophrenic Cerebral Cortices*. Neurochem. Res. 1992, 17, 983–990. [Google Scholar] [CrossRef]
- Weissman, A.D.; Casanova, M.F.; Kleinman, J.E.; London, E.D.; de Souza, E.B. Selective Loss of Cerebral Cortical Sigma, but Not PCP Binding Sites in Schizophrenia. Biol. Psychiatry 1991, 29, 41–54. [Google Scholar] [CrossRef]
- Pfeiffer, A.; Branti, V.; Herz, A.; Emrich, H.M. Psychotomimesis Mediated by K Opiate Receptors. Science 1986, 233, 774. [Google Scholar] [CrossRef]
- Tejeda, H.A.; Shippenberg, T.S.; Henriksson, R. The dynorphin/κ-opioid receptor system and its role in psychiatric disorders. Cell. Mol. Life Sci. 2012, 69, 857–896. [Google Scholar] [CrossRef]
- Marcel, L. De la Folie Causée par L’abus dês Boissons Alcooliques. Ph.D. Thesis, Imp. Rignoux, Paris, France, 1847. [Google Scholar]
- Soyka, M. 1990. Psychopathological characteristics in alcohol hallucinosis and paranoid schizophrenia. Acta Psychiatr. Scand. 1990, 81, 255–259. [Google Scholar] [CrossRef] [PubMed]
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, 4th ed.; American Psychiatric Association: Washington, DC, USA, 2000. [Google Scholar]
- Glass, I.B. Alcoholic Hallucinosis: A psychiatric enigma-1. The Development of an Idea. Br. J. Addict. 1989, 84, 29–41. [Google Scholar] [CrossRef] [PubMed]
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders; American Psychiatric Association: Washington, DC, USA, 1980. [Google Scholar]
- Kitabayashi, Y.; Narumoto, J.; Shibata, K.H.; Fukui, K. Neuropsychiatric Background of Alcohol Hallucinosis: A Spect Study. J. Neuropsychiatry 2007, 19, 85. [Google Scholar] [CrossRef] [PubMed]
- Soyka, M.; ZTDSTK. FDG-PET and IBZMSPECT suggest reduced thalamic activity but no dopaminergic dysfunction in chronic alcohol hallucinosis. J. Neuropsychiatry Clin. Neurosci. 2000, 12, 287–288. [Google Scholar] [CrossRef]
- Aliyev, N.A.; Aliyev, Z.N. Application of glycine in acute alcohol hallucinosis. Hum. Psychopharmacol. 2005, 20, 591–594. [Google Scholar] [CrossRef]
- Branchey, L.; Branchey, M.; Worner, T.M.; Zucker, D.; Shaw, S.; Lieber, C.S. Association between Amino Acid Alterations and Hallucinations in Alcoholic Patients. Biol. Psychiatry 1985, 20, 1167–1173. [Google Scholar] [CrossRef]
- Fadda, F.; Mosca, E.; Colombo, G.; Gessa, G.L. Effect of spontaneous ingestion of ethanol on brain dopamine metabolism. Life Sci. 1989, 44, 281–287. [Google Scholar] [CrossRef]
- Zuo, D.; Liu, Y.; Liu, Z.; Cui, J.; Zhou Liu, Y.; Li, Z.; Wu, Y. Alcohol aggravates ketamine-induced behavioral, morphological and neurochemical alterations in adolescent rats: The involvement of CREB-related pathways. Behav. Brain Res. 2018, 349, 80–90. [Google Scholar] [CrossRef]
- Liu, Y.; Lin, D.; Wu, B.; Zhou, W. Ketamine abuse potential and use disorder. Brain Res. Bull. 2016, 126, 68–73. [Google Scholar] [CrossRef] [Green Version]
- Tsai, G.; Gastfriend, D.R.; Coyle, J.T. The Glutamatergic Basis of Human Alcoholism. Am. J. Psychiatry 1995, 152, 332–340. [Google Scholar]
- Vengeliene, V.; Bilbao, A.; Molander, A.; Spanagel, R. Neuropharmacology of alcohol addiction. Br. J. Pharmacol. 2008, 154, 299–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, M.E.; Metzger, D.B. Alcohol withdrawal and brain injuries: Beyond classical mechanisms. Molecules 2010, 15, 4984–5011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Viviani, B.; Boraso, M.; Marchetti, N.; Marinovich, M. Perspectives on neuroinflammation and excitotoxicity: A neurotoxic conspiracy? NeuroToxicology 2014, 43, 10–20. [Google Scholar] [CrossRef]
- Miladinovic, T.; Nashed, M.G.; Singh, G. Overview of glutamatergic dysregulation in central pathologies. Biomolecules 2015, 5, 3112–3141. [Google Scholar] [CrossRef] [Green Version]
- Ménard, C.; Hodes, G.E.; Russo, S.J. Pathogenesis of depression: Insights from human and rodent studies. Neuroscience 2016, 321, 138–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beurel, E.; Toups, M.; Nemeroff, C.B. The Bidirectional Relationship of Depression and Inflammation: Double Trouble. Neuron 2020, 107, 234–256. [Google Scholar] [CrossRef]
- Réus, G.Z.; Fries, G.R.; Stertz, L.; Badawy, M.; Passos, I.C.; Barichello, T.; Kapczinski, F.; Quevedo, J. The role of inflammation and microglial activation in the pathophysiology of psychiatric disorders. Neuroscience 2015, 300, 141–154. [Google Scholar] [CrossRef] [PubMed]
- Volkow, N.D.; Michaelides, M.; Baler, R. The neuroscience of drug reward and addiction. Physiol. Rev. 2019, 99, 2115–2140. [Google Scholar] [CrossRef]
- Baik, J.H. Dopamine signaling in reward-related behaviors. Front. Neural Circuits 2013, 7, 152. [Google Scholar] [CrossRef] [Green Version]
- Lovinger, D.M. Excitotoxicity alcohol—Related Brain Damage. Alcohol ClinExp Res. 1993, 17, 19–27. [Google Scholar] [CrossRef]
- Schuckit, M.A.; Hesseibrock, V. Alcohol Dependence and Anxiety Disorders: What Is the Relationship? Focus 1994, 151, 1723–1734. [Google Scholar] [CrossRef]
- Yang, J.Y.; Xue, X.; Tian, H.; Wang, X.X.; Dong, Y.X.; Wang, F.; Zhao, Y.N.; Yao, X.C.; Cui, W.; Wu, C.F. Role of microglia in ethanol-induced neurodegenerative disease: Pathological and behavioral dysfunction at different developmental stages. Pharmacol. Ther. 2014, 144, 321–337. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Rabassa, M.; López, P.; Sánchez, R.; Hernández, C.; Rodríguez, C.; Rodríguez-santiago, R.E.; Orengo, J.C.; Green, V.; Yamamura, Y.; Rivera-Amill, V. Inflammatory biomarkers, microbiome, depression, and executive dysfunction in alcohol users. Int. J. Environ. Res. Public Health 2020, 17, 689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belém-Filho, I.J.A.; Ribera, P.C.; Nascimento, A.L.; Gomes, A.R.Q.; Lima, R.R.; Crespo-Lopez, M.E.; Monteiro, M.C.; Fontes-Júnior, E.A.; Lima, M.O.; Maia, C.S.F. Low doses of methylmercury intoxication solely or associated to ethanol binge drinking induce psychiatric-like disorders in adolescent female rats. Environ. Toxicol. Pharmacol. 2018, 60, 184–194. [Google Scholar] [CrossRef]
- Clayton, C.J.; Hicks, R.E. Ethanol, monoamines, and affect. J. Neural Transm. 1994, 98, 169–195. [Google Scholar] [CrossRef]
- Milio, C.; Hadfield, M.G. Ethanol Alters Monoamines in Mouse Brain Regions Specific. Brain Res. Bull. 1992, 29, 599–603. [Google Scholar] [CrossRef]
- Kaneyuki, T.; Morimasa, T.; Okada, H.; Shohmori, T. Acta Medica Okayama the effect of acute and repeated ethanol administration on monoamines and their metabolites in brain regions of rats. Acta Med. Okayama 1991, 45, 201–208. [Google Scholar]
- Costardi, J.V.V.; Nampo, R.A.T.; Silva, G.L.; FerreiraRibeiro, M.A.; Stella, H.J.; Stella, M.B.; Malheiros, S.V. A review on alcohol: From the central action mechanism to chemical dependency. Rev. Assoc. Med. Bras. 2015, 61, 381–387. [Google Scholar] [CrossRef] [Green Version]
- Dixon, M.L.; Thiruchselvam, R.; Todd, R.; Christoff, K. Emotion and the prefrontal cortex: An integrative review. Psychol. Bull. 2017, 143, 1033–1081. [Google Scholar] [CrossRef]
- Esperidião-Antonio, V.; Majeski-Colombo, M.; Toledo-Monteverde, D.; Moraes-Martins, G.; Fernandes, J.J.; Bauchiglioni de Assis, M.; Montenegro, S.; Siqueira-Batista, R. Neurobiology of emotions: An update. Int. Rev. Psychiatry 2017, 29, 293–307. [Google Scholar] [CrossRef]
- Li, Q.; Wu, H.R.; Fan, S.J.; Liu, D.X.; Jiang, H.; Zhang, Q.; Pan, F. The effects of sub-anesthetic ketamine plus ethanol behaviors and apoptosis in the prefrontal cortex and hippocampus of adolescent rats. Pharmacol. Biochem. Behav. 2019, 184, 1727–1742. [Google Scholar] [CrossRef] [PubMed]
- Fan, N.; Xu, K.; Ning, Y.; Rosenheck, R.; Wang, D.; Ke, X.; Ding, Y.; Sun, B.; Zhou, C.; Deng, X.; et al. Profiling the psychotic, depressive and anxiety symptoms in chronic ketamine users. Psychiatry Res. 2016, 237, 311–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Xu, Z.; Zhang, S.; Desrosiers, A.; Schottenfeld, R.S.; Chawarski, M.C. Profiles of psychiatric symptoms among amphetamine type stimulant and ketamine using inpatients in Wuhan, China. J. Psychiatr. Res. 2014, 53, 99–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silote, G.P.; de Oliveira, S.F.S.; Ribeiro, D.E.; Machado, M.S.; Andreatini, R.; Joca, S.R.L.; Beijamini, V. Ketamine effects on anxiety and fear-related behaviors: Current literature evidence and new findings. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2020, 100, 109878. [Google Scholar] [CrossRef] [PubMed]
- Perez-Caballero, L.; Torres-Sanchez, S.; Romero-López-Alberca, C.; González-Saiz, F.; Mico, J.A.; Berrocoso, E. Monoaminergic system and depression. Cell Tissue Res. 2019, 377, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Nicolaides, N.C.; Kyratzi, E.; Lamprokostopoulou, A.; Chrousos, G.P.; Charmandari, E. Stress, the stress system and the role of glucocorticoids. NeuroImmunoModulation 2014, 22, 6–19. [Google Scholar] [CrossRef]
- Ann Stephens, M.; Wand, G. Stress and the HPA Axis Role of Glucocorticoids in Alcohol Dependence. Alcohol Res. Curr. Rev. 2012, 4, 468–483. [Google Scholar]
- Allen, D.C.; Gonzales, S.W.; Grant, K.A. Effect of repeated abstinence on chronic ethanol self-administration in the rhesus monkey. Psychopharmacology 2018, 235, 109–120. [Google Scholar] [CrossRef]
- Rivier, C.; Imaki, T. Prolonged exposure to alcohol: Effect on CRF mRNA levels, and CRF-and stress-induced ACTH secretion in the rat. Brain Res. 1990, 520, 1–5. [Google Scholar] [CrossRef]
- Silvestre, J.S.; Pallarés, M.; Nadal, R.; Ferré, N. Opposite Effetc of Ethanol and Ketamine the Elevated plus-maze test Wistar rats undergoing a chronic oral voluntary consumption procedure. J. Psychopharmacol. 2002, 16, 305–312. [Google Scholar] [CrossRef]
- Sampaio, T.B.; de Oliveira, L.F.; Constantino, L.C.; Costa, A.P.; Poluceno, G.G.; Martins, W.C.; Dal-Cim, T.; de Oliveira, K.A.; Ludka, F.K.; Prediger, R.D.; et al. Long-Term Neurobehavioral Consequences of a Single Ketamine Neonatal Exposure in Rats: Effects on Cellular Viability and Glutamate Transport in Frontal Cortex and Hippocampus. Neurotox. Res. 2018, 34, 649–659. [Google Scholar] [CrossRef] [PubMed]
- Machado-Ferraro, K.M.; Soriano-de-Mello, D.S.; de Moura, I.P.; da Silveira, C.C.S.M.; de Farias, E.C.F.; Maia, M.L.F.; de Sales, S.C.D.; Carvalho, A.E.V.; Magno, I.M.N.; Fontes-Júnior, E.A.; et al. Long-lasting neurocognitive disorders: A case report of previously undescribed adverse effects after ketamine sedation and analgesia in a pediatric patient. Ann. Transl. Med. 2022, 10, 113. [Google Scholar] [CrossRef] [PubMed]
- Lisman, J.; Cooper, K.; Sehgal, M.; Silva, A.J. Memory formation depends on both synapse-specific modifications of synaptic strength and cell-specific increases in excitability. Nat. Neurosci. 2018, 21, 309–3014. [Google Scholar] [CrossRef] [Green Version]
- Asok, A.; Leroy, F.; Rayman, J.B.; Kandel, E.R. Molecular Mechanisms of the Memory Trace. Trends Neurosci. 2019, 42, 14–22. [Google Scholar] [CrossRef] [PubMed]
- Colucci-D’amato, L.; Speranza, L.; Volpicelli, F. Neurotrophic factor bdnf, physiological functions and therapeutic potential in depression, neurodegeneration and brain cancer. Int. J. Mol. Sci. 2020, 21, 1–29. [Google Scholar]
- Itoh, N.; Enomoto, A.; Nagai, T.; Takahashi, M.; Yamada, K. Molecular mechanism linking BDNF/TrkB signaling with the NMDA receptor in memory: The role of Girdin in the CNS. Rev. Neurosci. 2016, 27, 481–490. [Google Scholar] [CrossRef]
- Ortega-Martínez, S. A new perspective on the role of the CREB family of transcription factors in memory consolidation via adult hippocampal neurogenesis. Front. Mol. Neurosci. 2015, 8, 46. [Google Scholar] [CrossRef] [Green Version]
- Narendran, R.; Gordon Frankle, W.; Keefe, R.; Gil, R.; Martinez, D.; Slifstein, M.; Kegeles, L.S.; Talbot, P.S.; Huang, Y.; Hwang, D.R.; et al. Altered Prefrontal Dopaminergic Function in Chronic Recreational Ketamine Users. Am. J. Psychiatry 2005, 162, 2352–2359. Available online: http://ajp.psychiatryonline.org (accessed on 16 February 2022). [CrossRef] [Green Version]
- Brust, J.C.M. Ethanol and cognition: Indirect effects, neurotoxicity and neuroprotection: A review. Int. J. Environ. Res. Public Health 2010, 7, 1540–1557. [Google Scholar] [CrossRef]
- Pfisterer, U.; Khodosevich, K. Neuronal survival in the brain: Neuron type-specific mechanisms. Cell Death Dis. 2017, 8, e2643. [Google Scholar] [CrossRef]
- Reichardt, L.F. Neurotrophin-regulated signalling pathways. Philos. Trans. R. Soc. B Biol. Sci. 2006, 361, 1545–1564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Wang, Y.; Liu, X.; Dagda, R.K.; Zhang, Y. How AMPK and PKA interplay to regulate mitochondrial function and survival in models of ischemia and diabetes. Oxid. Med. Cell. Longev. 2017, 2017, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamat, P.K.; Kalani, A.; Rai, S.; Swarnkar, S.; Tota, S.; Nath, C.; Tyagi, N. Mechanism of Oxidative Stress and Synapse Dysfunction in the Pathogenesis of Alzheimer’s Disease: Understanding the Therapeutics Strategies. Mol. Neurobiol. 2016, 53, 648–661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, R.; Reddy, P.H. Role of Glutamate and NMDA Receptors in Alzheimer’s Disease. J. Alzheimer’s Dis. 2017, 57, 1041–1048. [Google Scholar] [CrossRef] [Green Version]
- Zádori, D.; Veres, G.; Szalárdy, L.; Klivényi, P.; Vécsei, L. Alzheimer’s Disease: Recent Concepts on the Relation of Mitochondrial Disturbances, Excitotoxicity, Neuroinflammation, and Kynurenines. J. Alzheimer’s Dis. 2018, 62, 523–547. [Google Scholar] [CrossRef] [Green Version]
- Aleksandrova, L.R.; Wang, Y.T.; Phillips, A.G. Hydroxynorketamine: Implications for the NMDA Receptor Hypothesis of Ketamine’s Antidepressant Action. Chronic Stress 2017, 1, 2470547017743511. [Google Scholar] [CrossRef] [Green Version]
- Hashimoto, K. Molecular mechanisms of the rapid-acting and long-lasting antidepressant actions of (R)-ketamine. Biochem. Pharmacol. 2020, 177, 113935. [Google Scholar] [CrossRef]
- Brum, G.F.; Rosa, H.Z.; Rossato, D.R.; Rosa, J.L.O.; Metz, V.G.; Milanesi, L.H.; Burger, M.E. Binge and Subchronic Exposure to Ketamine Promote Memory Impairments and Damages in the Hippocampus and Peripheral Tissues in Rats: Gallic Acid Protective Effects. Neurotox. Res. 2020, 38, 274–286. [Google Scholar] [CrossRef]
- Kunitoh, S.; Tanaka, T.; Imaoka, S.; Funaet, Y.; Monna, Y. Elsevier Science Ltd Medical Council on Alcoholism contribution of cytochrome p450s to meos (microsomal ethanol-oxidizing system): A specific and sensitive assay of meos activity by hplc with fluorescence labeling. Pergamon Alcohol Alcohol. 1993, 28, 63–68. [Google Scholar] [CrossRef]
- Kamp, J.; Jonkman, K.; van Velzen, M.; Aarts, L.; Niesters, M.; Dahan, A.; Olofsen, E. Pharmacokinetics of ketamine and its major metabolites norketamine, hydroxynorketamine, and dehydronorketamine: A model-based analysis. Br. J. Anaesth. 2020, 125, 750–761. [Google Scholar] [CrossRef]
- Mladěnka, P.; Applová, L.; Patočka, J.; Costa, V.M.; Remiao, F.; Pourová, J.; Mladěnka, A.; Karlíčková, J.; Jahodář, L.; Vopršalová, M.; et al. Comprehensive review of cardiovascular toxicity of drugs and related agents. Med. Res. Rev. 2018, 38, 1332–1403. [Google Scholar] [CrossRef] [PubMed]
- Suhail, M. Na+, K+-ATPase: Ubiquitous Multifunctional Transmembrane Protein and its Relevance to Various Pathophysiological Conditions. J. Clin. Med. Res. 2010, 2, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhatia, D.; Capili, A.; Choi, M.E. Mitochondrial dysfunction in kidney injury, inflammation, and disease: Potential therapeutic approaches. Kidney Res. Clin. Pract. 2020, 39, 244–258. [Google Scholar] [CrossRef] [PubMed]
- Patergnani, S.; Bouhamida, E.; Leo, S.; Pinton, P.; Rimessi, A. Mitochondrial oxidative stress and “mito-inflammation”: Actors in the diseases. Biomedicines 2021, 9, 216. [Google Scholar] [CrossRef]
- Strong, C.E.; Kabbaj, M. Neural Mechanisms Underlying the Rewarding and Therapeutic Effects of Ketamine as a Treatment for Alcohol Use Disorder. Front. Behav. Neurosci. 2020, 14, 593860. [Google Scholar] [CrossRef]
- Olsen, R.W.; Liang, J. Role of GABAA receptors in alcohol use disorders suggested by chronic intermittent ethanol (CIE) rodent model Tim Bliss. Mol. Brain 2017, 10, 1–20. [Google Scholar] [CrossRef]
- Nguyen, D.; Alavi, M.V.; Kim, K.Y.; Kang, T.; Scott, R.T.; Noh, Y.H.; Lindsey, J.D.; Wissinger, B.; Ellisman, M.H.; Weinreb, R.N.; et al. A new vicious cycle involving glutamate excitotoxicity, oxidative stress and mitochondrial dynamics. Cell Death Dis. 2011, 2, e240. [Google Scholar] [CrossRef]
- Tapia-Rojas, C.; Pérez, M.J.; Jara, C.; Vergara, E.H.; Quintanilla, R.A. Ethanol Consumption Affects Neuronal Function: Role of the Mitochondria. Mitochondrial Dis. 2017. Available online: https://www.intechopen.com/chapters/58876 (accessed on 26 June 2022).
- Oliveira, A.C.A.; Pereira, M.C.S.; Santana, L.N.D.S.; Fernandes, R.M.; Teixeira, F.B.; Oliveira, G.B.; Fernandes, L.M.; Fontes-Júnior, E.A.; Prediger, R.D.; Crespo-López, M.E.; et al. Chronic ethanol exposure during adolescence through early adulthood in female rats induces emotional and memory deficits associated with morphological and molecular alterations in hippocampus. J. Psychopharmacol. 2015, 29, 712–724. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Drug(s) | Evaluation Condition | Study Information | Organ(s) or Body(s) System(s) | Main Effects Described | Possible Mechanisms | Reference |
|---|---|---|---|---|---|---|
| Ketamine | Under drug effects | Literature review | Cardiovascular and urinary | Acute: hypertension and tachycardia. Chronic: risk of hemorrhagic/ulcerative cystitis and obstructive nephropathy | Not investigated | [46] |
| Ketamine | Under drug effects | Pattern of use: chronic Type: clinical study Finality of use: recreational Dose and use frequency: not informed | Liver, biliary, urinary, and cardiorespiratory system | Under drug: cholestasis and biliary dilatation; hypotension; tachycardia and tachypnoea; bilateral hydronephrosis; acute renal failure; and increase in hepatic transaminases and alkaline phosphatase | Not investigated | [51] |
| Ketamine | Under drug effects | Pattern of use: acute Type: pre-clinical study Finality of use: not informed Dose and use frequency: 10, 50, 100, and 200 mM for 1, 6, or 24 h | Liver (in vitro) | Apoptosis and decreased cell viability | DNA fragmentation, mitochondrial membrane potential and adenosine triphosphate levels decrease; cytosolic cytochrome and caspase-9, -3, and -6 activities increase | [55] |
| Ketamine | Under drug effects | Pattern of use: acute Type: pre-clinical study Finality of use: not informed Dose and use frequency: a single administration of 180 mmol/L | Hepatobiliary | Strength of phasic gallbladder contraction decrease | NMDA blockade? | [56] |
| Ketamine | Withdrawal and drug effects | Pattern of use: chronic Type: clinical study Finality of use: recreational Dose and use frequency: not informed | Urinary system | Intractable dysuria, painful urination, and gross hematuria | Not investigated | [62] |
| Ketamine | Under drug effects | Pattern of use: chronic Type: clinical study Finality of use: recreational Dose and use frequency: frequency not informed; dose 100–200 mg (i.m.)/ intranasal route dose not informed | Cardiovascular system | Chest pain, palpitations, tachycardia, and hypertension | Not investigated | [60] |
| Ketamine | Under drug and withdrawal effects | Literature review | Cardiorespiratory system | Hypertension, tachycardia, and palpitations; respiratory toxicity, including depression and apnea | Authors propose that cardiovascular toxicity can result from reflex sympathetic activation | [64] |
| Ketamine | Under drug effects | Pattern of use: acute Type: pre-clinical study Finality of use: not informed Dose and use frequency: single dose of 100 mg (i.p.) | Cardiovascular system | Cardiotoxicity | Increase of microRNA-208a, accompanied by increased inflammation and oxidative stress, suppression of CHD9 and Notch1, and induction of p65 protein expression in vitro model | [66] |
| Ketamine | Not informed | Pattern of use: not specified Type: clinical study Finality of use: recreational Dose and use frequency: not informed | Hepatobiliary | Dilated common bile ducts and increase of alkaline phosphatase | Not investigated | [48] |
| Ketamine | Not informed | Literature review | Urinary system | Severe dysuria, painful hematuria, nocturia, pelvic pain, epithelial inflammation similar to chronic interstitial cystitis, and unilateral or bilateral hydronephrosis | Not investigated | [73] |
| Ketamine | Under drug effects | Pattern of use: chronic misuse Type: clinical study Finality of use: recreational Dose and use frequency: dose not informed, daily consume | Urinary system | Dysuria, painful hematuria, and post-voiding pain | Not investigated | [74] |
| Ketamine | Withdrawal effects | Pattern of use: chronic Type: pre-clinical study Finality of use: non-therapeutical Dose and use frequency: 100 or 300 mg/kg/day for 4 weeks | Urinary system | Bladder inflammation, interstitial nephritis, and increased lymphocytes in bladder submucosal layer | Voiding dysfunction by neurogenic damage and dysregulation of purinergic transmission | [75] |
| Ketamine | Under drug effects | Pattern of use: acute Type: pre-clinical study Finality of use: not declared Dose and use frequency: 1000 µM/1x | Cardiovascular system (in vitro) | Cerebral vasoconstriction | Blockade of Ca2+ activated K+ channels | [77] |
| Ketamine | Under drug effects | Pattern of use: acute Type: pre-clinical study Finality of use: not declared Dose and use frequency: 1, 10, 100, 1000, and 10,000 µM/1x | Cardiovascular system (in vitro) | Not specified | Inhibition of sarcolemmal adenosine triphosphate-sensitive potassium (KATP) channels mediated by SUR subunit with specificity for cardiovascular KATP channels | [78] |
| Ketamine | Under drug effects | Pattern of use: chronic Type: pre-clinical study Finality of use: abuse Dose and use frequency: 100 mg/kg/day for 4, 8, and 16 weeks | Urinary system | Increased urination frequency and decreased bladder capacity after 8 weeks | Increased noncholinergic contractions and P2X1 receptor expression in bladder | [80] |
| Ketamine | Withdrawal effects | Pattern of use: chronic Type: pre-clinical study Finality of use: abuse Dose and use frequency: 30 mg/kg/day for 6 months | Urinary system | Relatively thinner bladder walls and infiltration of mononuclear cells, similar to clinical situation of interstitial cystitis | Decreased cholinergic neurons in urinary bladder by NMDA receptor overexpression | [81] |
| Ketamine plus polydrugs | Under drug and withdrawal effects | Pattern of use: chronic Type: clinical study Finality of use: recreational Dose and use frequency: not informed | Liver, biliary, and urinary system | Cystitis and urinary dysfunction; impaired liver function and biliary tree dilatation | Not investigated | [49] |
| Ketamine plus polydrug | Under drug effects | Pattern of use: chronic Type: clinical study Finality of use: recreational Dose and use frequency: not informed | Liver, biliary, cardiorespiratory, and urinary system | Common bile duct dilatation; increase in serum levels of hepatic transaminases; tachycardia; tachypnea and impaired respiratory function; overt hematuria associated with cramping abdominal pain, increased urinary frequency, dysuria, moderate bilateral hydronephrosis, and cystitis | Not investigated | [53] |
| Ketamine plus other drugs | Pattern of use: chronic Type: clinical study Finality of use: recreational Dose and use frequency: not informed | Liver and biliary system | Common bile duct dilatation, microscopic bile duct injury, and liver fibrosis | Not investigated | [54] | |
| Ketamine plus other drugs | Under drug effects | Pattern of use: not informed Type: clinical study Finality of use: recreational Dose and use frequency: not informed | Cardiovascular and urinary system | Palpitations and chest pain, renal colic, urine leak, hematuria, and stranguria | Not investigated | [36] |
| Ketamine plus other drugs | Under drug and withdrawal effects | Pattern of use: chronic misuse Type: clinical study Finality of use: recreational Dose and use frequency: not informed | Respiratory system | Authors report the effect of ketamine in producing impairment of pharyngeal and laryngeal reflexes, diaphragm rigidity, and transient respiratory depression | Not investigated | [44] |
| Ketamine plus other drugs | Not specified | Pattern of use: chronic misuse Type: clinical study Finality of use: recreational Dose and use frequency: dose 0.125–5 g, frequency 3–7 days/week | Urinary system | Pain in lower abdomen; burning during urination and urination frequency increase; incontinence and presence of blood in urine | Not investigated | [38] |
| Ketamine plus ethanol | Under drug and withdrawal effects | Pattern of use: chronic Type: clinical study Finality of use: recreational Dose and use frequency: not informed | Liver and biliary system | Impaired liver function and possible damage to bile ducts | Not investigated | [50] |
| Ketamine plus ethanol | Withdrawal effects | Literature review | Liver, biliary, urinary, and cardiovascular system | Rats: dysuria, increased collagen fibers in hepatic parenchyma, and tachycardia Humans: signs and symptoms of cystitis, tachycardia, dysuria, and increase of liver fibrosis | Not investigated | [61] |
| Ketamine plus ethanol | Withdrawal and drug effects | Pattern of use: chronic Type: clinical study Finality of use: recreational Dose and use frequency: dose not informed; occasional alcohol intake and daily ketamine inhalation | Liver and biliary system | Increase in alkaline phosphatase and gamma-glutamyl transpeptidase; concentric periductal fibrosis around bile ducts of varying sizes, consistent with primary or secondary sclerosing cholangitis; interlobular bile ducts with thickened basement membranes, mild lymphocytic infiltrates, and mild ductular reaction | Not investigated | [63] |
| Ethanol | Under drug effects | Literature review | Liver | Liver fibrosis and cirrhosis, as a well as synergic interaction to infectious agents to induce liver injury | Production of hepatotoxins, activation of redox-sensitive transcription factors (i.e., nuclear factor kappa B (NF-κB), neutrophils, and other immune cells’ recruitment), circulating pro-inflammatory cytokines levels increase, and intestinal dysbiosis with hepatic repercussions | [57] |
| Ethanol | Under drug effects | Literature review | Liver | Liver fibrosis and cirrhosis, as a well as synergic interaction to infectious agents to induce liver injury | Production of hepatotoxins, induction of oxidative stress, activation of immune cells and circulating pro-inflammatory cytokines levels increase, deviation of lipid metabolism pathways, alteration of liver tissue remodeling factors activity, and intestinal dysbiosis with hepatic repercussions | [58] |
| Ethanol | Under drug effects | Literature review | Liver | Liver fibrosis | Acetaldehyde leads to generation of reactive oxygen species and activation of AP-1 and NF-kB transcription factors, resulting in pro-inflammatory cytokines’ release and increased survival and remodeling activity of stellate liver cells | [59] |
| Ethanol | Under drug effects | Literature review | Cardiovascular system | Alcoholic cardiomyopathy and holiday heart syndrome (chronic); arrhythmias (chronic and acute); and acute atrial fibrillation (acute) | Cardiovascular toxicity is a result of dysregulation in calcium and sodium conductance; beyond harmful effects on cardiac contractile function, in general after acute ethanol exposure | [67] |
| Ethanol | Under drug effects | Literature review | Respiratory system | Chronic consumption increases the risk of developing lung diseases, such as acute lung injury, pneumonia, and pulmonary fibrosis | Glutathione levels decrease and TGF-1 expression increase; changes in tissue remodeling and extracellular matrix in lung tissue; activation of matrix metalloproteinases (MMPs) in lungs, particularly MMP-2 and MMP-9; and increase in reactive oxygen species generation | [71] |
| Ethanol | Not specified | Literature review | Urinary system | Lower urinary tract symptoms | Urothelium vulnerability to alcohol and its metabolites, increased permeability, and lower urinary tract symptoms | [82] |
| Ethanol | Withdrawal effects | Meta-analysis of prospective chronic studies | Urinary system | Kidney damage is proportional to time of consumption and amount of alcohol, and there is a high prevalence of proteinuria in chronic users | Not investigated | [83] |
| Ethanol | Not informed | Systematic review and meta-analysis | Urinary system | Statistically significant inverse relationship of CKD in male adults with high alcohol consumption; and no significant association between high alcohol consumption and risk of developing proteinuria or end-stage renal disease | Not investigated | [84] |
| Ethanol | Withdrawal effects | Pattern of use: chronic Type: pre-clinical study Finality of use: abuse Dose and use frequency: ethanol 20% (v/v) daily for 24 weeks | Urinary system | Increase of blood pressure, uric acid, and albumin; and kidney changes | Activation of renin–angiotensin system (RAS), oxidative stress and increased sympathetic nerve activity | [85] |
| Ethanol | Under drug effects | Pattern of use: chronic Type: pre-clinical study Finality of use: abuse Dose and use frequency: ethanol 10% (v/v) daily for 12 weeks | Urinary system | Reduction in creatinine clearance and urea in urine; and urea serum increase | Not investigated | [86] |
| Ethanol | Under drug effects | Pattern of use: chronic Type: pre-clinical study Finality of use: abuse Dose and use frequency: ethanol 20% (v/v) daily for 12 weeks | Cardiovascular system | Blood pressure increase | Increased aortic inflammation; elevated angiotensin II levels; induction of NADPH oxidase, leading to endothelial injury; CuZn–SOD depletion; downregulation of endothelial NO-generating system; and impaired vascular vasodilatation in rats | [87] |
| Ethanol | Withdrawal effects | Pattern of use: chronic Type: pre-clinical study Finality of use: abuse Dose and use frequency: ethanol 20% (v/v) daily for 6 weeks | Urinary system | Increase in nitrite and nitrate levels and tubular necrosis in proximal tubules that is consistent in humans (acute tubular necrosis) | Reactive oxidative species generation and infiltration of polymorphonuclear cells | [88] |
| Ethanol | Under drug effects | Pattern of use: chronic Type: pre-clinical study Finality of use: abuse Dose and use frequency: ethanol 2 g/kg daily for 10 and 30 weeks | Urinary system | Kidney weight increase and new protein band with high molecular weight after 30 weeks of ethanol treatment. | Reduction of reduced glutathione/oxidized glutathione, kidney alcohol dehydrogenase activities increase, and distinct effects under antioxidant enzymes after 10 and 30 weeks | [89] |
| Ethanol | Under drug effects | Pattern of use: chronic Type: pre-clinical study Finality of use: abuse Dose and use frequency: ethanol 30% (v/v) daily for 2 months | Cardiovascular and urinary system | Heart size augment and reduction of glomerular filtration rate | Increase of catalase activity and decrease of lipid peroxidation in heart; opposite effects in kidney | [91] |
| Drug(s) | Evaluation Condition | Study Information | CNS Function/ Disorder | Main Effects Described | Possible Mechanisms | Reference |
|---|---|---|---|---|---|---|
| Ketamine | Under drug and withdrawal effects | Pattern of use: acute Type: clinical study Finality of use: abuse Dose and use frequency: 2 mg/kg daily | Psychosis | Impaired on semantic memory tasks; higher levels of dissociation and schizotypal symptoms | Not investigated | [24] |
| Ketamine | Withdrawal effects | Pattern of use: chronic misuse Type: clinical study Finality of use: abuse Dose and use frequency: dose not informed; frequency— more than 4 times a week. | Cognition and depression | Short- and long-term memory impairments, vulnerability to dangers behavior, and depressive symptoms | Not investigated | [40] |
| Ketamine | Withdrawal effects | Pattern of use: chronic Type: clinical study Finality of use: abuse Dose and use frequency: dose not informed; frequent use | Cognition and depression. | Frequent use: reduced psychological well-being and broad range of cognitive impairments. Infrequent use: increased symptoms of thought disorder, delusions, and dissociation, but not cognitive impairment | Not investigated | [39] |
| Ketamine | Under drug effects | Pattern of use: acute Type: clinical study Finality of use: not specified Dose and use frequency: single administration of 0.4 mg/kg and 0.8 mg/kg. | Psychosis | Increased schizophrenic and dissociative symptoms after ketamine use; psychotomimetic effects of ketamine are detectable on clinical scales | Not investigated | [103] |
| Ketamine | Under drug effects | Pattern of use: acute Type: clinical study Finality of use: clinical Dose and use frequency: 0.5 mg/kg infused over 40 min | Emotionality | Subacute increase of prefrontal connectivity associated to antidepressant response | Ketamine seems activate prefrontal glutamate neurotransmission, contributing to transient psychotomimetic effects and delayed and sustained antidepressant effects | [114] |
| Ketamine | Under drug effects | Pattern of use: acute Type: clinical study Finality of use: subanesthetic Dose and use frequency: 0.3 mg/kg for 2 weeks | Psychosis | Increase in schizotypal symptoms | Flow activation on anterior cingulate and prefrontal cortex; decreased flow activation on visual cortex and hippocampus; and abnormal glutamatergic transmission involved on pathophysiology of psychotic symptoms | [117] |
| Ketamine | Not specified | Literature review | Cognition and psychosis | Cognitive impairment; psychotic and negative symptoms | Reduction of NMDA receptors related to negative symptoms | [108] |
| Ketamine | Under drug effects | Pattern of use: acute Type: clinical study Finality of use: non-therapeutical Dose and use frequency: 15 mg/kg for 5 min to induce psychopathological effects, followed by dose of 0.014 mg/kg/min for 90 min | Emotionality and psychosis | Affect changes, illusions, hallucinations, and ego dissolution | Inhibition of NMDA receptors to increase in DA levels in striatum, inducing euphoria- and mania-related features | [119] |
| Ketamine | Under drug and withdrawal effects | Pattern of use: acute Type: pre-clinical study Finality of use: not informed Dose and use frequency: 2, 10, and 20 mg/kg (i.p(i.p.), or once, or twice; or 25 mg/kg for 7 days | Cognition | Highest ketamine dose elevates levels of errors of omission in attentional tasks, whereas disrupted attentional performance during pre-treatment period | Increase in cortical acetylcholine release | [121] |
| Ketamine | Under drug effects | Pattern of use: acute Type: pre-clinical study Finality of use: subanesthetic Dose and use frequency: single dose 150 mg/kg | Schizotypal behaviors | Suppression of high-frequency EEG activity and disruption of cortical coherence | Increased concentrations of cortical ACh in active arousal systems in setting of unconscious state | [122] |
| Ketamine | Under drug effects | Pattern of use: not informed Type: pre-clinical study Finality of use: not informed Dose and use frequency: a single administration of 0.1 nM to 10,000 nM | Psychosis (in vitro) | Biological events related to schizophrenia | Ketamine binds to D2 receptors, increasing dopamine neuro-availability; and antagonism of NMDA receptors induces schizotypy | [123] |
| Ketamine | Under drug effects | Pattern of use: acute Type: pre-clinical study Finality of use: not specified Dose and use frequency: a single administration of 0.01 a 100 µM | Psychosis (in vitro) | Partial agonism on D2, and 5-HT2 (on a smaller scale) receptors | Ketamine binds to D2 receptors, increasing the neuro-availability of dopamine; small affinity to 5-HT2 receptors; and other mechanisms induce non-selective multi-system neurochemical perturbation | [124] |
| Ketamine | Under drug effects | Pattern of use: acute Type: pre-clinical study Finality of use: therapeutical Dose and use frequency: 30 mg/kg and local administration of 0.1 mM | Emotionality | Increased serotonin release in medial prefrontal cortex | Increase of serotonin release on medial prefrontal cortex through cholinergic neurons projected pedunculopontine tegmental nucleus to dorsal raphe nucleus | [125] |
| Ketamine | Not specified | Literature review | Psychosis | Schizotypal symptoms | Glutamatergic hypoactivity through antagonism of NMDA receptors may be associated with schizophrenia manifestation | [126] |
| Ketamine | Under drug effects | Literature review | Cognition | Abusive use of ketamine induces cognitive damage | NMDA receptors blockade on gamma-aminobutyric acid (GABA) neurons on thalamic reticular nucleus leads to disinhibition of dopaminergic neurons and increased dopamine release | [142] |
| Ketamine | Withdrawal effects | Pattern of use: acute Type: pre-clinical study Finality of use: abuse Dose and use frequency: 10 mg/kg for 3 days | Cognition and emotionality | Memory impairment, anxiogenic and depressive behavior | Oxidative stress in hippocampus | [25] |
| Ketamine | Withdrawal effects | Pattern of use: chronic Type: clinical study Finality of use: abuse Dose and use frequency: 3.4 g/day; frequency varied between 1 time per day, more than 4 times per week, and less than 4 times per week | Cognition and emotionality | Depressive, anxiogenic, and psychotic symptoms | Not investigated | [165] |
| Ketamine | Under drug and withdrawal effects | Pattern of use: acute Type: pre-clinical study Finality of use: anesthetic and subanesthetic Dose and use frequency: 2 mg/kg daily for 2 days | Anxiety | Doses employed have no effects on anxiety- or panic-related behaviors | Not investigated | [167] |
| Ketamine | Withdrawal effects | Pattern of use: chronic Type: clinical study Finality of use: abuse Dose and use frequency: not informed | Dissociative symptoms | Greater affective symptoms and perceptual disturbances | Not investigated | [93] |
| Ketamine | Withdrawal effects | Pattern of use: chronic Type: clinical study Finality of use: abuse Dose and use frequency: not informed | Cognition, emotionality and schizotypal symptoms | Semantic memory deficiencies decrease but are reversible with marked reduction in use; impairment in episodic memory and attentional functioning; schizotypal symptoms and perceptual distortions may persist after discontinuing ketamine use | Not investigated | [104] |
| Ketamine | Withdrawal effects | Pattern of use: acute Type: clinical study (case report) Finality of use: anesthetic Dose and use frequency 650 mg/kg to 15 mg/kg for 7 days | General behavioral alterations | Fever, sleep inversion, restlessness, and drooling (24 and 48 h after withdrawal); 17 days later, behavior and cognitive impairment, including aggression and language deficits, in addition to affecting motor skills | Not investigated | [175] |
| Ketamine | Withdrawal effects | Pattern of use: acute Type: pre-clinical study Finality of use: anesthetic Dose and use frequency: single dose of 20 mg/kg subcutaneously | Cognition | Impairment in short-term recognition memory | Decreased cellular viability in hippocampus, and long-term increase in hippocampal glutamate uptake. Prevention of vulnerability to glutamate-induced neurotoxicity in frontal cortex of adult rats | [174] |
| Ketamine | Under drug effects | Pattern of use: chronic Type: clinical study Finality of use: abuse Dose and use frequency: at least an average use of one vial per week or more over the last 3 months | Memory | Working memory not affected | Dorsolateral prefrontal cortex D1 receptor upregulation; binding potential upregulation significantly correlated to number of vials of ketamine used per week. | [181] |
| Ethanol | Withdrawal effects | Pattern of use: chronic Type: clinical study Finality of use: abuse Dose and use frequency: not informed | Psychosis | Hallucinations of schizophrenic origin and hallucinations from alcohol are very similar | Not investigated | [132] |
| Ethanol | Withdrawal effects | Pattern of use: chronic Type: clinical study Finality of use: abuse Dose and use frequency: 200 g/day for 30 years | General behavioral alterations | Finger tremor, mildly altered liver enzymes, and decreased regional cerebral blood flow | Not investigated | [136] |
| Ethanol | Withdrawal effects | Pattern of use: chronic Type: clinical study Finality of use: abuse Dose and use frequency: 400 g/day of alcohol for a long time (undefined) | Psychotic state | Hallucinations | Damage to thalamic structures associated with manifestation of hallucinations | [137] |
| Ethanol | Withdrawal effects | Pattern of use: chronic Type: clinical study Finality of use: abuse Dose and use frequency: unreported dose and consume frequency at least 10 years | Psychotic state | Verbal hallucinations, hallucinatory delusions, and affective frustration (mainly alarm and fear) in a state of clear consciousness | Acute alcoholic hallucinosis is linked to changes in excitatory and inhibitory transmission in the brain | [138] |
| Ethanol | Withdrawal effects | Pattern of use: acute Type: clinical study Finality of use: abuse Dose and use frequency: unreported dose; ex-user patients undergoing treatment | Psychotic state | Chronic alcohol consumption induces hallucinogenic events, and it alters plasmatic concentration of tyrosine, tryptophan, and phenylalanine | Amino acid imbalances result in decreased brain serotonin levels and increased brain dopamine, inducing hallucinatory experiences | [139] |
| Ethanol | Under drug or withdrawal effects | Pattern of use: chronic Type: pre-clinical study Finality of use: abuse Dose and use frequency: 2 g/kg/day or 10%; daily frequency | Rewarding system | Broader effects on dopaminergic system in voluntary ethanol intake model | Increased dopamine degradation, influencing the reward system | [140] |
| Ethanol | Under drug and withdrawal effects | Literature review | General behavioral alterations | Withdrawal symptoms, delirium tremens, Wernicke–Korsakoff syndrome, and fetal alcohol syndrome | Acute effects of ethanol disrupt glutamatergic neurotransmission by NMDA blockade. Prolonged inhibition of NMDA receptor results in development of supersensitivity. Acute absence of ethanol increases postsynaptic neurons activity (i.e., noradrenergic system and glutamate-induced excitotoxicity) | [143] |
| Ethanol | Under drug effects | Literature review | Reward system | Binge drinking induces decreased reward neurocircuitry function and recruitment of anti-reward/stress mechanisms | Ethanol interacts with NMDA, GABAA, glycine, 5-HT, and nicotinic receptors, as well as L-type Ca2+ channels and G protein-activated internal rectifier K+ channels of neurotransmitters/neuropeptides leading to typical acute behavioral effects of alcohol | [144] |
| Ethanol | Withdrawal effects | Literature review | General behavioral alterations | Anxiety, depression, tremors, rigidity, hyperactivity, convulsion, coma, and even death | Intense generation of reactive oxygen species and activation of stress-responding protein kinases | [145] |
| Ethanol | Under drug and withdrawal effects | Literature review | CNS adaptations | Brain excitotoxicity | Hyperexcitability after alcohol withdrawal may contribute to excitotoxicity. Alterations in function and/or expression of glutamate, GABA, and voltage-activated calcium channels contribute to hyperexcitability | [153] |
| Ethanol | Under drug and withdrawal effects | Literature review | General behavioral alterations | Depressive episodes, severe anxiety, insomnia, suicide, and abuse of other drugs | Not investigated | [154] |
| Ethanol | Under drug and withdrawal effects | Literature review | CNS impairments | Acute: excitement, ataxia, and lethargy. Chronic: cognitive, emotional, and motor disturbances. Withdrawal: autonomic hyperactivity, tremor, anxiety, restlessness, seizures, hallucinations, and delirium | Ethanol stimulates microglia, inducing neuroinflammation that triggers neuropathogenic processes | [155] |
| Ethanol | Withdrawal effects | Pattern of use: chronic Type: clinical study Finality of use: abuse Dose and use frequency: unreported dose and frequency | Cognition and emotionality | Depressive disorders, anxiety, sleep disturbance, impairment of cognitive ability, and increased plasma macrophage-derived chemokine | Macrophage-derived chemokine is associated with alcoholism, phobia, and interpersonal sensitivity | [156] |
| Ethanol | Withdrawal effects | Pattern of use: chronic Type: pre-clinical study Finality of use: abuse Dose and use frequency: 4% v.o. for 26 months | Anxiety | Social isolation and increased cortisol | Not investigated | [171] |
| Ethanol | Withdrawal effects | Pattern of use: acute Type: pre-clinical study Finality of use: abuse Dose and use frequency: vapor exposure for 7 days; blood alcohol levels 127 mg% | Endocrine regulation | Not specified | Hypothalamic–pituitary adrenal axis activity alterations | [172] |
| Ethanol | Under drug effects | Pattern of use: acute Type: clinical study Finality of use: abuse Dose and use frequency: a single dose of 0.8 g/kg | Cognition and emotionality | Negative effects on several dimensions of mood; impaired verbal memory; and dysphoria | Norepinephrine mediates behavioral alterations ethanol-induced (i.e., dysphoria), while serotonin provokes opposite activity | [158] |
| Ethanol | Under drug effects | Pattern of use: acute Type: pre-clinical study Finality of use: abuse Dose and use frequency: a single administration of 3.5 g/kg (i.p.) | CNS monoamine dependent functions | Not investigated | Mesolimbic and nigrostriatal dopaminergic systems’ hyperactivation, increasing DA release and catabolism | [159] |
| Ethanol | Under drug effects | Pattern of use: acute Type: pre-clinical study Finality of use: abuse Dose and use frequency: 1, 2, 3, and 4 g/kg for 7 days | CNS monoamine dependent functions | Not investigated | Dopamine, NE, and metabolite’s levels decrease on dorsal raphe and Locus coeruleus | [160] |
| Ethanol | Not informed | Literature review | General behavioral alterations | Psychomotor depression, difficulties in information storage and logical reasoning, motor incoordination, stimulation of reward system | Direct action on GABA, glutamate, and endocannabinoids systems; indirect action on limbic and opioid system; action on calcium channels, potent and proteins regulated by GABA hippocampus, in addition to central actions not mediated by vitamin B1 deficiency | [161] |
| Ethanol | Under drug and withdrawal effects | Literature review | Cognition | Nutritional disorders and dementia | Loss of hippocampal CA1 and CA3 pyramidal neurons, mossy fiber-CA3 synapses and dentate granule cells, and cholinergic neurons in basal forebrain; pathological neuroadaptations (i.e., excitatory/inhibitory neurotransmitters balance and oxidative stress) | [182] |
| Ketamine plus ethanol | Under drug effects | Pattern of use: sub-chronic Type: pre-clinical study Finality of use: abuse Dose and use frequency: 2 or 4 g/kg orally of ethanol plus 30 mg/kg i.p. ketamine for 14 days | Schizotypy | Increased activity, stereotyped behavior, ataxia, and morphological changes, as well as severe neurotoxicity | Altered behaviors were associated with alcohol-induced increases in ketamine-induced higher levels of Glu and DA in cortex and hippocampus | [141] |
| Ketamine plus ethanol | Under drug effects | Pattern of use: acute Type: pre-clinical study Finality of use: abuse Dose and use frequency: 10% ethanol solutions plus 0.28 mg/mL ketamine for 35 days, for 1 h | Anxiety | Ethanol: anxiogenic-like behavior. Ketamine: anxiolytic-like behavior. Ketamine + ethanol: absence of effect | Ketamine interfered in development of tolerance to anxiolytic effects of ethanol through modulation of several subsystems targeted by both drugs | [173] |
| Ethanol plus other drugs | Pattern of use: chronic Type: pre-clinical study Finality of use: abuse Dose and use frequency: 3 g/kg/day, 3 times a week for 5 weeks | Cognition and emotionality | Cognitive impairment; depressive and anxiety-like behavior | Alteration in peripheral markers of oxidative stress: decrease in nitrite levels; increase of sulfhydryl groups, MDA levels, superoxide dismutase, and catalase activity | [157] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kobayashi, N.H.C.; Farias, S.V.; Luz, D.A.; Machado-Ferraro, K.M.; Conceição, B.C.d.; Silveira, C.C.M.d.; Fernandes, L.M.P.; Cartágenes, S.d.C.; Ferreira, V.M.M.; Fontes-Júnior, E.A.; et al. Ketamine plus Alcohol: What We Know and What We Can Expect about This. Int. J. Mol. Sci. 2022, 23, 7800. https://doi.org/10.3390/ijms23147800
Kobayashi NHC, Farias SV, Luz DA, Machado-Ferraro KM, Conceição BCd, Silveira CCMd, Fernandes LMP, Cartágenes SdC, Ferreira VMM, Fontes-Júnior EA, et al. Ketamine plus Alcohol: What We Know and What We Can Expect about This. International Journal of Molecular Sciences. 2022; 23(14):7800. https://doi.org/10.3390/ijms23147800
Chicago/Turabian StyleKobayashi, Natalia Harumi Correa, Sarah Viana Farias, Diandra Araújo Luz, Kissila Márvia Machado-Ferraro, Brenda Costa da Conceição, Cinthia Cristina Menezes da Silveira, Luanna Melo Pereira Fernandes, Sabrina de Carvalho Cartágenes, Vânia Maria Moraes Ferreira, Enéas Andrade Fontes-Júnior, and et al. 2022. "Ketamine plus Alcohol: What We Know and What We Can Expect about This" International Journal of Molecular Sciences 23, no. 14: 7800. https://doi.org/10.3390/ijms23147800