
Ligand-Enhanced Negative Images Optimized for Docking Rescoring

 , , and
, , and
Abstract
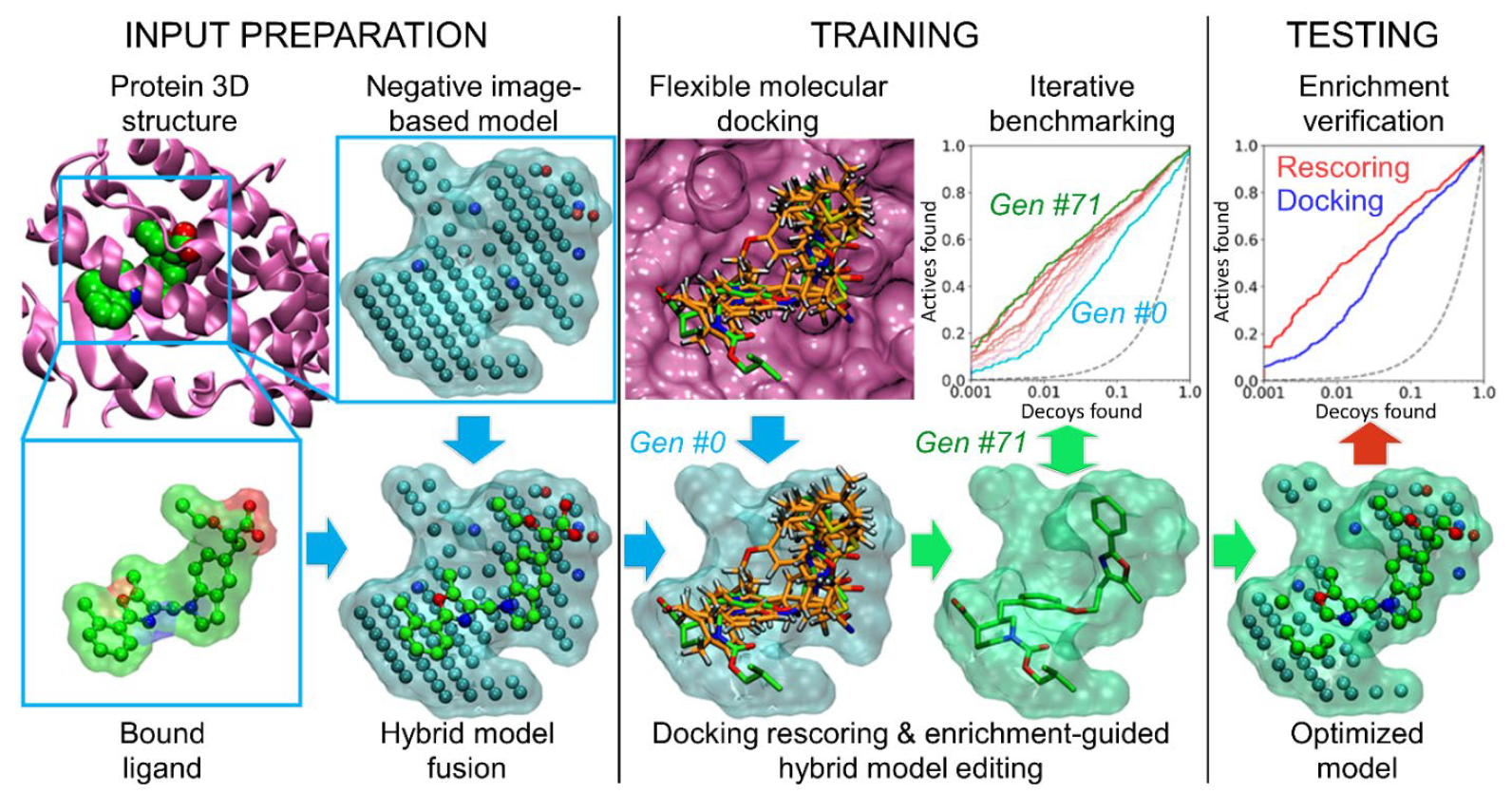
:1. Introduction
2. Results and Discussion
2.1. Fusion of Ligands into the Cavity-Based Negative Images
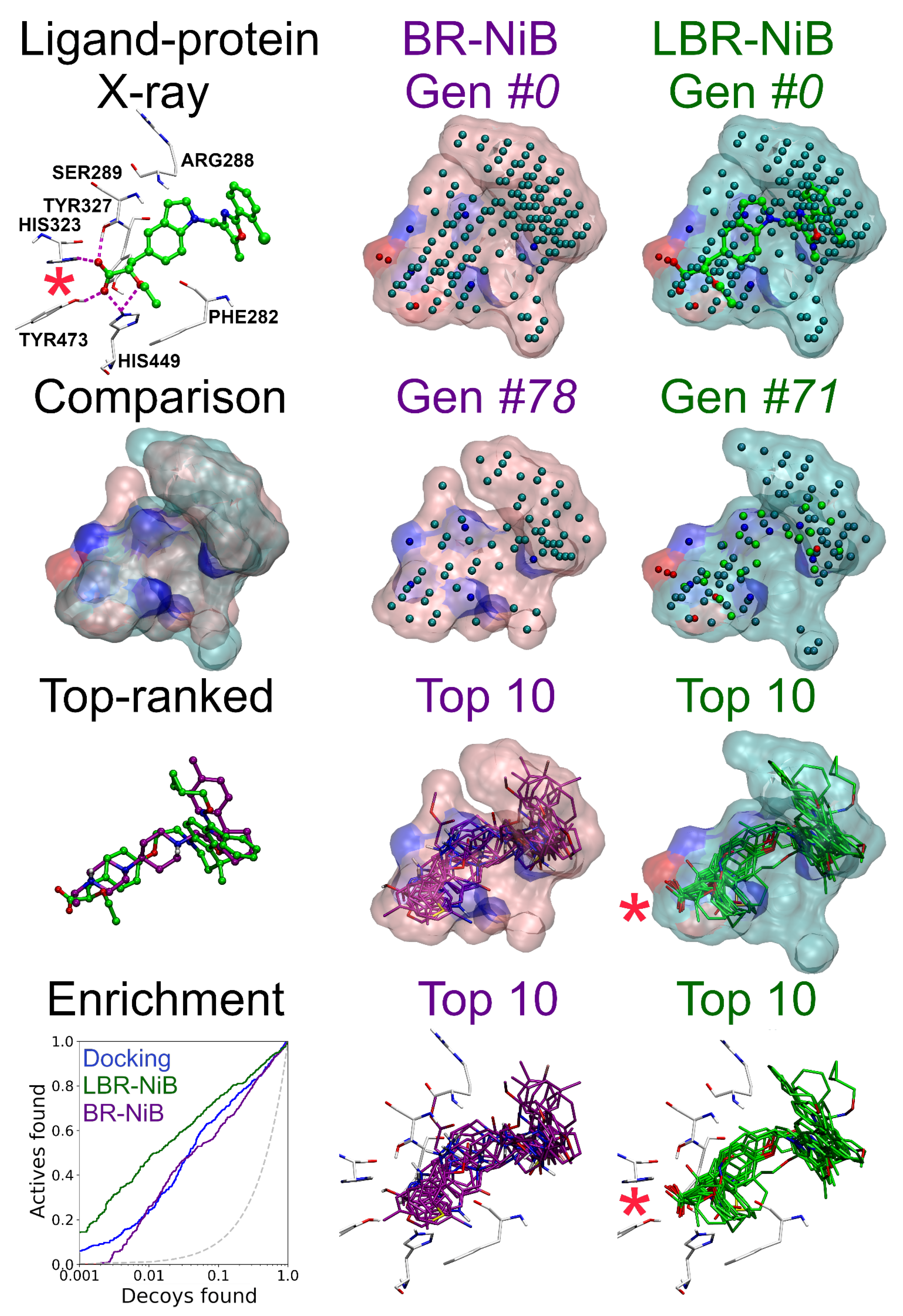
2.1.1. Ligands Boost Performance of Negative Image-Based Models via Optimization
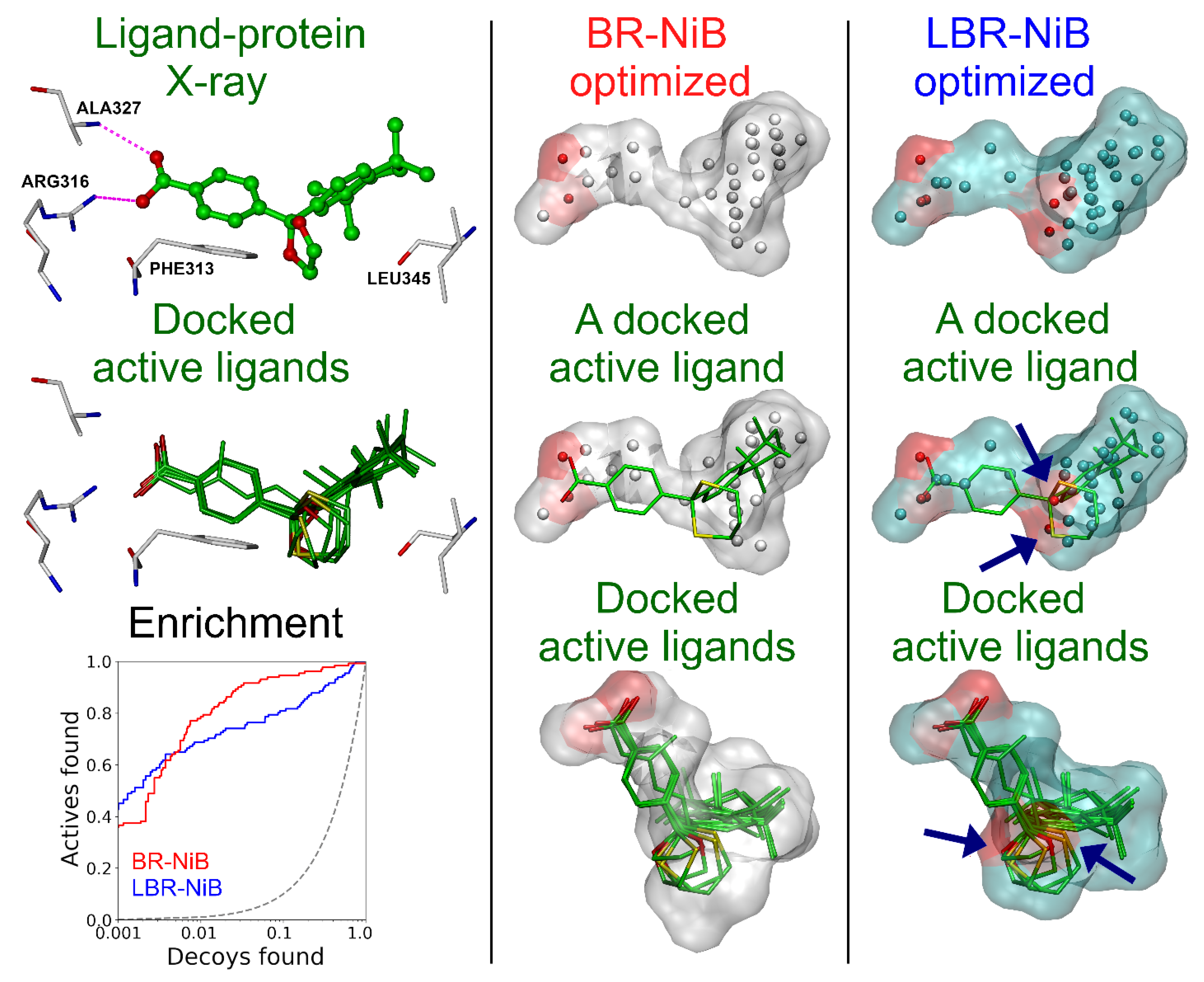
2.1.2. Two Ligands for Boosting Phosphodiesterase 5 Enrichment
2.1.3. Consistent Boost by Fusing Ligands into the Pre-Optimized Negative Image-Based Models
2.2. Dataset Curation Is Crucial for the Optimization
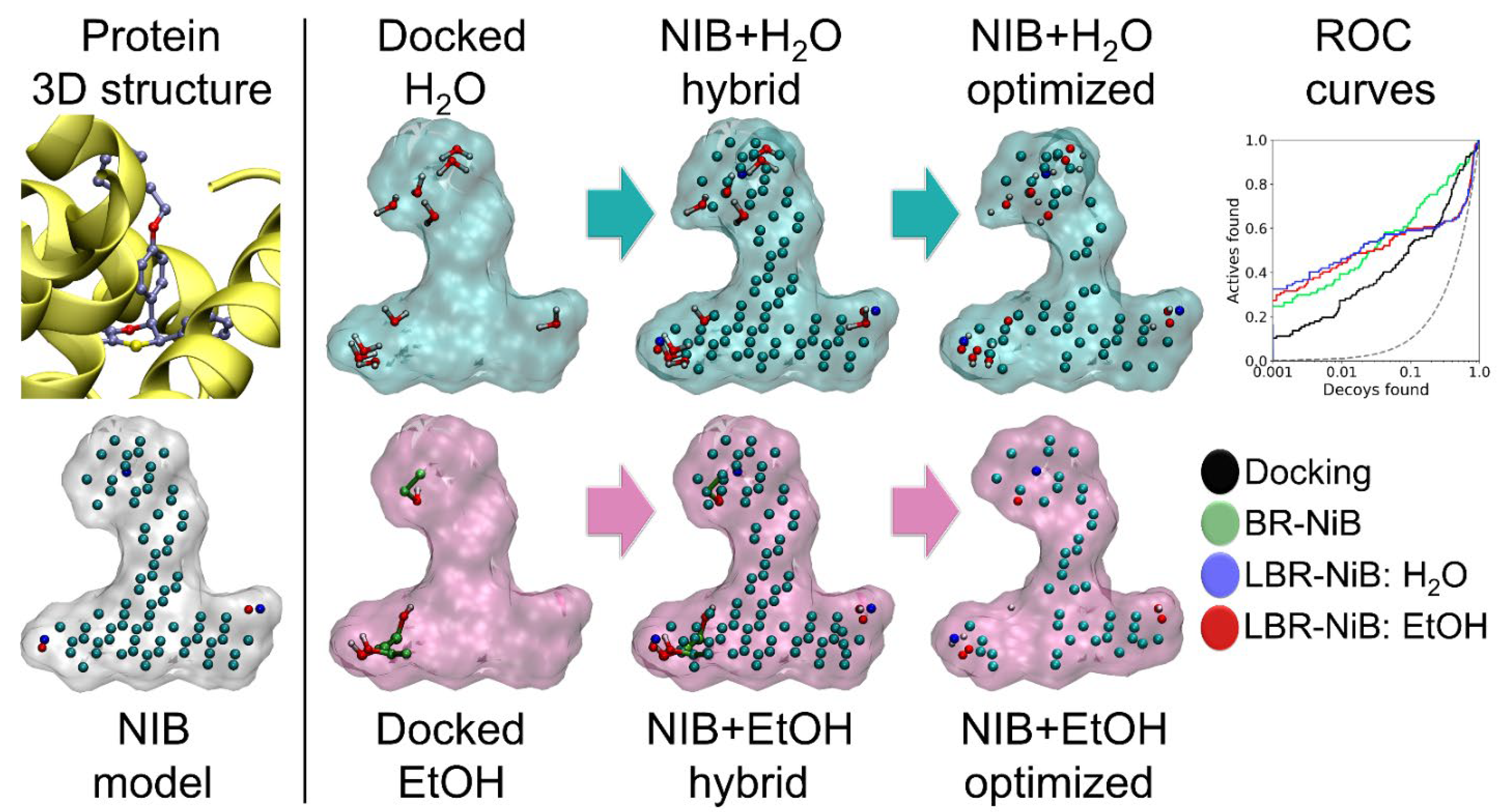
2.3. Docked Solvent Boosts the Negative Image-Based Models Performance via Optimization
2.4. Fused Ligands Reduce Chemotypic Diversity
2.5. Effect of Fused Ligands on the Negative Image-Based Model Performance
2.6. Practical Considerations of Using the Greedy Search Optimization
3. Materials and Methods
3.1. Protein Structure Preparation
3.2. Ligand Structure Preparation
3.3. Flexible Molecular Docking
3.4. Ligand-Negative Hybrid Model Generation
3.5. Negative Image-Based Rescoring
3.6. Brute Force Negative Image-Based Optimization
3.7. Data Analysis and Figure Preparation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bash, P.A.; Field, M.J.; Karplus, M. Free Energy Perturbation Method for Chemical Reactions in the Condensed Phase: A Dynamic Approach Based on a Combined Quantum and Molecular Mechanics Potential. J. Am. Chem. Soc. 1987, 109, 8092–8094. [Google Scholar] [CrossRef]
- Wang, R.; Lai, L.; Wang, S. Further Development and Validation of Empirical Scoring Functions for Structure-Based Binding Affinity Prediction. J. Comput. Aided Mol. Des. 2002, 16, 11–26. [Google Scholar] [CrossRef] [PubMed]
- Koes, D.R.; Baumgartner, M.P.; Camacho, C.J. Lessons Learned in Empirical Scoring with Smina from the CSAR 2011 Benchmarking Exercise. J. Chem. Inf. Model. 2013, 53, 1893–1904. [Google Scholar] [CrossRef] [PubMed]
- Naïm, M.; Bhat, S.; Rankin, K.N.; Dennis, S.; Chowdhury, S.F.; Siddiqi, I.; Drabik, P.; Sulea, T.; Bayly, C.I.; Jakalian, A.; et al. Solvated Interaction Energy (SIE) for Scoring Protein-Ligand Binding Affinities. 1. Exploring the Parameter Space. J. Chem. Inf. Model. 2007, 47, 122–133. [Google Scholar] [CrossRef]
- Kurkinen, S.T.; Niinivehmas, S.; Ahinko, M.; Lätti, S.; Pentikäinen, O.T.; Postila, P.A. Improving Docking Performance Using Negative Image-Based Rescoring. Front. Pharmacol. 2018, 9, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Ahinko, M.; Kurkinen, S.T.; Niinivehmas, S.P.; Pentikäinen, O.T.; Postila, P.A. A Practical Perspective: The Effect of Ligand Conformers on the Negative Image-Based Screening. Int. J. Mol. Sci. 2019, 20, 2779. [Google Scholar] [CrossRef] [Green Version]
- Kurkinen, S.T.; Lätti, S.; Pentikäinen, O.T.; Postila, P.A. Getting Docking into Shape Using Negative Image-Based Rescoring. J. Chem. Inf. Model. 2019, 59, 3584–3599. [Google Scholar] [CrossRef] [PubMed]
- Niinivehmas, S.P.; Salokas, K.; Lätti, S.; Raunio, H.; Pentikäinen, O.T. Ultrafast Protein Structure-Based Virtual Screening with Panther. J. Comput. Aided Mol. Des. 2015, 29, 989–1006. [Google Scholar] [CrossRef]
- Vainio, M.J.; Puranen, J.S.; Johnson, M.S. ShaEP: Molecular Overlay Based on Shape and Electrostatic Potential. J. Chem. Inf. Model. 2009, 49, 492–502. [Google Scholar] [CrossRef]
- Kurkinen, S.T.; Lehtonen, J.V.; Pentikäinen, O.T.; Postila, P.A. Optimization of Cavity-Based Negative Images to Boost Docking Enrichment in Virtual Screening. J. Chem. Inf. Model. 2021, 62, 1100–1112. [Google Scholar] [CrossRef]
- Huang, N.; Shoichet, B.K.; Irwin, J.J. Benchmarking Sets for Molecular Docking. J. Med. Chem. 2006, 49, 6789–6801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mysinger, M.M.; Carchia, M.; Irwin, J.J.; Shoichet, B.K. Directory of Useful Decoys, Enhanced (DUD-E): Better Ligands and Decoys for Better Benchmarking. J. Med. Chem. 2012, 55, 6582–6594. [Google Scholar] [CrossRef] [PubMed]
- Stein, R.M.; Yang, Y.; Balius, T.E.; O’Meara, M.J.; Lyu, J.; Young, J.; Tang, K.; Shoichet, B.K.; Irwin, J.J. Property-Unmatched Decoys in Docking Benchmarks. J. Chem. Inf. Model. 2021, 61, 699–714. [Google Scholar] [CrossRef] [PubMed]
- Rohrer, S.G.; Baumann, K. Maximum Unbiased Validation (MUV) Data Sets for Virtual Screening Based on PubChem Bioactivity Data. J. Chem. Inf. Model. 2009, 49, 169–184. [Google Scholar] [CrossRef]
- Halgren, T.A.; Murphy, R.B.; Friesner, R.A.; Beard, H.S.; Frye, L.L.; Pollard, W.T.; Banks, J.L. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 2. Enrichment Factors in Database Screening. J. Med. Chem. 2004, 47, 1750–1759. [Google Scholar] [CrossRef]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 1. Method and Assessment of Docking Accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef]
- Allen, W.J.; Balius, T.E.; Mukherjee, S.; Brozell, S.R.; Moustakas, D.T.; Lang, P.T.; Case, D.A.; Kuntz, I.D.; Rizzo, R.C. DOCK 6: Impact of New Features and Current Docking Performance. J. Comput. Chem. 2015, 36, 1132–1156. [Google Scholar] [CrossRef] [Green Version]
- Jones, G.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and Validation of a Genetic Algorithm for Flexible Docking. J. Mol. Biol. 1997, 267, 727–748. [Google Scholar] [CrossRef] [Green Version]
- Korb, O.; Stützle, T.; Exner, T.E. Empirical Scoring Functions for Advanced Protein-Ligand Docking with PLANTS. J. Chem. Inf. Model. 2009, 49, 84–96. [Google Scholar] [CrossRef]
- Lätti, S.; Niinivehmas, S.; Pentikäinen, O.T. Rocker: Open Source, Easy-to-Use Tool for AUC and Enrichment Calculations and ROC Visualization. J. Cheminform. 2016, 8, 45. [Google Scholar] [CrossRef] [Green Version]
- Jokinen, E.M.; Postila, P.A.; Ahinko, M.; Niinivehmas, S.; Pentikäinen, O.T. Fragment- and Negative Image-Based Screening of Phosphodiesterase 10A Inhibitors. Chem. Biol. Drug Des. 2019, 94, 1799–1812. [Google Scholar] [CrossRef] [PubMed]
- Núñez, V.; Alamedaa, D.; Rico, D.; Mota, R.; Gonzalo, P.; Cedenilla, M.; Fischer, T.; Boscá, L.; Glass, C.K.; Arroyo, A.G.; et al. Retinoid X Receptor α Controls Innate Inflammatory Responses through the Up-Regulation of Chemokine Expression. Proc. Natl. Acad. Sci. USA 2010, 107, 10626–10631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vignozzi, L.; Gacci, M.; Cellai, I.; Morelli, A.; Maneschi, E.; Comeglio, P.; Santi, R.; Filippi, S.; Sebastianelli, A.; Nesi, G.; et al. PDE5 Inhibitors Blunt Inflammation in Human BPH: A Potential Mechanism of Action for PDE5 Inhibitors in LUTS. Prostate 2013, 73, 1391–1402. [Google Scholar] [CrossRef]
- Martin, H. Role of PPAR-Gamma in Inflammation. Prospects for Therapeutic Intervention by Food Components. Mutat. Res. Fundam. Mol. Mech. Mutagen. 2009, 669, 1–7. [Google Scholar] [CrossRef]
- Karuppuchamy, T.; Behrens, E.H.; González-Cabrera, P.; Sarkisyan, G.; Gima, L.; Boyer, J.D.; Bamias, G.; Jedlicka, P.; Veny, M.; Clark, D.; et al. Sphingosine-1-Phosphate Receptor-1 (S1P 1) Is Expressed by Lymphocytes, Dendritic Cells, and Endothelium and Modulated during Inflammatory Bowel Disease. Mucosal Immunol. 2017, 10, 162–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geppert, H.; Vogt, M.; Bajorath, J. Current Trends in Ligand-Based Virtual Screening: Molecular Representations, Data Mining Methods, New Application Areas, and Performance Evaluation. J. Chem. Inf. Model. 2010, 50, 205–216. [Google Scholar] [CrossRef]
- Ripphausen, P.; Nisius, B.; Bajorath, J. State-of-the-Art in Ligand-Based Virtual Screening. Drug Discov. Today 2011, 16, 372–376. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, B.; Hilpert, H.; Benz, J.; Binggeli, A.; Grether, U.; Humm, R.; Märki, H.P.; Meyer, M.; Mohr, P. Structure-Based Design of Indole Propionic Acids as Novel PPARα/γ Co-Agonists. Bioorg. Med. Chem. Lett. 2006, 16, 4016–4020. [Google Scholar] [CrossRef]
- Mobley, D.L.; Dill, K.A. Binding of Small-Molecule Ligands to Proteins: “What You See” Is Not Always “What You Get”. Structure 2009, 17, 489–498. [Google Scholar] [CrossRef] [Green Version]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Word, J.M.; Lovell, S.C.; Richardson, J.S.; Richardson, D.C. Asparagine and Glutamine: Using Hydrogen Atom Contacts in the Choice of Side-Chain Amide Orientation. J. Mol. Biol. 1999, 285, 1735–1747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lehtonen, J.V.; Still, D.-J.; Rantanen, V.-V.; Ekholm, J.; Björklund, D.; Iftikhar, Z.; Huhtala, M.; Repo, S.; Jussila, A.; Jaakkola, J.; et al. BODIL: A Molecular Modeling Environment for Structure-Function Analysis and Drug Design. J. Comput. Aided Mol. Des. 2004, 18, 401–419. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, M.; Nishimura, T. Mersenne Twister: A 623-Dimensionally Equidistributed Uniform Pseudo-Random Number Generator. ACM Trans. Model. Comput. Simul. 1998, 8, 3–30. [Google Scholar] [CrossRef] [Green Version]
- Harder, E.; Damm, W.; Maple, J.; Wu, C.; Reboul, M.; Xiang, J.Y.; Wang, L.; Lupyan, D.; Dahlgren, M.K.; Knight, J.L.; et al. OPLS3: A Force Field Providing Broad Coverage of Drug-like Small Molecules and Proteins. J. Chem. Theory Comput. 2016, 12, 281–296. [Google Scholar] [CrossRef]
- Rarey, M.; Kramer, B.; Lengauer, T.; Klebe, G. A Fast Flexible Docking Method Using an Incremental Construction Algorithm. J. Mol. Biol. 1996, 261, 470–489. [Google Scholar] [CrossRef] [Green Version]
- Truchon, J.F.; Bayly, C.I. Evaluating Virtual Screening Methods: Good and Bad Metrics for the “Early Recognition” Problem. J. Chem. Inf. Model. 2007, 47, 488–508. [Google Scholar] [CrossRef]
- Lätti, S.T.; Niinivehmas, S.; Pentikäinen, O.T. Sdfconf: A Novel, Flexible, and Robust Molecular Data Management Tool. J. Chem. Inf. Model. 2022, 62, 9–15. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38, 27–28. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Train/ Test (1) | Method (2) | Yield | COX2 + CEL | RXRα + HXA | RXRα + BM6 | MR + AS4 | NEU + RA2 | PDE5 + VIA/CIA | ER + E4D | PPARγ + 208 |
|---|---|---|---|---|---|---|---|---|---|---|
| 100:100 | LBR-NiB | AUC | 0.85 ± 0.01 | 0.92 ± 0.02 | 0.91 ± 0.02 | 0.86 ± 0.02 | 0.97 ± 0.01 | 0.87 ± 0.01 | 0.74 ± 0.01 (↔) | 0.88 ± 0.01 |
| EFd 1% | 44.8 | 65.7 | 78.6 | 27.7 | 81.6 | 28.6 | 35.8 | 46.9 | ||
| EFd 5% | 63.5 | 77.1 | 85.5 | 57.5 | 92.9 | 53.8 | 51.7 | 65.9 | ||
| BR20 | 0.61 | 0.77 | 0.84 | 0.51 | 0.90 | 0.48 | 0.50 | 0.63 | ||
| LBR-NiB + shape only | AUC | 0.85 ± 0.01 | 0.90 ± 0.02 | 0.93 ± 0.02 | 0.81 ± 0.03 | 0.97 ± 0.01 | 0.88 ± 0.01 | 0.67 ± 0.02 (↓) | 0.85 ± 0.01 (↔) | |
| EFd 1% | 50.6 | 67.2 | 69.5 | 30.9 | 81.6 | 29.4 | 36.3 | 42.4 | ||
| EFd 5% | 64.4 | 77.9 | 84.7 | 48.9 | 93.9 | 53.3 | 45.2 | 59.5 | ||
| BR20 | 0.63 | 0.76 | 0.82 | 0.47 | 0.90 | 0.49 | 0.46 | 0.58 | ||
| 70:30 | LBR-NiB | AUC | 0.84 ± 0.02 | 0.93 ± 0.03 | 0.94 ± 0.03 | 0.84 ± 0.05 | 0.95 ± 0.03 | 0.90 ± 0.02 | 0.77 ± 0.03 (↔) | 0.86 ± 0.02 (↔) |
| EFd 1% | 43.0 | 70.0 | 72.5 | 31.0 | 70.0 | 17.5 | 37.6 | 42.5 | ||
| EFd 5% | 61.5 | 80.0 | 87.5 | 55.2 | 86.7 | 55.0 | 49.6 | 63.7 | ||
| BR20 | 0.60 | 0.81 | 0.84 | 0.53 | 0.82 | 0.46 | 0.51 | 0.61 | ||
| LBR-NiB + shape only | AUC | 0.83 ± 0.02 | 0.93 ± 0.03 | 0.92 ± 0.03 | 0.82 ± 0.05 | 0.94 ± 0.03 | 0.90 ± 0.02 | 0.74 ± 0.03 (↔) | 0.84 ± 0.02 (↔) | |
| EFd 1% | 31.1 | 30.0 | 72.5 | 31.0 | 70.0 | 27.5 | 38.5 | 38.4 | ||
| EFd 5% | 59.3 | 65.0 | 87.5 | 48.3 | 80.0 | 54.2 | 47.0 | 56.2 | ||
| BR20 | 0.53 | 0.59 | 0.83 | 0.47 | 0.82 | 0.49 | 0.49 | 0.55 | ||
| 10:90 | LBR-NiB | AUC | 0.83 ± 0.01 | 0.91 ± 0.02 | 0.91 ± 0.02 | 0.76 ± 0.03 | 0.93 ± 0.02 | 0.83 ± 0.01 | 0.69 ± 0.02 (↔) | 0.86 ± 0.01 (↔) |
| EFd 1% | 35.2 | 41.5 | 56.8 | 20.0 | 48.3 | 16.7 | 29.1 | 35.8 | ||
| EFd 5% | 57.9 | 58.5 | 75.4 | 36.5 | 73.0 | 38.2 | 40.9 | 57.8 | ||
| BR20 | 0.55 | 0.60 | 0.71 | 0.36 | 0.68 | 0.36 | 0.42 | 0.55 | ||
| LBR-NiB + shape only | AUC | 0.83 ± 0.01 | 0.71 ± 0.03 | 0.88 ± 0.02 | 0.78 ± 0.03 | 0.94 ± 0.02 | 0.83 ± 0.01 | 0.73 ± 0.02 (↔) | 0.84 ± 0.01 (↔) | |
| EFd 1% | 45.2 | 21.2 | 61.0 | 21.2 | 50.6 | 17.8 | 28.8 | 25.9 | ||
| EFd 5% | 59.7 | 36.4 (↓) | 76.3 | 38.8 | 71.9 | 37.9 | 39.5 | 51.8 (↓) | ||
| BR20 | 0.59 | 0.39 | 0.73 | 0.37 | 0.69 | 0.36 | 0.40 | 0.48 (↓) |
| Train/ Test (1) | Method (2) | Yield | COX2 + CEL | RXRα + HXA | RXRα + BM6 | MR + AS4 | NEU + RA2 | PDE5 + VIA/CIA | ER + E4D | PPARγ + 208 |
|---|---|---|---|---|---|---|---|---|---|---|
| 100:100 | BR-NiB + LBR-NiB | AUC | 0.80 ± 0.01 | 0.92 ± 0.02 | 0.93 ± 0.02 | 0.85 ± 0.02 | 0.97 ± 0.01 | 0.87 ± 0.01 | 0.75 ± 0.02 (↔) | 0.87 ± 0.01 (↔) |
| EFd 1% | 43.0 | 70.2 | 75.6 | 30.9 | 85.7 | 25.4 | 39.2 | 38.4 | ||
| EFd 5% | 59.5 | 80.9 | 84.0 | 54.3 | 91.8 | 50.8 | 52.0 | 61.4 | ||
| BR20 | 0.59 | 0.79 | 0.83 | 0.51 | 0.90 | 0.46 | 0.52 | 0.58 | ||
| BR-NiB + LBR-NiB + shape only | AUC | 0.85 ± 0.01 | 0.90 ± 0.02 | 0.91 ± 0.02 | 0.80 ± 0.03 | 0.97 ± 0.01 | 0.88 ± 0.01 | 0.68 ± 0.02 (↓) | 0.84 ± 0.01 (↔) | |
| EFd 1% | 50.3 | 65.6 | 73.3 | 31.9 | 83.7 | 31.4 | 37.1 | 43.0 | ||
| EFd 5% | 63.7 | 78.6 | 83.2 | 45.7 | 91.8 | 55.3 | 47.3 | 58.9 | ||
| BR20 | 0.63 | 0.76 | 0.82 | 0.46 | 0.90 | 0.50 | 0.47 | 0.58 | ||
| 70:30 | BR-NiB + LBR-NiB | AUC | 0.78 ± 0.02 | 0.93 ± 0.03 | 0.98 ± 0.01 | 0.86 ± 0.04 | 0.93 ± 0.03 | 0.88 ± 0.02 | 0.80 ± 0.02 | 0.86 ± 0.02(↔) |
| EFd 1% | 48.1 | 70.0 | 80.0 | 31.0 | 63.3 | 20.0 | 30.8 | 37.0 | ||
| EFd 5% | 56.3 | 85.0 | 85.0 | 55.2 | 83.3 | 48.3 | 46.2 | 62.3 | ||
| BR20 | 0.58 | 0.82 | 0.84 | 0.54 | 0.78 | 0.43 | 0.46 | 0.57 | ||
| BR-NiB + LBR-NiB + shape only | AUC | 0.84 ± 0.02 | 0.90 ± 0.03 | 0.92 ± 0.03 | 0.82 ± 0.05 | 0.94 ± 0.03 | 0.90 ± 0.02 | 0.72 ± 0.03 | 0.84 ± 0.02 (↔) | |
| EFd 1% | 46.7 | 70.0 | 75.0 | 20.1 | 83.3 | 20.8 | 38.5 | 40.4 | ||
| EFd 5% | 61.5 | 80.0 | 87.5 | 48.3 | 93.3 | 50.8 | 49.6 | 54.8 | ||
| BR20 | 0.61 | 0.77 | 0.84 | 0.46 | 0.81 | 0.46 | 0.50 | 0.56 | ||
| 10:90 | BR-NiB + LBR-NiB | AUC | 0.83 ± 0.01 | 0.89 ± 0.02 | 0.88 ± 0.02 | 0.82 ± 0.09 | 0.96 ± 0.01 | 0.83 ± 0.01 | 0.68 ± 0.02 | 0.88 ± 0.01 |
| EFd 1% | 32.4 | 48.3 | 65.3 | 23.9 | 64.9 | 19.2 | 29.1 | 33.9 | ||
| EFd 5% | 57.1 | 67.8 | 73.7 | 39.8 | 85.1 | 40.5 | 42.1 | 62.4 | ||
| BR20 | 0.53 | 0.66 | 0.75 | 0.39 | 0.80 | 0.37 | 0.42 | 0.56 | ||
| BR-NiB + LBR-NiB + shape only | AUC | 0.83 ± 0.01 | 0.80 ± 0.02(↔) | 0.86 ± 0.02 | 0.77 ± 0.03 | 0.95 ± 0.02 | 0.84 ± 0.01 | 0.73 ± 0.02 | 0.85 ± 0.01 | |
| EFd 1% | 46.7 | 39.8 | 55.9 | 22.7 | 61.7 | 17.4 | 29.8 | 28.3 | ||
| EFd 5% | 57.9 | 52.5 | 71.2 | 39.8 | 81.9 | 41.3 | 45.1 | 53.2(↓) | ||
| BR20 | 0.59 | 0.53 | 0.70 | 0.38 | 0.78 | 0.38 | 0.45 | 0.50 |
| Train/ Test (1) | Method (2) | Yield | COX2 | RXRα | MR | NEU | PDE5 | ER | PPARγ |
|---|---|---|---|---|---|---|---|---|---|
| water 70:30 | LBR-NiB | AUC | 0.78 ± 0.01 | 0.95 ± 0.02 | 0.73 ± 0.05 | 0.93 ± 0.03 | 0.83 ± 0.02 | 0.72 ± 0.03 (↔) | 0.84 ± 0.02 (↔) |
| EFd 1% | 29.1 | 82.5 | 17.2 | 73.3 | 15.8 | 46.2 | 38.4 | ||
| EFd 5% | 51.0 | 89.0 | 44.8 | 86.7 | 38.3 | 57.3 | 56.2 (↓) | ||
| BR20 | 0.48 | 0.87 | 0.39 | 0.81 | 0.35 | 0.57 | 0.54 | ||
| LBR-NiB + shape only | AUC | 0.83 ± 0.02 | 0.90 ± 0.03 | 0.70 ± 0.05 | 0.94 ± 0.03 | 0.89 ± 0.02 | 0.73 ± 0.03 (↔) | 0.82 ± 0.02 (↔) | |
| EFd 1% | 40.7 | 67.5 | 24.1 | 66.7 | 23.3 | 44.4 | 41.1 | ||
| EFd 5% | 58.5 | 80.0 | 48.3 | 86.7 | 47.5 | 49.6 | 52.7 (↓) | ||
| BR20 | 0.58 | 0.77 | 0.42 | 0.82 | 0.46 | 0.52 | 0.54 | ||
| ethanol 70:30 | LBR-NiB | AUC | 0.79 ± 0.02 | 0.97 ± 0.02 | 0.77 ± 0.05 | 0.94 ± 0.03 | 0.82 ± 0.02 | 0.72 ± 0.03 (↔) | 0.82 ± 0.02 (↔) |
| EFd 1% | 38.5 | 77.5 | 31.0 | 73.3 | 18.3 | 43.6 | 32.9 | ||
| EFd 5% | 50.4 | 87.5 | 44.8 | 86.7 | 39.2 | 53.0 | 47.9 (↓) | ||
| BR20 | 0.54 | 0.86 | 0.45 | 0.83 | 0.36 | 0.55 | 0.47 (↓) | ||
| LBR-NiB + shape only | AUC | 0.82 ± 0.02 | 0.90 ± 0.03 | 0.79 ± 0.05 | 0.94 ± 0.03 | 0.88 ± 0.02 | 0.71 ± 0.03 (↓) | 0.84 ± 0.02 (↔) | |
| EFd 1% | 43.7 | 45.0 | 27.6 | 60.0 | 19.2 | 33.3 | 37.7 | ||
| EFd 5% | 60.0 | 85.0 | 48.3 | 83.3 | 45.8 | 49.6 | 52.7 (↓) | ||
| BR20 | 0.59 | 0.74 | 0.45 | 0.80 | 0.42 | 0.49 | 0.54 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kurkinen, S.T.; Lehtonen, J.V.; Pentikäinen, O.T.; Postila, P.A. Ligand-Enhanced Negative Images Optimized for Docking Rescoring. Int. J. Mol. Sci. 2022, 23, 7871. https://doi.org/10.3390/ijms23147871
Kurkinen ST, Lehtonen JV, Pentikäinen OT, Postila PA. Ligand-Enhanced Negative Images Optimized for Docking Rescoring. International Journal of Molecular Sciences. 2022; 23(14):7871. https://doi.org/10.3390/ijms23147871
Chicago/Turabian StyleKurkinen, Sami T., Jukka V. Lehtonen, Olli T. Pentikäinen, and Pekka A. Postila. 2022. "Ligand-Enhanced Negative Images Optimized for Docking Rescoring" International Journal of Molecular Sciences 23, no. 14: 7871. https://doi.org/10.3390/ijms23147871
APA StyleKurkinen, S. T., Lehtonen, J. V., Pentikäinen, O. T., & Postila, P. A. (2022). Ligand-Enhanced Negative Images Optimized for Docking Rescoring. International Journal of Molecular Sciences, 23(14), 7871. https://doi.org/10.3390/ijms23147871