
Structure Prediction, Evaluation, and Validation of GPR18 Lipid Receptor Using Free Programs


Abstract
:
1. Introduction
2. Results
2.1. Selection of Generated Models
2.2. Structural Motifs Comparison
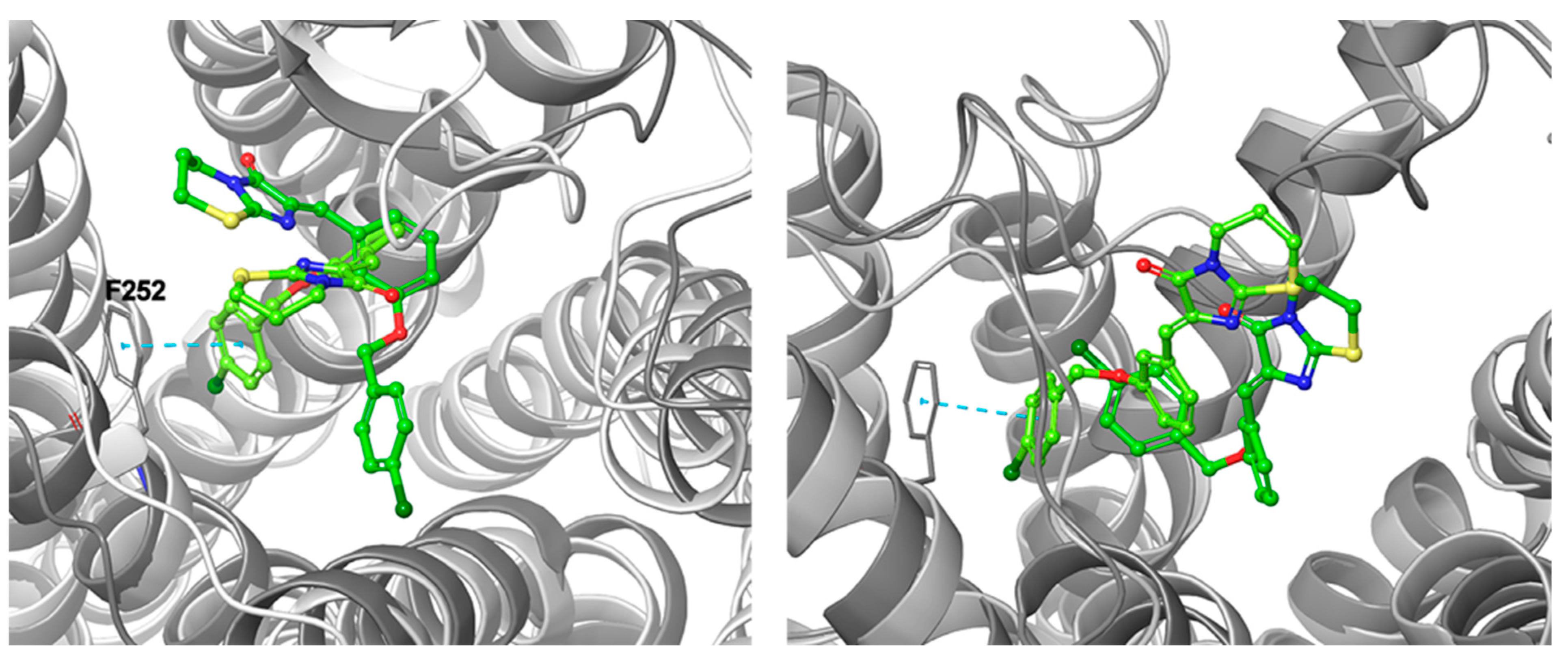
2.3. Binding Site/Mode Comparison
2.4. Docking Comparison
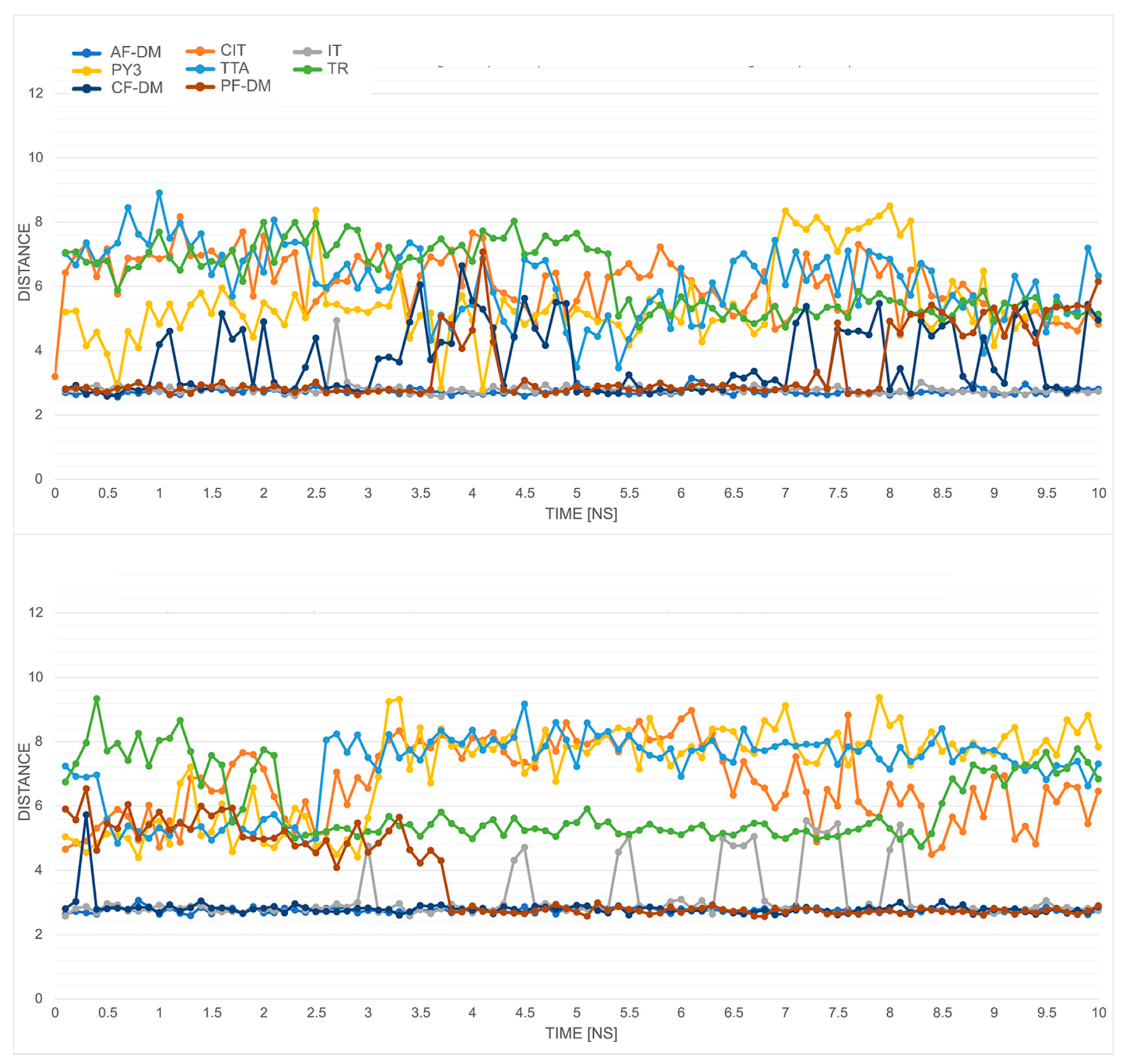
2.5. Molecular Dynamics Simulations
3. Discussion
4. Materials and Methods
4.1. GPR18 Structure Prediction
4.1.1. Classical Method
4.1.2. Threading Method
4.1.3. Ab Initio Method
4.2. Preliminary Structure Evaluation
4.3. Model Refinement
4.4. Enrichment Test
4.5. Models’ Comparison
4.5.1. Preliminary Ranking System
4.5.2. Secondary Ranking System
4.6. GPCR Lipid Receptors Structure Comparison
4.7. Actual Docking Procedure
- DockThor (DT) as described in Section 4.4—main docking procedure;
- GWO Vina 1.0 (Computational Biology and Bioinformatics Lab, University of Macau, China) [96], using mostly default parameters (except of exhaustiveness = 32)—additional, pose accuracy validation docking procedure.
4.8. Molecular Dynamics (MD) Simulations
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Katritch, V.; Cherezov, V.; Stevens, R.C. Structure-function of the G protein-coupled receptor superfamily. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 531–556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Isberg, V.; Mordalski, S.; Munk, C.; Rataj, K.; Harpsøe, K.; Hauser, A.S.; Vroling, B.; Bojarski, A.J.; Vriend, G.; Gloriam, D.E. GPCRdb: An information system for G protein-coupled receptors. Nucleic Acids Res. 2016, 44, D356–D364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Civelli, O.; Reinscheid, R.K.; Zhang, Y.; Wang, Z.; Fredriksson, R.; Schiöth, H.B. G protein-coupled receptor deorphanizations. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 127–146. [Google Scholar] [CrossRef] [Green Version]
- Gantz, I.; Muraoka, A.; Yang, Y.-K.; Samuelson, L.C.; Zimmerman, E.M.; Cook, H.; Yamada, T. Cloning and Chromosomal Localization of a Gene (GPR18) Encoding a Novel Seven Transmembrane Receptor Highly Expressed in Spleen and Testis. Genomics 1997, 42, 462–466. [Google Scholar] [CrossRef]
- Kohno, M.; Hasegawa, H.; Inoue, A.; Muraoka, M.; Miyazaki, T.; Oka, K.; Yasukawa, M. Identification of N-arachidonylglycine as the endogenous ligand for orphan G-protein-coupled receptor GPR18. Biochem. Biophys. Res. Commun. 2006, 347, 827–832. [Google Scholar] [CrossRef]
- Mchugh, D.; Hu, S.S.; Rimmerman, N.; Juknat, A.; Vogel, Z.; Walker, J.M.; Bradshaw, H.B. N-arachidonoyl glycine, an abundant endogenous lipid, potently drives directed cellular migration through GPR18, the putative abnormal cannabidiol receptor. BMC Neurosci. 2010, 11, 44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McHugh, D.; Page, J.; Dunn, E.; Bradshaw, H.B. $Δ$ 9-tetrahydrocannabinol and N-arachidonyl glycine are full agonists at GPR18 receptors and induce migration in human endometrial HEC-1B cells. Br. J. Pharmacol. 2012, 165, 2414–2424. [Google Scholar] [CrossRef] [Green Version]
- Chiang, N.; Dalli, J.; Colas, R.A.; Serhan, C.N. Identification of resolvin D2 receptor mediating resolution of infections and organ protection. J. Exp. Med. 2015, 212, 1203–1217. [Google Scholar] [CrossRef] [Green Version]
- Zhang, T.; Zuo, G.; Zhang, H. GPR18 Agonist Resolvin D2 Reduces Early Brain Injury in a Rat Model of Subarachnoid Hemorrhage by Multiple Protective Mechanisms. Cell. Mol. Neurobiol. 2021, 41, 1–14. [Google Scholar] [CrossRef]
- Alexander, S.P.H.; Battey, J.; Benson, H.E.; Benya, R.V.; Bonner, T.I.; Davenport, A.P.; Dhanachandra Singh, K.; Eguchi, S.; Harmar, A.; Holliday, N.; et al. Class A Orphans (version 2020.5) in the IUPHAR/BPS Guide to Pharmacology Database. CITE 2020, 1–45. [Google Scholar] [CrossRef]
- Yin, H.; Chu, A.; Li, W.; Wang, B.; Shelton, F.; Otero, F.; Nguyen, D.G.; Caldwell, J.S.; Chen, Y.A. Lipid G protein-coupled receptor ligand identification using $β$-arrestin PathHunterTM assay. J. Biol. Chem. 2009, 284, 12328–12338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Southern, C.; Cook, J.M.; Neetoo-Isseljee, Z.; Taylor, D.L.; Kettleborough, C.A.; Merritt, A.; Bassoni, D.L.; Raab, W.J.; Quinn, E.; Wehrman, T.S.; et al. Screening $β$-arrestin recruitment for the identification of natural ligands for orphan G-protein-coupled receptors. J. Biomol. Screen. 2013, 18, 599–609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, V.B.; Puhl, H.L.; Ikeda, S.R. N-arachidonyl glycine does not activate G protein-coupled receptor 18 signaling via canonical pathways. Mol. Pharmacol. 2013, 83, 267–282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finlay, D.B.; Joseph, W.R.; Grimsey, N.L.; Glass, M. GPR18 undergoes a high degree of constitutive trafficking but is unresponsive to N-Arachidonoyl Glycine. PeerJ 2016, 4, e1835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, Y.; Verdegaal, E.M.E.; Siderius, M.; Bebelman, J.P.; Smit, M.J.; Leurs, R.; Willemze, R.; Tensen, C.P.; Osanto, S. Quantitative expression profiling of G-protein-coupled receptors (GPCRs) in metastatic melanoma: The constitutively active orphan GPCR GPR18 as novel drug target. Pigment Cell Melanoma Res. 2011, 24, 207–218. [Google Scholar] [CrossRef]
- Morales, P.; Lago-Fernandez, A.; Hurst, D.P.; Sotudeh, N.; Brailoiu, E.; Reggio, P.H.; Abood, M.E.; Jagerovic, N. Therapeutic Exploitation of GPR18: Beyond the Cannabinoids? J. Med. Chem. 2020, 63, 14216–14227. [Google Scholar] [CrossRef]
- Biringer, R.G. Endocannabinoid signaling pathways: Beyond CB1R and CB2R. J. Cell Commun. Signal. 2021, 15, 335–360. [Google Scholar] [CrossRef]
- Morales, P.; Reggio, P.H. An Update on Non-CB 1, Non-CB 2 Cannabinoid Related G-Protein-Coupled Receptors. Cannabis Cannabinoid Res. 2017, 2, 265–273. [Google Scholar] [CrossRef] [Green Version]
- Reyes-Resina, I.; Navarro, G.; Aguinaga, D.; Canela, E.I.; Schoeder, C.T.; Załuski, M.; Kieć-Kononowicz, K.; Saura, C.A.; Müller, C.E.; Franco, R. Molecular and functional interaction between GPR18 and cannabinoid CB2 G-protein-coupled receptors. Relevance in neurodegenerative diseases. Biochem. Pharmacol. 2018, 157, 169–179. [Google Scholar] [CrossRef] [Green Version]
- McHugh, D. GPR18 in microglia: Implications for the CNS and endocannabinoid system signalling. Br. J. Pharmacol. 2012, 167, 1575–1582. [Google Scholar] [CrossRef] [Green Version]
- Kothandan, G.; Cho, S.J. Homology Modeling of GPR18 Receptor, an Orphan G-protein-coupled Receptor. J. Chosun Nat. Sci. 2013, 6, 16–20. [Google Scholar] [CrossRef] [Green Version]
- Schoeder, C.T. Identification, Optimization and Characterization of Pharmacological Tools for the Cannabinoid-Activated Orphan G Protein-Coupled Receptor GPR18 and Related Receptors. Doctoral Dissertation, Universitäts- und Landesbibliothek Bonn, Bonn, Germany, 2017. [Google Scholar]
- Kuder, K.J.; Karcz, T.; Kaleta, M.; Kiec-Kononowicz, K. Molecular modeling of an orphan GPR18 receptor. Lett. Drug Des. Discov. 2019, 16, 1167–1174. [Google Scholar] [CrossRef]
- Sotudeh, N.; Morales, P.; Hurst, D.P.; Lynch, D.L.; Reggio, P.H. Towards a molecular understanding of the cannabinoid related orphan receptor gpr18: A focus on its constitutive activity. Int. J. Mol. Sci. 2019, 20, 2300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neumann, A.; Engel, V.; Mahardhika, A.B.; Schoeder, C.T.; Namasivayam, V.; Kieć-Kononowicz, K.; Müller, C.E. Computational investigations on the binding mode of ligands for the cannabinoid-activated g protein-coupled receptor GPR18. Biomolecules 2020, 10, 686. [Google Scholar] [CrossRef]
- Lupas, A.N.; Pereira, J.; Alva, V.; Merino, F.; Coles, M.; Hartmann, M.D. The breakthrough in protein structure prediction. Biochem. J. 2021, 478, 1885–1890. [Google Scholar] [CrossRef]
- Heo, L.; Feig, M. Multi-State Modeling of G-protein Coupled Receptors at Experimental Accuracy. Proteins Struct. Funct. Bioinform. 2021, 1–22. [Google Scholar] [CrossRef]
- Munk, C.; Isberg, V.; Mordalski, S.; Harpsøe, K.; Rataj, K.; Hauser, A.S.; Kolb, P.; Bojarski, A.J.; Vriend, G.; Gloriam, D.E. GPCRdb: The G protein-coupled receptor database-an introduction. Br. J. Pharmacol. 2016, 173, 2195–2207. [Google Scholar] [CrossRef]
- Dror, R.O.; Arlow, D.H.; Maragakis, P.; Mildorf, T.J.; Pan, A.C.; Xu, H.; Borhani, D.W.; Shaw, D.E. Activation mechanism of the $β$2-adrenergic receptor. Proc. Natl. Acad. Sci. USA 2011, 108, 18684. [Google Scholar] [CrossRef] [Green Version]
- Latorraca, N.R.; Venkatakrishnan, A.J.; Dror, R.O. GPCR Dynamics: Structures in Motion. Chem. Rev. 2016, 117, 139–155. [Google Scholar] [CrossRef]
- Schmeisser, M.G. Creation of a GPR18 Homology Model Using Conformational Memories. Doctoral Dissertation, University of North Carolina at Greensboro, Greensboro, NC, USA, 2013. [Google Scholar]
- Weis, W.I.; Kobilka, B.K. The Molecular Basis of G Protein-Coupled Receptor. Annu. Rev. Biochem. 2018, 87, 897–919. [Google Scholar] [CrossRef]
- Gacasan, S.B.; Baker, D.L.; Parrill, A.L. G protein-coupled receptors: The evolution of structural insight. AIMS Biophys. 2017, 4, 491–527. [Google Scholar] [CrossRef] [PubMed]
- Cao, C.; Tan, Q.; Xu, C.; He, L.; Yang, L.; Zhou, Y.; Zhou, Y.; Qiao, A.; Lu, M.; Yi, C.; et al. Structural basis for signal recognition and transduction by platelet-activating-factor receptor. Nat. Struct. Mol. Biol. 2018, 25, 488–495. [Google Scholar] [CrossRef] [PubMed]
- Luginina, A.; Gusach, A.; Marin, E.; Mishin, A.; Brouillette, R.; Popov, P.; Shiriaeva, A.; Besserer-Offroy, É.; Longpré, J.M.; Lyapina, E.; et al. Structure-based mechanism of cysteinyl leukotriene receptor inhibition by antiasthmatic drugs. Sci. Adv. 2019, 5, eaax2518. [Google Scholar] [CrossRef] [Green Version]
- Kumar, P.; Bansal, M. Dissecting π-helices: Sequence, structure and function. FEBS J. 2015, 282, 4415–4432. [Google Scholar] [CrossRef] [PubMed]
- Van der Kant, R.; Vriend, G. Alpha-bulges in G protein-coupled receptors. Int. J. Mol. Sci. 2014, 15, 7841–7864. [Google Scholar] [CrossRef] [Green Version]
- Zimmermann, L.; Stephens, A.; Nam, S.Z.; Rau, D.; Kübler, J.; Lozajic, M.; Gabler, F.; Söding, J.; Lupas, A.N.; Alva, V. A Completely Reimplemented MPI Bioinformatics Toolkit with a New HHpred Server at its Core. J. Mol. Biol. 2018, 430, 2237–2243. [Google Scholar] [CrossRef]
- Gabler, F.; Nam, S.Z.; Till, S.; Mirdita, M.; Steinegger, M.; Söding, J.; Lupas, A.N.; Alva, V. Protein Sequence Analysis Using the MPI Bioinformatics Toolkit. Curr. Protoc. Bioinform. 2020, 72, e108. [Google Scholar] [CrossRef]
- Jendele, L.; Krivak, R.; Skoda, P.; Novotny, M.; Hoksza, D. PrankWeb: A web server for ligand binding site prediction and visualization. Nucleic Acids Res. 2019, 47, W345–W349. [Google Scholar] [CrossRef] [Green Version]
- Du, Z.; Su, H.; Wang, W.; Ye, L.; Wei, H.; Peng, Z.; Anishchenko, I.; Baker, D.; Yang, J. The trRosetta server for fast and accurate protein structure prediction. Nat. Protoc. 2021, 16, 5634–5651. [Google Scholar] [CrossRef]
- Baek, M.; Dimaio, F.; Anishchenko, I.; Dauparas, J.; Ovchinnikov, S.; Lee, G.R.; Wang, J.; Cong, Q.; Kinch, L.N.; Schaeffer, R.D.; et al. Accurate prediction of protein structures and interactions using a 3-track network. Science 2021, 373, 871–876. [Google Scholar] [CrossRef]
- Millán, C.; Keegan, R.M.; Pereira, J.; Sammito, M.D.; Simpkin, A.J.; McCoy, A.J.; Lupas, A.N.; Hartmann, M.D.; Rigden, D.J.; Read, R.J. Assessing the utility of CASP14 models for molecular replacement. Proteins Struct. Funct. Bioinform. 2021, 89, 1752–1769. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Schwede, T.; Kopp, J.; Guex, N.; Peitsch, M.C. SWISS-MODEL: An automated protein homology-modeling server. Nucleic Acids Res. 2003, 31, 3381–3385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fenalti, G.; Giguere, P.M.; Katritch, V.; Huang, X.-P.; Thompson, A.A.; Cherezov, V.; Roth, B.L.; Stevens, R.C. Molecular control of δ-opioid receptor signalling. Nature 2014, 506, 191–196. [Google Scholar] [CrossRef] [Green Version]
- Warren Lyford DeLano. The PyMOL Molecular Graphics System; Version 1.8.2.0; Schrödinger, LLC: New York, NY, USA, 2018. [Google Scholar]
- Sievers, F.; Higgins, D.G. The Clustal Omega Multiple Alignment Package. Methods Mol. Biol. 2021, 2231, 3–16. [Google Scholar] [CrossRef]
- Bramucci, E.; Paiardini, A.; Bossa, F.; Pascarella, S. PyMod: Sequence similarity searches, multiple sequence-structure alignments, and homology modeling within PyMOL. BMC Bioinform. 2012, 13, S2. [Google Scholar] [CrossRef] [Green Version]
- Šali, A.; Blundell, T.L. Comparative Protein Modelling by Satisfaction of Spatial Restraints. J. Mol. Biol. 1993, 234, 779–815. [Google Scholar] [CrossRef]
- Shapovalov, M.V.; Dunbrack, R.L., Jr. A smoothed backbone-dependent rotamer library for proteins derived from adaptive kernel density estimates and regressions. Structure 2011, 19, 844. [Google Scholar] [CrossRef] [Green Version]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera? A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [Green Version]
- Schoeder, C.T.; Kaleta, M.; Mahardhika, A.B.; Olejarz-Maciej, A.; Łażewska, D.; Kieć-Kononowicz, K.; Müller, C.E. Structure-activity relationships of imidazothiazinones and analogs as antagonists of the cannabinoid-activated orphan G protein-coupled receptor GPR18. Eur. J. Med. Chem. 2018, 155, 381–397. [Google Scholar] [CrossRef]
- Brooks, B.R.; Bruccoleri, R.E.; Olafson, B.D.; States, D.J.; Swaminathan, S.; Karplus, M. CHARMM: A program for macromolecular energy, minimization, and dynamics calculations. J. Comput. Chem. 1983, 4, 187–217. [Google Scholar] [CrossRef]
- Brooks, B.R.; Brooks, C.L.; Mackerell, A.D.; Nilsson, L.; Petrella, R.J.; Roux, B.; Won, Y.; Archontis, G.; Bartels, C.; Boresch, S.; et al. CHARMM: The biomolecular simulation program. J. Comput. Chem. 2009, 30, 1545–1614. [Google Scholar] [CrossRef] [PubMed]
- MacKerell, A.D., Jr.; Brooks, B.; Brooks, I.I.I.C.L.; Nilsson, L.; Roux, B.; Won, Y.; Karplus, M. The Encyclopedia of Computational Chemistry; Schleyer, R., Ed.; John Wiley & Sons: Chichester, UK, 1998; Volume 1. [Google Scholar]
- Wu, E.L.; Cheng, X.; Jo, S.; Rui, H.; Song, K.C.; Dávila-Contreras, E.M.; Qi, Y.; Lee, J.; Monje-Galvan, V.; Venable, R.M.; et al. CHARMM-GUI Membrane Builder toward realistic biological membrane simulations. J. Comput. Chem. 2014, 35, 1997–2004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. CHARMM-GUI: A web-based graphical user interface for CHARMM. J. Comput. Chem. 2008, 29, 1859–1865. [Google Scholar] [CrossRef] [PubMed]
- Jo, S.; Cheng, X.; Islam, S.M.; Huang, L.; Rui, H.; Zhu, A.; Lee, H.S.; Qi, Y.; Han, W.; Vanommeslaeghe, K.; et al. CHARMM-GUI PDB Manipulator for Advanced Modeling and Simulations of Proteins Containing Nonstandard Residues. Adv. Protein Chem. Struct. Biol. 2014, 96, 235–265. [Google Scholar] [CrossRef] [Green Version]
- S AVESv6.0-Structure Validation Server. Available online: https://saves.mbi.ucla.edu (accessed on 18 May 2022).
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [Green Version]
- Thompson, J.D.; Higgins, D.G.; Gibson, T.J. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994, 22, 4673–4680. [Google Scholar] [CrossRef] [Green Version]
- Marti-Renom, M.A.; Madhusudhan, M.S.; Sali, A. Alignment of protein sequences by their profiles. Protein Sci. 2004, 13, 1071. [Google Scholar] [CrossRef] [Green Version]
- Wu, H.; Wacker, D.; Mileni, M.; Katritch, V.; Han, G.W.; Vardy, E.; Liu, W.; Thompson, A.A.; Huang, X.-P.; Carroll, F.I.; et al. Structure of the human κ-opioid receptor in complex with JDTic. Nature 2012, 485, 327–332. [Google Scholar] [CrossRef]
- Miller, R.L.; Thompson, A.A.; Trapella, C.; Guerrini, R.; Malfacini, D.; Patel, N.; Han, G.W.; Cherezov, V.; Caló, G.; Katritch, V.; et al. The Importance of Ligand-Receptor Conformational Pairs in Stabilization: Spotlight on the N/OFQ G Protein-Coupled Receptor. Structure 2015, 23, 2291–2299. [Google Scholar] [CrossRef] [Green Version]
- Guedes, I.A.; Barreto, A.M.S.; Marinho, D.; Krempser, E.; Kuenemann, M.A.; Sperandio, O.; Dardenne, L.E.; Miteva, M.A. New machine learning and physics-based scoring functions for drug discovery. Sci. Rep. 2021, 11, 3198. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Yan, R.; Roy, A.; Xu, D.; Poisson, J.; Zhang, Y. The I-TASSER suite: Protein structure and function prediction. Nat. Methods 2014, 12, 7–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Zhang, Y. I-TASSER server: New development for protein structure and function predictions. Nucleic Acids Res. 2015, 43, W174–W181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, W.; Zhang, C.; Li, Y.; Pearce, R.; Bell, E.W.; Zhang, Y. Folding non-homologous proteins by coupling deep-learning contact maps with I-TASSER assembly simulations. Cell Rep. Methods 2021, 1, 100014. [Google Scholar] [CrossRef]
- The Yang Zhang Lab. Available online: https://zhanggroup.org (accessed on 18 May 2022).
- Zheng, W.; Zhang, C.; Wuyun, Q.; Pearce, R.; Li, Y.; Zhang, Y. LOMETS2: Improved meta-threading server for fold-recognition and structure-based function annotation for distant-homology proteins. Nucleic Acids Res. 2019, 47, W429–W436. [Google Scholar] [CrossRef] [Green Version]
- Wu, S.; Zhang, Y. LOMETS: A local meta-threading-server for protein structure prediction. Nucleic Acids Res. 2007, 35, 3375–3382. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Anishchenko, I.; Park, H.; Peng, Z.; Ovchinnikov, S.; Baker, D. Improved protein structure prediction using predicted interresidue orientations. Proc. Natl. Acad. Sci. USA 2020, 117, 1496–1503. [Google Scholar] [CrossRef]
- Protein Structure Prediction by TrRosetta. Available online: https://yanglab.nankai.edu.cn/trRosetta/ (accessed on 18 May 2022).
- Robetta (Protein Structure Prediction Service). Available online: https://robetta.bakerlab.org (accessed on 18 May 2022).
- Kanz, C.; Aldebert, P.; Althorpe, N.; Baker, W.; Baldwin, A.; Bates, K.; Browne, P.; van den Broek, A.; Castro, M.; Cochrane, G.; et al. The EMBL Nucleotide Sequence Database. Nucleic Acids Res. 2005, 33, D29–D33. [Google Scholar] [CrossRef] [Green Version]
- Bhattacharya, D.; Nowotny, J.; Cao, R.; Cheng, J. 3Drefine: An interactive web server for efficient protein structure refinement. Nucleic Acids Res. 2016, 44, W406–W409. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, Y. A Novel Side-Chain Orientation Dependent Potential Derived from Random-Walk Reference State for Protein Fold Selection and Structure Prediction. PLoS ONE 2010, 5, e15386. [Google Scholar] [CrossRef] [Green Version]
- Chen, V.B.; Arendall, W.B.; Headd, J.J.; Keedy, D.A.; Immormino, R.M.; Kapral, G.J.; Murray, L.W.; Richardson, J.S.; Richardson, D.C. MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 12–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shuvo, M.H.; Gulfam, M.; Bhattacharya, D. DeepRefiner: High-accuracy protein structure refinement by deep network calibration. Nucleic Acids Res. 2021, 49, W147–W152. [Google Scholar] [CrossRef] [PubMed]
- Davies, M.; Nowotka Michałand Papadatos, G.; Dedman, N.; Gaulton, A.; Atkinson, F.; Bellis, L.; Overington, J.P. ChEMBL web services: Streamlining access to drug discovery data and utilities. Nucleic Acids Res. 2015, 43, W612–W620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mendez, D.; Gaulton, A.; Bento, A.P.; Chambers, J.; De Veij, M.; Félix, E.; Magariños, M.P.; Mosquera, J.F.; Mutowo, P.; Nowotka Michałand Gordillo-Marañón, M.; et al. ChEMBL: Towards direct deposition of bioassay data. Nucleic Acids Res. 2019, 47, D930–D940. [Google Scholar] [CrossRef]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminformatics 2011, 3, 1–14. [Google Scholar] [CrossRef] [Green Version]
- RStudio Team. RStudio: Integrated Development for R; RStudio, PBC: Boston, MA, USA, 2020. [Google Scholar]
- Microsoft 365: Microsoft Excel, Microsoft Corporation 2021. Available online: https://office.microsoft.com/excel (accessed on 2 June 2022).
- Colovos, C.; Yeates, T.O. Verification of protein structures: Patterns of nonbonded atomic interactions. Protein Sci. 1993, 2, 1511. [Google Scholar] [CrossRef] [Green Version]
- Bowie, J.U.; Luthy, R.; Eisenberg, D. A method to identify protein sequences that fold into a known three-dimensional structure. Science 1991, 253, 164–170. [Google Scholar] [CrossRef] [Green Version]
- Lüthy, R.; Bowie, J.U.; Eisenberg, D. Assessment of protein models with three-dimensional profiles. Nature 1992, 356, 83–85. [Google Scholar] [CrossRef]
- Pontius, J.; Richelle, J.; Wodak, S.J. Deviations from standard atomic volumes as a quality measure for protein crystal structures. J. Mol. Biol. 1996, 264, 121–136. [Google Scholar] [CrossRef] [Green Version]
- Laskowski, R.A.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Alford, R.F.; Leaver-Fay, A.; Jeliazkov, J.R.; O’Meara, M.J.; DiMaio, F.P.; Park, H.; Shapovalov, M.V.; Renfrew, P.D.; Mulligan, V.K.; Kappel, K.; et al. The Rosetta all-atom energy function for macromolecular modeling and design. J. Chem. Theory Comput. 2017, 13, 3031. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Zhou, Y. Distance-scaled, finite ideal-gas reference state improves structure-derived potentials of mean force for structure selection and stability prediction. Protein Sci. 2009, 11, 2714–2726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, H.; Skolnick, J. GOAP: A Generalized Orientation-Dependent, All-Atom Statistical Potential for Protein Structure Prediction. Biophys. J. 2011, 101, 2043. [Google Scholar] [CrossRef] [Green Version]
- Lu, M.; Dousis, A.D.; Ma, J. OPUS-PSP: An Orientation-dependent Statistical All-atom Potential Derived from Side-chain Packing. J. Mol. Biol. 2008, 376, 288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schrödinger Release 2021-1: Protein Preparation Wizard; Epik, Schrödinger, LLC: New York, NY, USA; Impact, Schrödinger, LLC: New York, NY, USA; Prime, Schrödinger, LLC: New York, NY, USA, 2021.
- Wong, K.M.; Tai, H.K.; Siu, S.W.I. GWOVina: A grey wolf optimization approach to rigid and flexible receptor docking. Chem. Biol. Drug Des. 2021, 97, 97–110. [Google Scholar] [CrossRef] [PubMed]
- Phillips, J.C.; Hardy, D.J.; Maia, J.D.C.; Stone, J.E.; Ribeiro, J.V.; Bernardi, R.C.; Buch, R.; Fiorin, G.; Hénin, J.; Jiang, W.; et al. Scalable molecular dynamics on CPU and GPU architectures with NAMD. J. Chem. Phys. 2020, 153, 044130. [Google Scholar] [CrossRef]
- Miller, B.T.; Singh, R.P.; Klauda, J.B.; Hodošček, M.; Brooks, B.R.; Lee, H.; Iii, W. CHARMMing: A New, Flexible Web Portal for CHARMM. J. Chem. Inf. Modeling 2008, 48, 1920–1929. [Google Scholar] [CrossRef] [Green Version]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Model | ERRAT [%] | VERIFY 3D [%] | PROVE [%] | Ramachandran (Core, Disall [%]; Labell. Residues) | RW+ [kcal/mol] | Mol Prob | Rosetta Energy Scores | DFIRE Scores | GOAP Scores | OPUS-PSP Scores | Pred. Global Quality | |||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| AF-DM | - | 99.3 | 53.06 | 2.9% | 95.2 | 0.0 | 0 | −7935 | 0.650 | - | −656.4 | - | - | - |
| CF-DM | - | 88.38 | 52.04 | 4.6% | 94.1 | 0.0 | 10 | −7691 | 1.550 | - | −634.9 | - | - | - |
| PF-DM | - | 95.71 | 44.56 | 5.4% | 93.0 | 0.4 | 11 | −7673 | 1.460 | - | −633.5 | - | - | - |
| TR_1 | 5 | 100.0 | 70.75 | 2.9% | 95.6 | 0.0 | 1 | −7987 | 0.880 | −768.2 | −654.3 | −3713 | −5913 | 0.255 |
| TTA_5 | 1 | 98.25 | 60.88 | 2.8% | 93.7 | 1.1 | 10 | −7769 | 1.223 | −829.4 | −639.2 | −3606 | −5426 | 0.270 |
| CIT_1 | 4 | 100.0 | 70.41 | 3.1% | 92.6 | 0.4 | 2 | −7829 | 1.297 | −670.2 | −642.0 | −3487 | −5876 | 0.237 |
| IT_1 | 4 | 100.0 | 60.20 | 3.1% | 93.3 | 1.1 | 5 | −7770 | 1.449 | −589.7 | −637.5 | −3434 | −5678 | 0.214 |
| PY3_10 | 1 | 98.60 | 66.33 | 3.6% | 90.7 | 1.5 | 11 | −7614 | 1.494 | −595.3 | −624.5 | −3467 | −5459 | 0.222 |
| Model | Total Stars | V1 | R119-S230 Distance [Å] 1 | V2 | V1 + V2 | AUC | V3 | V1 + V2 + V3 | |
|---|---|---|---|---|---|---|---|---|---|
| CIT_1 | 4 | 22.0 | ** | 2.09 | *** | 5 | 0.811 | *** | 8 |
| TTA_5 | 1 | 16.0 | * | 2.97 | ** | 3 | 0.808 | *** | 6 |
| IT_1 | 4 | 11.5 | - | 2.41 | *** | 3 | 0.797 | *** | 6 |
| PY3_10 | 1 | 4.0 | - | 1.94 | *** | 3 | 0.791 | *** | 6 |
| TR_1 | 5 | 32.0 | *** | 2.95 | ** | 5 | 0.763 | * | 6 |
| CF-DM | - | 11.5 | - | 3.40 | ** | 2 | 0.834 | *** | 5 |
| PF-DM | - | 8.5 | - | 3.27 | ** | 2 | 0.783 | ** | 4 |
| AF-DM | - | 19.0 | * | 4.28 | * | 2 | 0.752 | * | 3 |
| *** | ** | * | None | ||
|---|---|---|---|---|---|
| ERRAT [86] [%] | 100 | ≥99 | ≥98 | <98 | |
| VERIFY 3D [87,88] [%] | ≥73 | ≥67 | ≥61 | <61 | |
| PROVE [89] [%] | ≤2.8 | ≤3.2 | ≤3.6 | >3.6 | |
| Ramachandran plot and χ1–χ2 [90] | Core [%]; Disall [%] | ≥94.4; 0.0 | ≥93.7; ≤0.4 | ≥93.0; ≤0.8 | <93.0; >0.8 |
| Labelled residues | ≤3 | ≤5 | ≤7 | >7 | |
| RWplus [78] | ≤−79,000 | ≤−78,250 | ≤−77,500 | >−77,500 | |
| MolProbity [82] | ≤1.0 | ≤1.2 | ≤1.4 | >1.4 | |
| Rosetta energy scores [91] | ≤−750 | ≤−675 | ≤−600 | >−600 | |
| DFIRE scores [92] | ≤−649 | ≤−641 | ≤−633 | >−633 | |
| GOAP scores [93] | ≤−37,200 | ≤−36,000 | ≤−34,800 | ≤−34,800 | |
| OPUS-PSP scores [94] | ≤−5770 | ≤−5650 | ≤−5530 | ≤−5530 | |
| Predicted global quality scores [80] | ≥0.26 | ≥0.24 | ≥0.22 | <0.22 | |
| *** | ** | * | (None) | |
|---|---|---|---|---|
| V1 | ≥27 | ≥20 | ≥13 | <13 |
| V2 | ≤2.5 Å | ≤3.5 Å | ≤4.5 Å | >4.5 Å |
| V3 | ≥0.79 | ≥0.77 | ≥0.75 | <0.75 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Michalik, I.; Kuder, K.J.; Kieć-Kononowicz, K.; Handzlik, J. Structure Prediction, Evaluation, and Validation of GPR18 Lipid Receptor Using Free Programs. Int. J. Mol. Sci. 2022, 23, 7917. https://doi.org/10.3390/ijms23147917
Michalik I, Kuder KJ, Kieć-Kononowicz K, Handzlik J. Structure Prediction, Evaluation, and Validation of GPR18 Lipid Receptor Using Free Programs. International Journal of Molecular Sciences. 2022; 23(14):7917. https://doi.org/10.3390/ijms23147917
Chicago/Turabian StyleMichalik, Ilona, Kamil J. Kuder, Katarzyna Kieć-Kononowicz, and Jadwiga Handzlik. 2022. "Structure Prediction, Evaluation, and Validation of GPR18 Lipid Receptor Using Free Programs" International Journal of Molecular Sciences 23, no. 14: 7917. https://doi.org/10.3390/ijms23147917
APA StyleMichalik, I., Kuder, K. J., Kieć-Kononowicz, K., & Handzlik, J. (2022). Structure Prediction, Evaluation, and Validation of GPR18 Lipid Receptor Using Free Programs. International Journal of Molecular Sciences, 23(14), 7917. https://doi.org/10.3390/ijms23147917