
Drug Discovery Using Evolutionary Similarities in Chemical Binding to Inhibit Patient-Derived Hepatocellular Carcinoma

 , , , , and
, , , , and
Abstract
:1. Introduction
2. Results
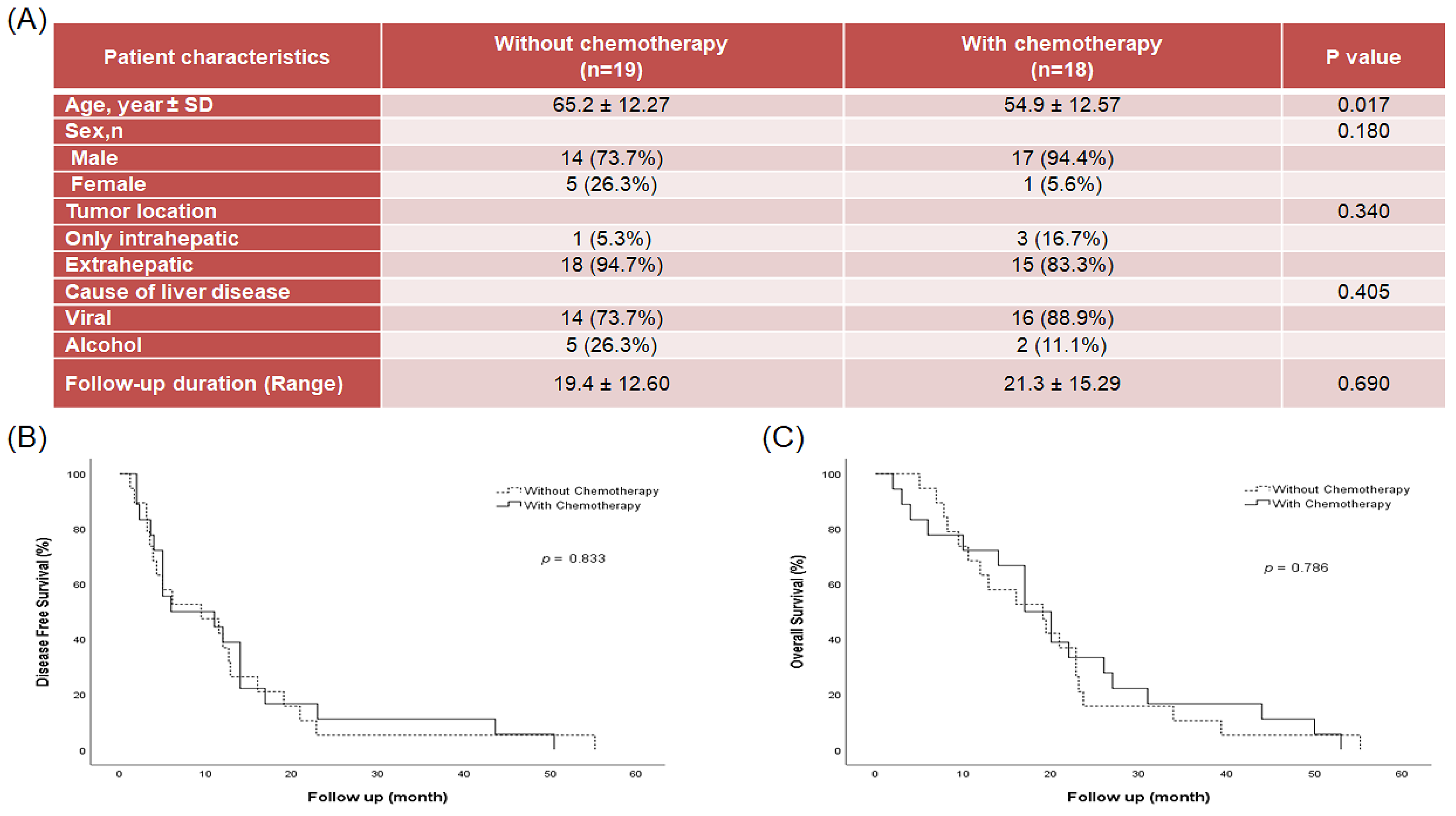
2.1. Patient Disease Characteristics
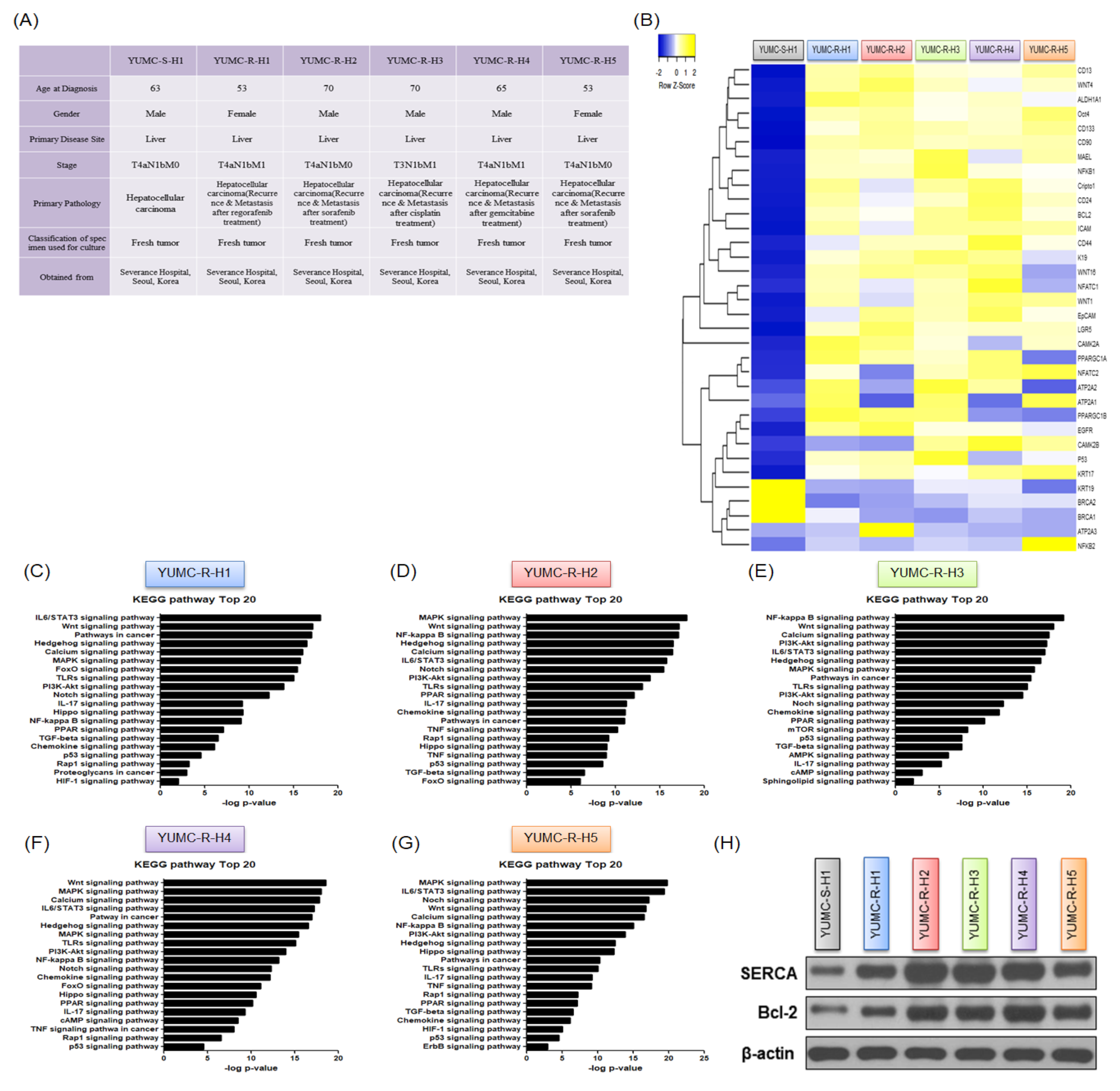
2.2. Contrasting Gene Expression and Signaling Stimulation between Patient-Derived Anti-Cancer Drug-Sensitive and Drug-Resistant HCC Cells
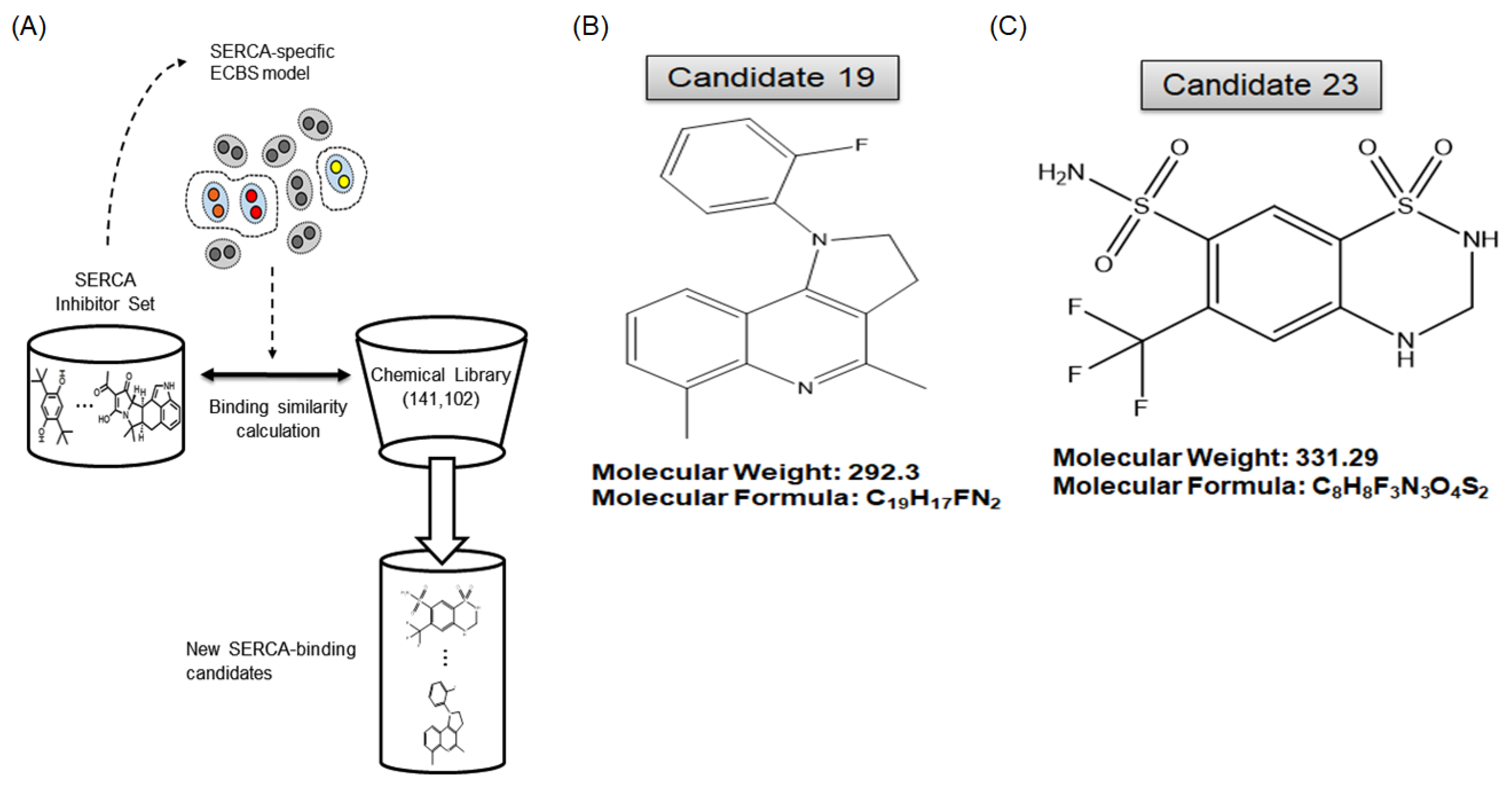
2.3. A Novel Therapeutic Trial of Candidates 19 and 23, SERCA Inhibitors, for Patient-Derived Drug-Resistant HCC Treatment via in Silico Screening
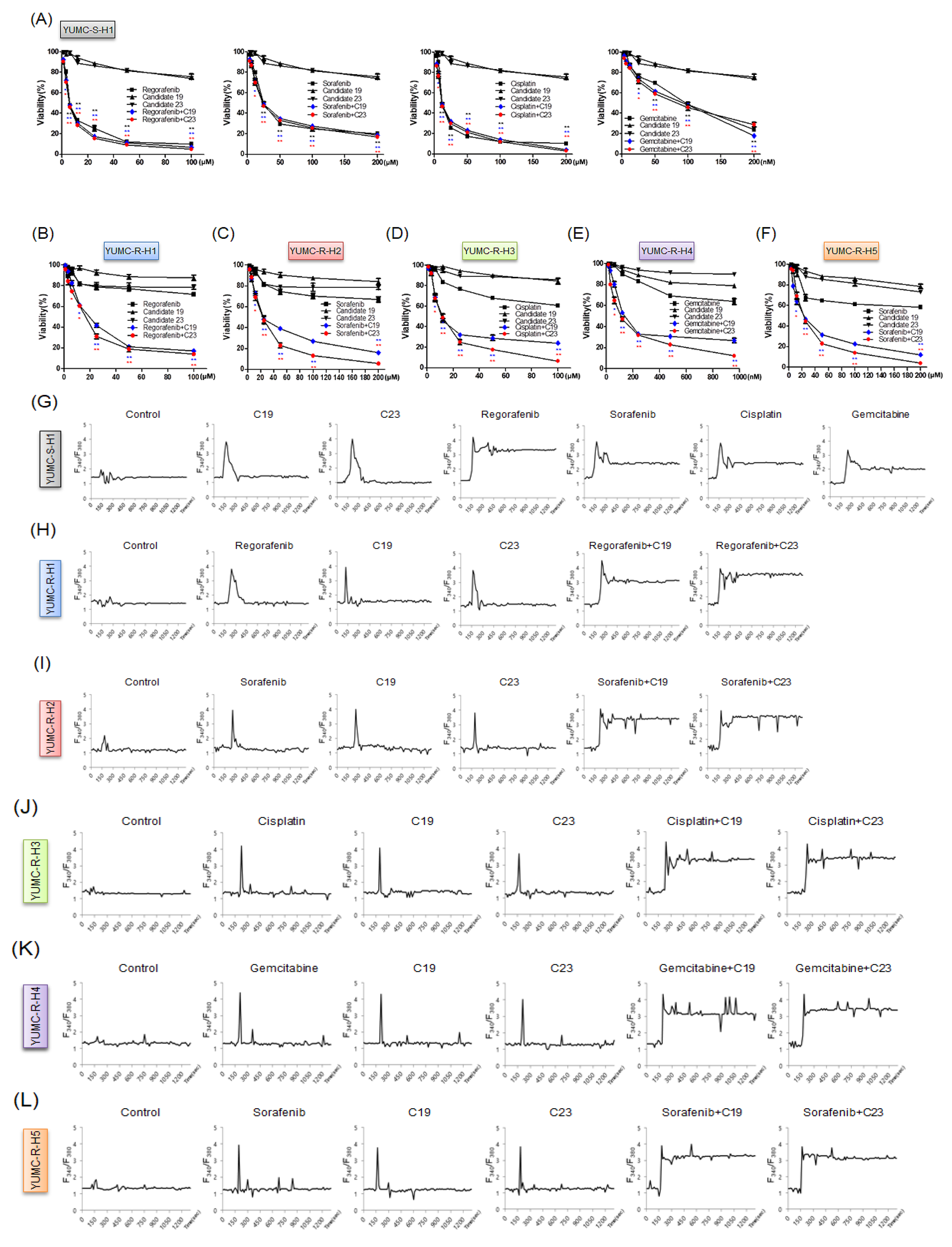
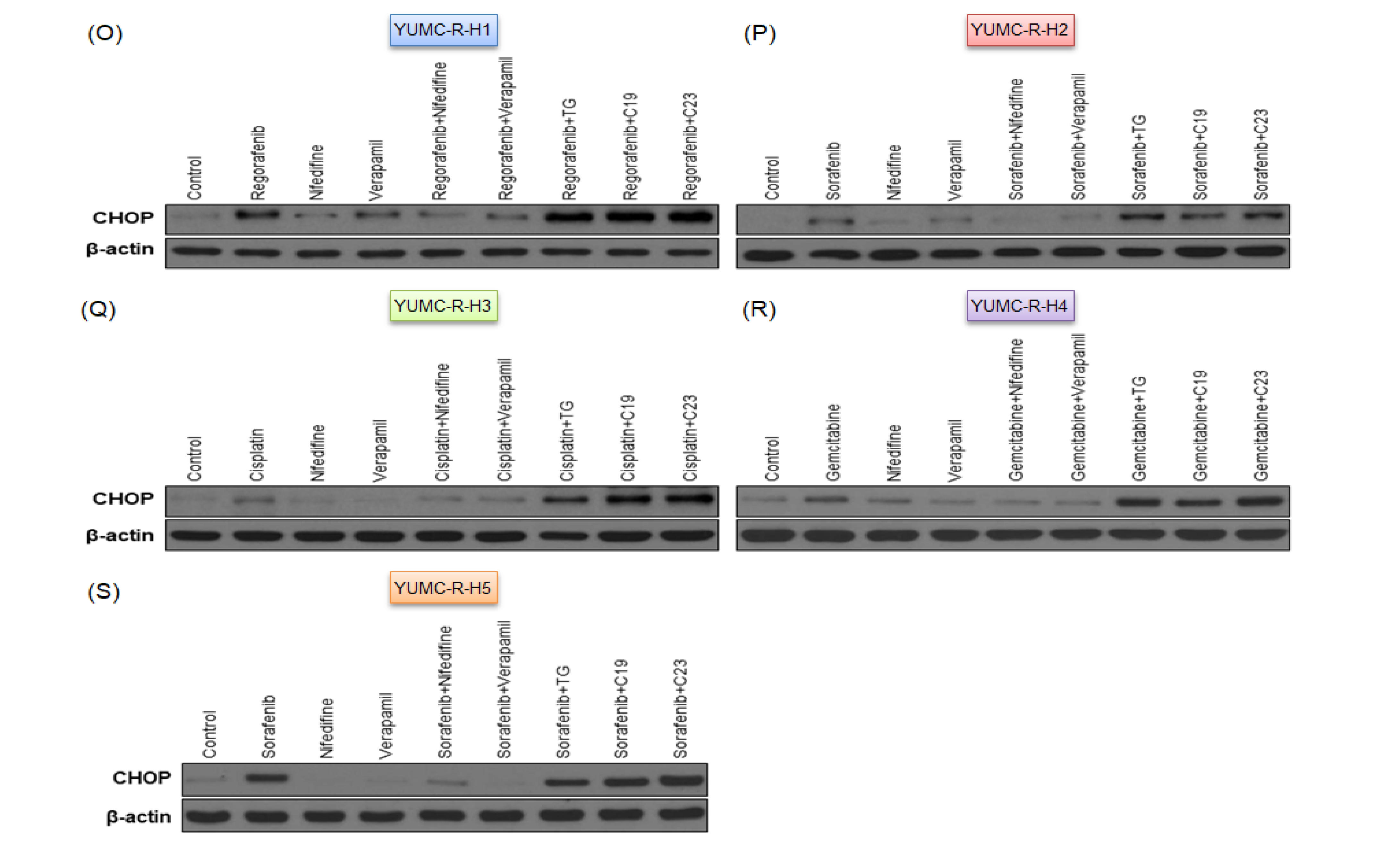
2.4. Novel Pharmacophore Candidates 19 and 23 Suppressed the Survival of Drug-Resistant HCC Cells
2.5. SERCA Increased the Anti-Apoptotic Activity of Severe ER Stress- and Drug-Resistant HCC Cells upon Prolonged Anti-Cancer Drug Treatment
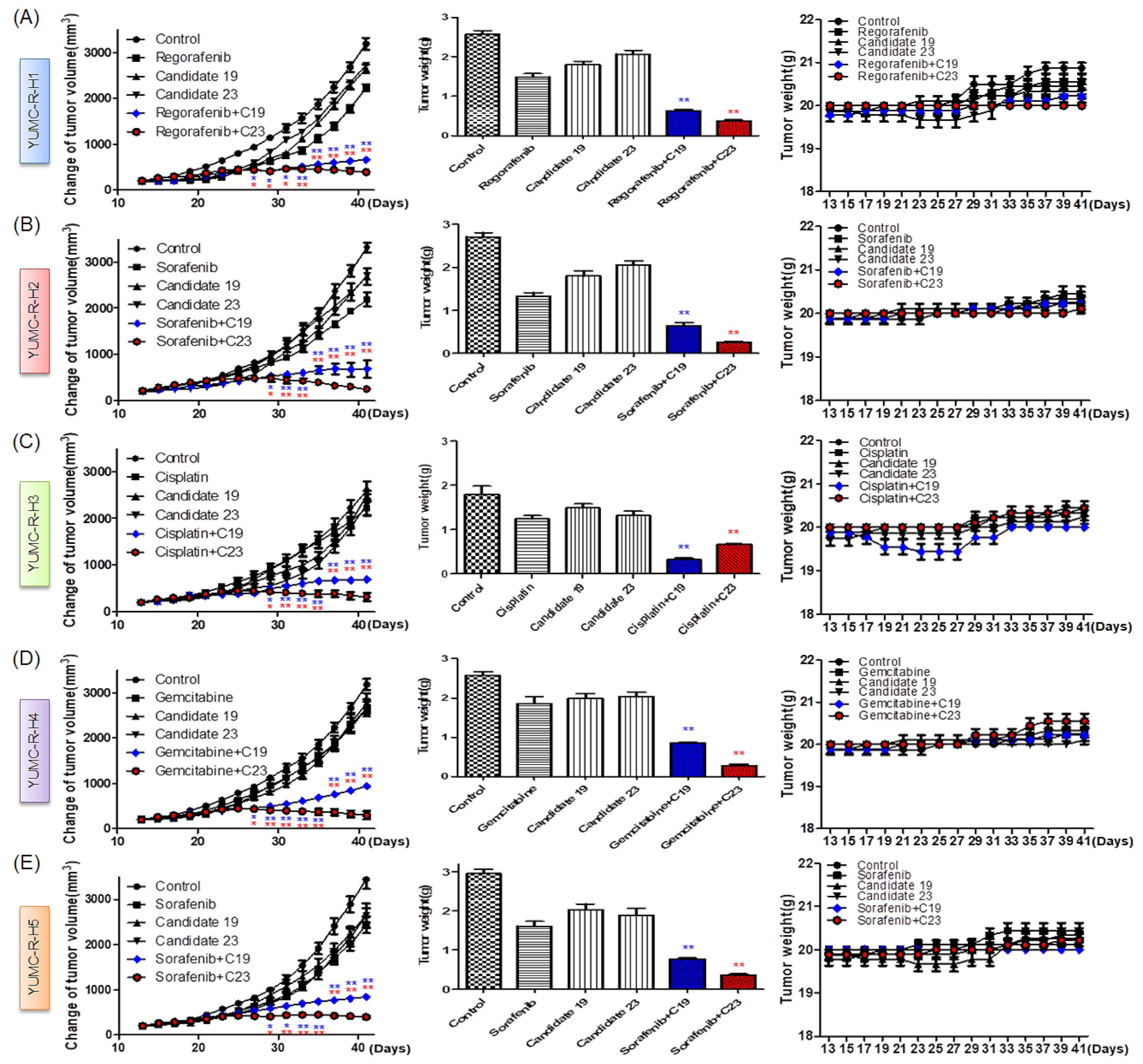
2.6. Novel Therapeutic Trials of Candidates 19 and 23 in Patient-Derived Drug-Resistant HCC Cell-Treated Mouse Xenograft Model
3. Discussion
4. Materials and Methods
4.1. Study Design and Ethical Considerations
4.2. Patient Characteristics
4.2.1. Patient for Sample 1
4.2.2. Patient for Samples 2 and 6
4.2.3. Patient for Samples 3 and 4
4.2.4. Patient for Sample 5
4.3. Patient Tissue Specimens
4.4. Ethical Considerations
4.5. Tumor Cell Isolation and Primary Culture
4.6. mRNA-Seq Data
4.7. Statistical Analysis of Gene Expression Level
4.8. Hierarchical Clustering
4.9. Whole RNA Extraction and Quantitative Real-Time Reverse Transcription PCR (qRT-PCR)
4.10. Cell Culture
4.11. Cell Viability Assay
4.12. Cytosolic Free Calcium Measurements by Microspectrofluorimetry
4.13. Immunoblot Analysis
4.14. Cell Cycle Analysis Using Flow Cytometry
4.15. Pharmacophore- and Docking-Based Sequential Virtual Screening for the Identification of a Novel SERCA Inhibitor
4.16. Patient-Derived Hepatocellular Carcinoma (HCC) Cell Xenograft
4.17. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ikeda, M.; Mitsunaga, S.; Ohno, I.; Hashimoto, Y.; Takahashi, H.; Watanabe, K.; Umemoto, K.; Okusaka, T. Systemic chemotherapy for advanced hepatocellular carcinoma: Past, present, and future. Diseases 2015, 3, 360–381. [Google Scholar] [CrossRef] [Green Version]
- Kishi, Y.; Hasegawa, K.; Sugawara, Y.; Kokudo, N. Hepatocellular carcinoma: Current management and future development-improved outcomes with surgical resection. Int. J. Hepatol. 2011, 2011, 728103. [Google Scholar] [CrossRef] [Green Version]
- Le Grazie, M.; Biagini, M.R.; Tarocchi, M.; Polvani, S.; Galli, A. Chemotherapy for hepatocellular carcinoma: The present and the future. World J. Hepatol. 2017, 9, 907–920. [Google Scholar] [CrossRef]
- Wu, T.C.; Shen, Y.C.; Cheng, A.L. Evolution of systemic treatment for advanced hepatocellular carcinoma. Kaohsiung J. Med. Sci. 2021, 37, 643–653. [Google Scholar] [CrossRef]
- Llovet, J.M.; Montal, R.; Sia, D.; Finn, R.S. Molecular therapies and precision medicine for hepatocellular carcinoma. Nat. Rev. Clin. Oncol. 2018, 15, 599–616. [Google Scholar] [CrossRef]
- Faubert, B.; Solmonson, A.; DeBerardinis, R.J. Metabolic reprogramming and cancer progression. Science 2020, 368, eaaw5473. [Google Scholar] [CrossRef]
- Yoshida, G.J. Metabolic reprogramming: The emerging concept and associated therapeutic strategies. J. Exp. Clin. Cancer Res. 2015, 34, 111. [Google Scholar] [CrossRef] [Green Version]
- Vaupel, P.; Schmidberger, H.; Mayer, A. The warburg effect: Essential part of metabolic reprogramming and central contributor to cancer progression. Int. J. Radiat. Biol. 2019, 95, 912–919. [Google Scholar] [CrossRef]
- Chatterjee, N.; Bivona, T.G. Polytherapy and targeted cancer drug resistance. Trends Cancer 2019, 5, 170–182. [Google Scholar] [CrossRef]
- Furtado, C.L.M.; Dos Santos Luciano, M.C.; Silva Santos, R.D.; Furtado, G.P.; Moraes, M.O.; Pessoa, C. Epidrugs: Targeting epigenetic marks in cancer treatment. Epigenetics 2019, 14, 1164–1176. [Google Scholar] [CrossRef]
- Shaffer, S.M.; Dunagin, M.C.; Torborg, S.R.; Torre, E.A.; Emert, B.; Krepler, C.; Beqiri, M.; Sproesser, K.; Brafford, P.A.; Xiao, M.; et al. Rare cell variability and drug-induced reprogramming as a mode of cancer drug resistance. Nature 2017, 546, 431–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krol, M.; Pawlowski, K.M.; Majchrzak, K.; Szyszko, K.; Motyl, T. Why chemotherapy can fail? Pol. J. Vet. Sci. 2010, 13, 399–406. [Google Scholar] [PubMed]
- Wang, X.; Zhang, H.; Chen, X. Drug resistance and combating drug resistance in cancer. Cancer Drug Resist. 2019, 2, 141–160. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Ao, X.; Ji, G.; Zhang, Y.; Yu, W.; Wang, J. Mechanisms of action and clinical implications of micrornas in the drug resistance of gastric cancer. Front. Oncol. 2021, 11, 768918. [Google Scholar] [CrossRef]
- Si, W.; Shen, J.; Zheng, H.; Fan, W. The role and mechanisms of action of micrornas in cancer drug resistance. Clin. Epigenet. 2019, 11, 25. [Google Scholar] [CrossRef] [PubMed]
- Brini, M.; Carafoli, E. Calcium pumps in health and disease. Physiol. Rev. 2009, 89, 1341–1378. [Google Scholar] [CrossRef] [Green Version]
- Periasamy, M.; Huke, S. Serca pump level is a critical determinant of ca(2+)homeostasis and cardiac contractility. J. Mol. Cell. Cardiol. 2001, 33, 1053–1063. [Google Scholar] [CrossRef]
- Arbabian, A.; Brouland, J.P.; Gelebart, P.; Kovacs, T.; Bobe, R.; Enouf, J.; Papp, B. Endoplasmic reticulum calcium pumps and cancer. Biofactors 2011, 37, 139–149. [Google Scholar] [CrossRef]
- Ma, Y.; Hendershot, L.M. Er chaperone functions during normal and stress conditions. J. Chem. Neuroanat. 2004, 28, 51–65. [Google Scholar] [CrossRef]
- VanHouten, J.; Sullivan, C.; Bazinet, C.; Ryoo, T.; Camp, R.; Rimm, D.L.; Chung, G.; Wysolmerski, J. Pmca2 regulates apoptosis during mammary gland involution and predicts outcome in breast cancer. Proc. Natl. Acad. Sci. USA 2010, 107, 11405–11410. [Google Scholar] [CrossRef] [Green Version]
- Chemaly, E.R.; Troncone, L.; Lebeche, D. Serca control of cell death and survival. Cell Calcium 2018, 69, 46–61. [Google Scholar] [CrossRef] [PubMed]
- Shin, D.H.; Leem, D.G.; Shin, J.S.; Kim, J.I.; Kim, K.T.; Choi, S.Y.; Lee, M.H.; Choi, J.H.; Lee, K.T. Compound k induced apoptosis via endoplasmic reticulum Ca(2+) release through ryanodine receptor in human lung cancer cells. J. Ginseng Res. 2018, 42, 165–174. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.G.; Pathan, N.; Ethell, I.M.; Krajewski, S.; Yamaguchi, Y.; Shibasaki, F.; McKeon, F.; Bobo, T.; Franke, T.F.; Reed, J.C. Ca2+-induced apoptosis through calcineurin dephosphorylation of bad. Science 1999, 284, 339–343. [Google Scholar] [CrossRef] [PubMed]
- Dolai, S.; Pal, S.; Yadav, R.K.; Adak, S. Endoplasmic reticulum stress-induced apoptosis in leishmania through Ca2+-dependent and caspase-independent mechanism. J. Biol. Chem. 2011, 286, 13638–13646. [Google Scholar] [CrossRef] [Green Version]
- Kerr, A.J.; Dodwell, D.; McGale, P.; Holt, F.; Duane, F.; Mannu, G.; Darby, S.C.; Taylor, C.W. Adjuvant and neoadjuvant breast cancer treatments: A systematic review of their effects on mortality. Cancer Treat. Rev. 2022, 105, 102375. [Google Scholar] [CrossRef]
- Ettrich, T.J.; Sturm, N.; Güthle, M.; Hüttner, F.J.; Perkhofer, L. Pancreatic cancer: Current multimodality treatment options and the future impact of molecular biological profiling. Visc. Med. 2022, 38, 20–29. [Google Scholar] [CrossRef]
- Heinemann, V.; Stintzing, S. Neoadjuvant and adjuvant therapy of resectable colon cancer—Current standards and developments. Dtsch. Med. Wochenschr. 2021, 146, 1457–1467. [Google Scholar]
- Friedlaender, A.; Naidoo, J.; Banna, G.L.; Metro, G.; Forde, P.; Addeo, A. Role and impact of immune checkpoint inhibitors in neoadjuvant treatment for nsclc. Cancer Treat. Rev. 2022, 104, 102350. [Google Scholar] [CrossRef]
- Akateh, C.; Black, S.M.; Conteh, L.; Miller, E.D.; Noonan, A.; Elliott, E.; Pawlik, T.M.; Tsung, A.; Cloyd, J.M. Neoadjuvant and adjuvant treatment strategies for hepatocellular carcinoma. World J. Gastroenterol. 2019, 25, 3704–3721. [Google Scholar] [CrossRef]
- Foerster, F.; Galle, P.R. The current landscape of clinical trials for systemic treatment of hcc. Cancers 2021, 13, 1962. [Google Scholar] [CrossRef]
- Sahin, I.H.; Khalil, L.; Millett, R.; Kaseb, A. Neoadjuvant and adjuvant treatment approaches for hepatocellular carcinoma: Future outlook. Chin. Clin. Oncol. 2021, 10, 7. [Google Scholar] [CrossRef] [PubMed]
- Vasan, N.; Baselga, J.; Hyman, D.M. A view on drug resistance in cancer. Nature 2019, 575, 299–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xavier, C.P.; Pesic, M.; Vasconcelos, M.H. Understanding cancer drug resistance by developing and studying resistant cell line models. Curr. Cancer Drug Targets 2016, 16, 226–237. [Google Scholar] [CrossRef] [PubMed]
- Du, B.; Shim, J.S. Targeting epithelial-mesenchymal transition (emt) to overcome drug resistance in cancer. Molecules 2016, 21, 965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alasiri, G.; Jiramongkol, Y.; Zona, S.; Fan, L.Y.; Mahmud, Z.; Gong, G.; Lee, H.J.; Lam, E.W. Regulation of perk expression by foxo3: A vulnerability of drug-resistant cancer cells. Oncogene 2019, 38, 6382–6398. [Google Scholar] [CrossRef] [Green Version]
- Fernandes, C.; Prabhu, P.; Juvale, K.; Suares, D.; Yc, M. Cancer cell fusion: A potential target to tackle drug-resistant and metastatic cancer cells. Drug Discov. Today 2019, 24, 1836–1844. [Google Scholar] [CrossRef]
- Nedeljkovic, M.; Damjanovic, A. Mechanisms of chemotherapy resistance in triple-negative breast cancer-how we can rise to the challenge. Cells 2019, 8, 957. [Google Scholar] [CrossRef] [Green Version]
- O’Donnell, J.S.; Teng, M.W.L.; Smyth, M.J. Cancer immunoediting and resistance to t cell-based immunotherapy. Nat. Rev. Clin. Oncol. 2019, 16, 151–167. [Google Scholar] [CrossRef]
- Pilie, P.G.; Tang, C.; Mills, G.B.; Yap, T.A. State-of-the-art strategies for targeting the DNA damage response in cancer. Nat. Rev. Clin. Oncol. 2019, 16, 81–104. [Google Scholar] [CrossRef]
- Nikolaou, M.; Pavlopoulou, A.; Georgakilas, A.G.; Kyrodimos, E. The challenge of drug resistance in cancer treatment: A current overview. Clin. Exp. Metastasis 2018, 35, 309–318. [Google Scholar] [CrossRef]
- Mashouri, L.; Yousefi, H.; Aref, A.R.; Ahadi, A.M.; Molaei, F.; Alahari, S.K. Exosomes: Composition, biogenesis, and mechanisms in cancer metastasis and drug resistance. Mol. Cancer 2019, 18, 75. [Google Scholar] [CrossRef] [PubMed]
- Berger, M.F.; Mardis, E.R. The emerging clinical relevance of genomics in cancer medicine. Nat. Rev. Clin. Oncol. 2018, 15, 353–365. [Google Scholar] [CrossRef] [PubMed]
- van Zyl, B.; Tang, D.; Bowden, N.A. Biomarkers of platinum resistance in ovarian cancer: What can we use to improve treatment. Endocr. Relat. Cancer 2018, 25, R303–R318. [Google Scholar] [CrossRef] [PubMed]
- Ashworth, A.; Lord, C.J. Synthetic lethal therapies for cancer: What’s next after parp inhibitors? Nat. Rev. Clin. Oncol. 2018, 15, 564–576. [Google Scholar] [CrossRef]
- Torre, L.A.; Bray, F.; Siegel, R.L.; Ferlay, J.; Lortet-Tieulent, J.; Jemal, A. Global cancer statistics, 2012. CA Cancer J. Clin. 2015, 65, 87–108. [Google Scholar] [CrossRef] [Green Version]
- Llovet, J.M.; Zucman-Rossi, J.; Pikarsky, E.; Sangro, B.; Schwartz, M.; Sherman, M.; Gores, G. Hepatocellular carcinoma. Nat. Rev. Dis. Primers 2016, 2, 16018. [Google Scholar] [CrossRef]
- Vogel, A.; Martinelli, E.; ESMO Guidelines Committee. Updated treatment recommendations for hepatocellular carcinoma (hcc) from the esmo clinical practice guidelines. Ann. Oncol. 2021, 32, 801–805. [Google Scholar] [CrossRef]
- Ayuso, C.; Rimola, J.; Vilana, R.; Burrel, M.; Darnell, A.; Garcia-Criado, A.; Bianchi, L.; Belmonte, E.; Caparroz, C.; Barrufet, M.; et al. Diagnosis and staging of hepatocellular carcinoma (hcc): Current guidelines. Eur. J. Radiol. 2018, 101, 72–81. [Google Scholar] [CrossRef]
- Forner, A.; Reig, M.; Bruix, J. Hepatocellular carcinoma. Lancet 2018, 391, 1301–1314. [Google Scholar] [CrossRef]
- Li, L.; Wang, H. Heterogeneity of liver cancer and personalized therapy. Cancer Lett. 2016, 379, 191–197. [Google Scholar] [CrossRef]
- Lee, E.; Yang, J.; Ku, M.; Kim, N.H.; Park, Y.; Park, C.B.; Suh, J.S.; Park, E.S.; Yook, J.I.; Mills, G.B.; et al. Metabolic stress induces a wnt-dependent cancer stem cell-like state transition. Cell Death Dis. 2015, 6, e1805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, K.C.; Kim, S.W.; Jeon, J.Y.; Jo, A.R.; Choi, H.J.; Kim, J.; Lee, H.G.; Kim, Y.; Mills, G.B.; Noh, S.H.; et al. Survival of cancer stem-like cells under metabolic stress via camk2alpha-mediated upregulation of sarco/endoplasmic reticulum calcium atpase expression. Clin. Cancer Res. 2018, 24, 1677–1690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, L.Y.; Chen, S.J.; Xiao, S.H.; Sun, Q.J.; Ding, C.H.; Zheng, B.N.; Zhu, X.Y.; Liu, S.Q.; Yang, F.; Yang, Y.X.; et al. Targeting p300/cbp attenuates hepatocellular carcinoma progression through epigenetic regulation of metabolism. Cancer Res. 2021, 81, 860–872. [Google Scholar] [CrossRef]
- Roti, G.; Carlton, A.; Ross, K.N.; Markstein, M.; Pajcini, K.; Su, A.H.; Perrimon, N.; Pear, W.S.; Kung, A.L.; Blacklow, S.C.; et al. Complementary genomic screens identify serca as a therapeutic target in notch1 mutated cancer. Cancer Cell 2013, 23, 390–405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pagliaro, L.; Marchesini, M.; Roti, G. Targeting oncogenic notch signaling with serca inhibitors. J. Hematol. Oncol. 2021, 14, 8. [Google Scholar] [CrossRef]
- Riera Leal, A.; Ortiz-Lazareno, P.C.; Jave-Suarez, L.F.; Ramirez De Arellano, A.; Aguilar-Lemarroy, A.; Ortiz-Garcia, Y.M.; Barron-Gallardo, C.A.; Solis-Martinez, R.; Luquin De Anda, S.; Munoz-Valle, J.F.; et al. 17betaestradiolinduced mitochondrial dysfunction and warburg effect in cervical cancer cells allow cell survival under metabolic stress. Int. J. Oncol. 2020, 56, 33–46. [Google Scholar]
- Li, M.; Wu, C.; Muhammad, J.S.; Yan, D.; Tsuneyama, K.; Hatta, H.; Cui, Z.G.; Inadera, H. Melatonin sensitises shikonin-induced cancer cell death mediated by oxidative stress via inhibition of the sirt3/sod2-akt pathway. Redox Biol. 2020, 36, 101632. [Google Scholar] [CrossRef]
- Bleeker, N.P.; Cornea, R.L.; Thomas, D.D.; Xing, C. A novel serca inhibitor demonstrates synergy with classic serca inhibitors and targets multidrug-resistant aml. Mol. Pharm. 2013, 10, 4358–4366. [Google Scholar] [CrossRef] [Green Version]
- Zhai, X.; Sterea, A.M.; Hiani, Y.E. Lessons from the endoplasmic reticulum Ca2+ transporters-a cancer connection. Cells 2020, 9, 1536. [Google Scholar] [CrossRef]
- Park, K.; Ko, Y.J.; Durai, P.; Pan, C.H. Machine learning-based chemical binding similarity using evolutionary relationships of target genes. Nucleic Acids Res. 2019, 47, e128. [Google Scholar] [CrossRef]
- Foufelle, F.; Fromenty, B. Role of endoplasmic reticulum stress in drug-induced toxicity. Pharmacol. Res. Perspect. 2016, 4, e00211. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.; Kim, B. Anti-cancer natural products and their bioactive compounds inducing er stress-mediated apoptosis: A review. Nutrients 2018, 10, 1021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munoz-Guardiola, P.; Casas, J.; Megias-Roda, E.; Sole, S.; Perez-Montoyo, H.; Yeste-Velasco, M.; Erazo, T.; Dieguez-Martinez, N.; Espinosa-Gil, S.; Munoz-Pinedo, C.; et al. The anti-cancer drug abtl0812 induces er stress-mediated cytotoxic autophagy by increasing dihydroceramide levels in cancer cells. Autophagy 2021, 17, 1349–1366. [Google Scholar] [CrossRef] [PubMed]
- Zhang, I.X.; Raghavan, M.; Satin, L.S. The endoplasmic reticulum and calcium homeostasis in pancreatic beta cells. Endocrinology 2020, 161, bqz028. [Google Scholar] [CrossRef] [PubMed]
- Krebs, J.; Agellon, L.B.; Michalak, M. Ca2+ homeostasis and endoplasmic reticulum (er) stress: An integrated view of calcium signaling. Biochem. Biophys. Res. Commun. 2015, 460, 114–121. [Google Scholar] [CrossRef]
- Kania, E.; Pajak, B.; Orzechowski, A. Calcium homeostasis and er stress in control of autophagy in cancer cells. Biomed. Res. Int. 2015, 2015, 352794. [Google Scholar] [CrossRef] [Green Version]
- Rizzuto, R.; De Stefani, D.; Raffaello, A.; Mammucari, C. Mitochondria as sensors and regulators of calcium signalling. Nat. Rev. Mol. Cell Biol. 2012, 13, 566–578. [Google Scholar] [CrossRef]
- Moreau, B.; Nelson, C.; Parekh, A.B. Biphasic regulation of mitochondrial ca2+ uptake by cytosolic ca2+ concentration. Curr. Biol. 2006, 16, 1672–1677. [Google Scholar] [CrossRef] [Green Version]
- Mattson, M.P.; Chan, S.L. Calcium orchestrates apoptosis. Nat. Cell Biol. 2003, 5, 1041–1043. [Google Scholar] [CrossRef] [Green Version]
- Gao, L.L.; Li, F.R.; Jiao, P.; Yao, S.T.; Sang, H.; Si, Y.H. Apoptosis of human ovarian cancer cells induced by paris chinensis dioscin via a ca(2+)-mediated mitochondrion pathway. Asian Pac. J. Cancer Prev. 2011, 12, 1361–1366. [Google Scholar]
- Pinton, P.; Giorgi, C.; Siviero, R.; Zecchini, E.; Rizzuto, R. Calcium and apoptosis: Er-mitochondria ca2+ transfer in the control of apoptosis. Oncogene 2008, 27, 6407–6418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, J.H.; Choi, K.H.; Kim, S.Y.; Park, C.S.; Kim, S.M.; Park, K.C. Patient-derived, drug-resistant colon cancer cells evade chemotherapeutic drug effects via the induction of epithelial-mesenchymal transition-mediated angiogenesis. Int. J. Mol. Sci. 2020, 21, 7469. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. Hisat: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [Green Version]
- Han, J.Y.; Ahn, K.S.; Kim, T.S.; Kim, Y.H.; Cho, K.B.; Shin, D.W.; Baek, W.K.; Suh, S.I.; Jang, B.C.; Kang, K.J. Liquid biopsy from bile-circulating tumor DNA in patients with biliary tract cancer. Cancers 2021, 13, 4581. [Google Scholar] [CrossRef] [PubMed]
- Yun, H.J.; Kim, M.; Kim, S.Y.; Fang, S.; Kim, Y.; Chang, H.S.; Chang, H.J.; Park, K.C. Effects of anti-cancer drug sensitivity-related genetic differences on therapeutic approaches in refractory papillary thyroid cancer. Int. J. Mol. Sci. 2022, 23, 699. [Google Scholar] [CrossRef] [PubMed]
- Yun, H.J.; Kim, H.J.; Kim, J.; Kim, S.Y.; Chang, H.S.; Park, C.S.; Chang, H.J.; Park, K.C. Synergistic anticancer activity of n-hydroxy-7-(2-naphthylthio) heptanomide, sorafenib, and radiation therapy in patient-derived anaplastic thyroid cancer models. Int. J. Mol. Sci. 2021, 22, 536. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cells name | PatientsNumber | Age | Sex | First Operation | Second Operation | Third Operation | Fourth Operation | Chemotherapy Regimen before Specimen Was Obtained |
|---|---|---|---|---|---|---|---|---|
| YUMC-S-H1 | 1 | 63 | male | laparoscopic left lateral sectionectomy * | □ | □ | □ | none |
| YUMC-R-H1 | 2 | 53 | female | left trisectionectomy and caudate segmentectomy with radical resection of common bile duct | palliative abdominal wall mass excision * | Sorafenib | ||
| YUMC-R-H5 | 2 | 53 | female | left trisectionectomy and caudate segmentectomy with radical resection of common bile duct | palliative abdominal wall mass excision | palliative chest wall mass excision * | sorafenib, regorafenib | |
| YUMC-R-H2 | 3 | 70 | male | laparoscopic right anterior sectionectomy | laparoscopic aortocaval lymph node dissection | superior mesenterioc lymph node dissection * | Sorafenib regorafenib | |
| YUMC-R-H3 | 3 | 70 | male | laparoscopic right anterior sectionectomy | laparoscopic aortocaval lymph node dissection | superior mesenterioc lymph node dissection | Multiple lymph node dissection * | Sorafenib regorafenib Foluorouracil and Cisplatin |
| YUMC-R-H4 | 4 | 63 | male | wedge resection of segment 5 | right hepatectomy * | □ | □ | Gemcitabine and Cisplatin |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lim, J.H.; Park, K.; Choi, K.H.; Kim, C.W.; Lee, J.H.; Weicker, R.; Pan, C.-H.; Kim, S.-M.; Park, K.C. Drug Discovery Using Evolutionary Similarities in Chemical Binding to Inhibit Patient-Derived Hepatocellular Carcinoma. Int. J. Mol. Sci. 2022, 23, 7971. https://doi.org/10.3390/ijms23147971
Lim JH, Park K, Choi KH, Kim CW, Lee JH, Weicker R, Pan C-H, Kim S-M, Park KC. Drug Discovery Using Evolutionary Similarities in Chemical Binding to Inhibit Patient-Derived Hepatocellular Carcinoma. International Journal of Molecular Sciences. 2022; 23(14):7971. https://doi.org/10.3390/ijms23147971
Chicago/Turabian StyleLim, Jin Hong, Keunwan Park, Kyung Hwa Choi, Chan Wung Kim, Jae Ha Lee, Raymond Weicker, Cheol-Ho Pan, Seok-Mo Kim, and Ki Cheong Park. 2022. "Drug Discovery Using Evolutionary Similarities in Chemical Binding to Inhibit Patient-Derived Hepatocellular Carcinoma" International Journal of Molecular Sciences 23, no. 14: 7971. https://doi.org/10.3390/ijms23147971