
Cancer-Associated Dysregulation of Sumo Regulators: Proteases and Ligases


Abstract
:1. Introduction
2. The Sumoylation Pathway
2.1. SUMO Proteases
2.2. SUMO Ligases

{kind=link}
{kind=link}
{kind=link}
| Enzymes | Family | Proteins | References |
|---|---|---|---|
| Proteases | SENP | SENP1, SENP2, SENP3, SENP5, SENP6, SENP7 | [21] |
| USPL1 | USPL1 | [23] | |
| DeSI | DESI1, DESI2 | [22] | |
| Ligases | SP-RING | PIAS1, PIAS2, PIAS3, PIAS4 | [28,30,31] |
| NSMCE2 (NSE2/MMS21) | |||
| ZMIZ1 (ZIMP10), ZMIZ2 | |||
| TRIM | TRIM1, TRIM11, TRIM19 (PML), TRIM22, TRIM27, TRIM28 (KAP1), TRIM32, TRIM33 (TIF1g), TRIM36, TRIM38, TRIM39, TRIML2 | [37,41,42,43,44,45] | |
| HDACs IIa | HDAC4, HDAC5, HDAC7, HDAC9 (MITR) | [46] | |
| Elongases | ZNF451 | [39,40] | |
| KIAA 1586 | |||
| Others | RanBP2 | [18,28,31,47,48,49] | |
| CBX4 (PC2) | |||
| TOPORS | |||
| RSUME | |||
| MUL1 (MAPL) | |||
| RHES | |||
| ARF (P14) | |||
| SF2 (ASF) | |||
| SLX4 | |||
| KROX20 | |||
| RNF212 | |||
| UHRF2 (RNF107) | |||
| TRAF7 (RNF119) | |||
| BCA2 (RNF115) | |||
| ZBED1 (DREF) | |||
| MDM2 |
3. SUMO in Cancer
4. SUMO Proteases in Cancer
4.1. SENP1
| Proteases | Cancer Types * | References |
|---|---|---|
| SENP1 | breast | [60,84,90,94] |
| CRC | [83,98,99,100,101,102] | |
| HCC | [92,93] | |
| lung | [85,94] | |
| MM | [103] | |
| neuroblastoma | [91] | |
| ovarian | [104] | |
| pancreas | [87] | |
| PCa | [82,97,105] | |
| thyroid | [86] | |
| SENP2 | bladder | [106] |
| breast | [107] | |
| GC | [108] | |
| HCC | [109] | |
| MM | [72] | |
| PCa | [110] | |
| SENP3 | AML | [111] |
| breast | [112,113] | |
| CRC | [114,115] | |
| GC | [116] | |
| HCC | [117,118] | |
| HNC | [119] | |
| laryngeal | [120] | |
| OSCC | [121] | |
| osteosarcoma | [122] | |
| ovarian | [123] | |
| SENP5 | breast | [124] |
| HCC | [125,126] | |
| OSCC | [127,128] | |
| osteosarcoma | [129] | |
| SENP6 | AML | [130] |
| B-cell lymphoma | [131] | |
| SENP7 | breast | [26,132] |
| CRC | [133,134] | |
| HCC | [135] | |
| PCa | [136] |
4.2. SENP3 and SENP7
4.3. Other SENP Proteins
5. SUMO Ligases in Cancer
| Families | Proteins | Cancer Types * | References |
|---|---|---|---|
| SP-RING | PIAS1 | APL | [154] |
| breast | [155,156,157,158] | ||
| CRC | [159] | ||
| EC | [160] | ||
| GC | [161] | ||
| GBM | [162] | ||
| lung | [154,163] | ||
| lymphoma | [61,164] | ||
| melanoma, renal | [165] | ||
| MM | [103,166] | ||
| oncoviruses | [167,168,169] | ||
| OSSC | [170] | ||
| PCa | [171,172,173] | ||
| thyroid | [174] | ||
| PIAS2 | breast | [175,176] | |
| GC, HCC, leukemia, ovarian, renal, sarcoma, testicular | [165] | ||
| osteosarcoma | [177] | ||
| PCa | [171] | ||
| thyroid | [174] | ||
| PIAS3 | adrenocortical, mesothelioma, renal, sarcoma | [165] | |
| breast | [176,178,179,180,181,182] | ||
| cervical | [183] | ||
| CRC | [184,185,186] | ||
| esophageal, glioma, lung | [187] | ||
| GC | [169] | ||
| GBM | [162,188,189] | ||
| HCC | [190] | ||
| OSCC | [162,188,189] | ||
| PCa | [171,172,191,192,193] | ||
| PIAS4 | adrenocortical, mesothelioma, thyoma | [165] | |
| breast | [176,194,195] | ||
| CRC | [196] | ||
| GC | [197] | ||
| HCC | [198] | ||
| lung | [199,200] | ||
| ovarian | [201] | ||
| pancreas | [202] | ||
| Others | CBX4 | bladder, cholangiocarcinoma, EC, esophageal, melanoma, mesothelioma, pancreatic, renal, sarcoma, thyoma, thyroid | [165] |
| breast | [203] | ||
| cervical | [204,205] | ||
| CRC | [206] | ||
| GC | [207] | ||
| HCC | [208] | ||
| lung | [209] | ||
| osteosarcoma | [210] | ||
| PCa | [211,212] | ||
| RanBP2 | cervical | [213] | |
| cholangiocarcinoma | [214] | ||
| GBM | [162] | ||
| HCC | [215] | ||
| lung | [216] | ||
| OSCC | [170] | ||
| PCa | [217] | ||
| BCA2 | breast | [47] | |
| ZNF451 | breast | [218] | |
| HCC | [219] | ||
| pancreas | [220] | ||
| PCa | [221] | ||
| TRIM family | APL | [222] | |
| breast | [170,223,224,225] | ||
| cervical | [226] | ||
| CRC | [227,228,229] | ||
| EC | [230] | ||
| esophageal squamous cell | [231] | ||
| GC | [232,233,234,235,236,237] | ||
| HCC | [238] | ||
| lung | [227,239,240,241,242,243] | ||
| ovarian | [244,245,246] | ||
| pancreas | [247] | ||
| PCa | [248] | ||
| renal | [249] | ||
| thyroid | [250] |
5.1. PIAS1
5.2. PIAS2
5.3. PIAS3
5.4. PIAS4
5.5. Other Ligases
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ALT | Alternative lengthening of telomeres |
| AML | Acute myeloid leukemia |
| APL | Acute promyelocytic leukemia |
| AR | Androgen receptor |
| CRC | Colorectal cancer |
| DCs | Dendritic cells |
| DDR | DNA damage response |
| DSBs | Double strand breaks |
| EBV | Epstein-Barr virus |
| EC | Endometrial cancer |
| ER | Estrogen receptor |
| EMT | Epithelial to mesenchymal transition |
| GBM | Glioblastoma |
| GC | Gastric cancer |
| HCC | Hepatocellular carcinoma |
| HDAC | Histone deacetylase |
| HNC | Head and neck cancer |
| KO | Knock out |
| MM | Multiple myeloma |
| MI | Momordin Ic |
| NBs | Nuclear bodies |
| NPC | Nuclear pore complex |
| NSCLC | Non-small-cell lung cancer |
| OGD | Oxygen and glucose deprivation |
| OSCC | Oral squamous cell carcinoma |
| PCa | Prostate cancer |
| PTM | Post-translational modification |
| ROS | Reactive oxygen species |
| SIM | SUMO interacting motif |
| TNBC | Triple-negative breast cancer |
| UBLs | Ubiquitin-like modifiers |
References
- Sahin, U.; de The, H.; Lallemand-Breitenbach, V. Sumoylation in Physiology, Pathology and Therapy. Cells 2022, 11, 814. [Google Scholar] [CrossRef] [PubMed]
- Celen, A.B.; Sahin, U. Sumoylation on its 25th anniversary: Mechanisms, pathology, and emerging concepts. FEBS J. 2020, 287, 3110–3140. [Google Scholar] [CrossRef] [PubMed]
- Hendriks, I.A.; Lyon, D.; Su, D.; Skotte, N.H.; Daniel, J.A.; Jensen, L.J.; Nielsen, M.L. Site-specific characterization of endogenous SUMOylation across species and organs. Nat. Commun. 2018, 9, 2456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hendriks, I.A.; Lyon, D.; Young, C.; Jensen, L.J.; Vertegaal, A.C.; Nielsen, M.L. Site-specific mapping of the human SUMO proteome reveals co-modification with phosphorylation. Nat. Struct. Mol. Biol. 2017, 24, 325–336. [Google Scholar] [CrossRef] [PubMed]
- Hendriks, I.A.; Vertegaal, A.C. A comprehensive compilation of SUMO proteomics. Nat. Rev. Mol. Cell Biol. 2016, 17, 581–595. [Google Scholar] [CrossRef]
- Bernstock, J.D.; Yang, W.; Ye, D.G.; Shen, Y.; Pluchino, S.; Lee, Y.J.; Hallenbeck, J.M.; Paschen, W. SUMOylation in brain ischemia: Patterns, targets, and translational implications. J. Cereb. Blood Flow Metab. 2018, 38, 5–16. [Google Scholar] [CrossRef] [Green Version]
- Enserink, J.M. Sumo and the cellular stress response. Cell Div. 2015, 10, 4. [Google Scholar] [CrossRef] [Green Version]
- Niskanen, E.A.; Palvimo, J.J. Chromatin SUMOylation in heat stress: To protect, pause and organise?: SUMO stress response on chromatin. Bioessays 2017, 39, 1600263. [Google Scholar] [CrossRef]
- Mahajan, R.; Delphin, C.; Guan, T.; Gerace, L.; Melchior, F. A small ubiquitin-related polypeptide involved in targeting RanGAP1 to nuclear pore complex protein RanBP2. Cell 1997, 88, 97–107. [Google Scholar] [CrossRef] [Green Version]
- Matunis, M.J.; Coutavas, E.; Blobel, G. A novel ubiquitin-like modification modulates the partitioning of the Ran-GTPase-activating protein RanGAP1 between the cytosol and the nuclear pore complex. J. Cell Biol. 1996, 135, 1457–1470. [Google Scholar] [CrossRef]
- Lascorz, J.; Codina-Fabra, J.; Reverter, D.; Torres-Rosell, J. SUMO-SIM interactions: From structure to biological functions. Semin. Cell Dev. Biol. 2021, S1084-9521(21)00283-4. [Google Scholar] [CrossRef] [PubMed]
- Yau, T.Y.; Sander, W.; Eidson, C.; Courey, A.J. SUMO Interacting Motifs: Structure and Function. Cells 2021, 10, 2825. [Google Scholar] [CrossRef] [PubMed]
- Sriramachandran, A.M.; Dohmen, R.J. SUMO-targeted ubiquitin ligases. Biochim. Biophys. Acta 2014, 1843, 75–85. [Google Scholar] [CrossRef] [Green Version]
- Sahin, U.; Ferhi, O.; Jeanne, M.; Benhenda, S.; Berthier, C.; Jollivet, F.; Niwa-Kawakita, M.; Faklaris, O.; Setterblad, N.; de The, H.; et al. Oxidative stress-induced assembly of PML nuclear bodies controls sumoylation of partner proteins. J. Cell Biol. 2014, 204, 931–945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jentsch, S.; Psakhye, I. Control of nuclear activities by substrate-selective and protein-group SUMOylation. Annu. Rev. Genet. 2013, 47, 167–186. [Google Scholar] [CrossRef]
- Hay, R.T. SUMO: A history of modification. Mol. Cell 2005, 18, 1–12. [Google Scholar] [CrossRef]
- Nacerddine, K.; Lehembre, F.; Bhaumik, M.; Artus, J.; Cohen-Tannoudji, M.; Babinet, C.; Pandolfi, P.P.; Dejean, A. The SUMO pathway is essential for nuclear integrity and chromosome segregation in mice. Dev. Cell 2005, 9, 769–779. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Gutierrez, P.; Garcia-Dominguez, M. SUMO control of nervous system development. Semin. Cell Dev. Biol. 2021, S1084-9521(21)00304-9. [Google Scholar] [CrossRef]
- Wang, L.; Wansleeben, C.; Zhao, S.; Miao, P.; Paschen, W.; Yang, W. SUMO2 is essential while SUMO3 is dispensable for mouse embryonic development. EMBO Rep. 2014, 15, 878–885. [Google Scholar] [CrossRef] [Green Version]
- Chymkowitch, P.; Nguea, P.A.; Enserink, J.M. SUMO-regulated transcription: Challenging the dogma. Bioessays 2015, 37, 1095–1105. [Google Scholar] [CrossRef]
- Kunz, K.; Piller, T.; Muller, S. SUMO-specific proteases and isopeptidases of the SENP family at a glance. J. Cell Sci. 2018, 131, jcs211904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, E.J.; Shin, H.M.; Nam, E.; Kim, W.S.; Kim, J.H.; Oh, B.H.; Yun, Y. DeSUMOylating isopeptidase: A second class of SUMO protease. EMBO Rep. 2012, 13, 339–346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schulz, S.; Chachami, G.; Kozaczkiewicz, L.; Winter, U.; Stankovic-Valentin, N.; Haas, P.; Hofmann, K.; Urlaub, H.; Ovaa, H.; Wittbrodt, J.; et al. Ubiquitin-specific protease-like 1 (USPL1) is a SUMO isopeptidase with essential, non-catalytic functions. EMBO Rep. 2012, 13, 930–938. [Google Scholar] [CrossRef] [Green Version]
- Cortes-Montero, E.; Rodriguez-Munoz, M.; Sanchez-Blazquez, P.; Garzon, J. The Axonal Motor Neuropathy-Related HINT1 Protein Is a Zinc- and Calmodulin-Regulated Cysteine SUMO Protease. Antioxid. Redox Signal. 2019, 31, 503–520. [Google Scholar] [CrossRef] [PubMed]
- Romeo, K.; Louault, Y.; Cantaloube, S.; Loiodice, I.; Almouzni, G.; Quivy, J.P. The SENP7 SUMO-Protease Presents a Module of Two HP1 Interaction Motifs that Locks HP1 Protein at Pericentric Heterochromatin. Cell Rep. 2015, 10, 771–782. [Google Scholar] [CrossRef] [PubMed]
- Bawa-Khalfe, T.; Lu, L.S.; Zuo, Y.; Huang, C.; Dere, R.; Lin, F.M.; Yeh, E.T. Differential expression of SUMO-specific protease 7 variants regulates epithelial-mesenchymal transition. Proc. Natl. Acad. Sci. USA 2012, 109, 17466–17471. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.J.; Wu, D.; Khan, F.A.; Huo, L.J. DeSUMOylation: An Important Therapeutic Target and Protein Regulatory Event. DNA Cell Biol. 2015, 34, 652–660. [Google Scholar] [CrossRef]
- Salas-Lloret, D.; Gonzalez-Prieto, R. Insights in Post-Translational Modifications: Ubiquitin and SUMO. Int. J. Mol. Sci. 2022, 23, 3281. [Google Scholar] [CrossRef]
- Flotho, A.; Melchior, F. Sumoylation: A regulatory protein modification in health and disease. Annu. Rev. Biochem. 2013, 82, 357–385. [Google Scholar] [CrossRef]
- Rabellino, A.; Andreani, C.; Scaglioni, P.P. The Role of PIAS SUMO E3-Ligases in Cancer. Cancer Res. 2017, 77, 1542–1547. [Google Scholar] [CrossRef] [Green Version]
- Shi, X.; Du, Y.; Li, S.; Wu, H. The Role of SUMO E3 Ligases in Signaling Pathway of Cancer Cells. Int. J. Mol. Sci. 2022, 23, 3639. [Google Scholar] [CrossRef] [PubMed]
- Pichler, A.; Knipscheer, P.; Saitoh, H.; Sixma, T.K.; Melchior, F. The RanBP2 SUMO E3 ligase is neither HECT- nor RING-type. Nat. Struct. Mol. Biol. 2004, 11, 984–991. [Google Scholar] [CrossRef] [PubMed]
- Reverter, D.; Lima, C.D. Insights into E3 ligase activity revealed by a SUMO-RanGAP1-Ubc9-Nup358 complex. Nature 2005, 435, 687–692. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, Y.; Kikuchi, Y. Yeast PIAS-type Ull1/Siz1 is composed of SUMO ligase and regulatory domains. J. Biol. Chem. 2005, 280, 35822–35828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Dominguez, M.; March-Diaz, R.; Reyes, J.C. The PHD domain of plant PIAS proteins mediates sumoylation of bromodomain GTE proteins. J. Biol. Chem. 2008, 283, 21469–21477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ivanov, A.V.; Peng, H.; Yurchenko, V.; Yap, K.L.; Negorev, D.G.; Schultz, D.C.; Psulkowski, E.; Fredericks, W.J.; White, D.E.; Maul, G.G.; et al. PHD domain-mediated E3 ligase activity directs intramolecular sumoylation of an adjacent bromodomain required for gene silencing. Mol. Cell 2007, 28, 823–837. [Google Scholar] [CrossRef] [Green Version]
- Chu, Y.; Yang, X. SUMO E3 ligase activity of TRIM proteins. Oncogene 2011, 30, 1108–1116. [Google Scholar] [CrossRef] [Green Version]
- Takayama, K.I.; Suzuki, T.; Tanaka, T.; Fujimura, T.; Takahashi, S.; Urano, T.; Ikeda, K.; Inoue, S. TRIM25 enhances cell growth and cell survival by modulating p53 signals via interaction with G3BP2 in prostate cancer. Oncogene 2018, 37, 2165–2180. [Google Scholar] [CrossRef]
- Cappadocia, L.; Pichler, A.; Lima, C.D. Structural basis for catalytic activation by the human ZNF451 SUMO E3 ligase. Nat. Struct. Mol. Biol. 2015, 22, 968–975. [Google Scholar] [CrossRef] [Green Version]
- Eisenhardt, N.; Chaugule, V.K.; Koidl, S.; Droescher, M.; Dogan, E.; Rettich, J.; Sutinen, P.; Imanishi, S.Y.; Hofmann, K.; Palvimo, J.J.; et al. A new vertebrate SUMO enzyme family reveals insights into SUMO-chain assembly. Nat. Struct. Mol. Biol. 2015, 22, 959–967. [Google Scholar] [CrossRef]
- Hannoun, Z.; Maarifi, G.; Chelbi-Alix, M.K. The implication of SUMO in intrinsic and innate immunity. Cytokine Growth Factor Rev. 2016, 29, 3–16. [Google Scholar] [CrossRef]
- Hu, M.M.; Yang, Q.; Xie, X.Q.; Liao, C.Y.; Lin, H.; Liu, T.T.; Yin, L.; Shu, H.B. Sumoylation Promotes the Stability of the DNA Sensor cGAS and the Adaptor STING to Regulate the Kinetics of Response to DNA Virus. Immunity 2016, 45, 555–569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ikeuchi, Y.; Dadakhujaev, S.; Chandhoke, A.S.; Huynh, M.A.; Oldenborg, A.; Ikeuchi, M.; Deng, L.; Bennett, E.J.; Harper, J.W.; Bonni, A.; et al. TIF1gamma protein regulates epithelial-mesenchymal transition by operating as a small ubiquitin-like modifier (SUMO) E3 ligase for the transcriptional regulator SnoN1. J. Biol. Chem. 2014, 289, 25067–25078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kung, C.P.; Khaku, S.; Jennis, M.; Zhou, Y.; Murphy, M.E. Identification of TRIML2, a novel p53 target, that enhances p53 SUMOylation and regulates the transactivation of proapoptotic genes. Mol. Cancer Res. 2015, 13, 250–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, G.; Harischandra, D.S.; Ghaisas, S.; Zhang, P.; Prall, W.; Huang, L.; Maghames, C.; Guo, L.; Luna, E.; Mack, K.L.; et al. TRIM11 Prevents and Reverses Protein Aggregation and Rescues a Mouse Model of Parkinson’s Disease. Cell Rep. 2020, 33, 108418. [Google Scholar] [CrossRef]
- Gregoire, S.; Yang, X.J. Association with class IIa histone deacetylases upregulates the sumoylation of MEF2 transcription factors. Mol. Cell. Biol. 2005, 25, 2273–2287. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y.; Castro-Gonzalez, S.; Chen, Y.; Serra-Moreno, R. Effects of the SUMO Ligase BCA2 on Metabolic Activity, Cell Proliferation, Cell Migration, Cell Cycle, and the Regulation of NF-kappaB and IRF1 in Different Breast Epithelial Cellular Contexts. Front. Cell Dev. Biol. 2021, 9, 711481. [Google Scholar] [CrossRef]
- Stindt, M.H.; Carter, S.; Vigneron, A.M.; Ryan, K.M.; Vousden, K.H. MDM2 promotes SUMO-2/3 modification of p53 to modulate transcriptional activity. Cell Cycle 2011, 10, 3176–3188. [Google Scholar] [CrossRef] [Green Version]
- Talamillo, A.; Barroso-Gomila, O.; Giordano, I.; Ajuria, L.; Grillo, M.; Mayor, U.; Barrio, R. The role of SUMOylation during development. Biochem. Soc. Trans. 2020, 48, 463–478. [Google Scholar] [CrossRef] [Green Version]
- Kunz, K.; Wagner, K.; Mendler, L.; Holper, S.; Dehne, N.; Muller, S. SUMO Signaling by Hypoxic Inactivation of SUMO-Specific Isopeptidases. Cell Rep. 2016, 16, 3075–3086. [Google Scholar] [CrossRef] [Green Version]
- Henley, J.M.; Craig, T.J.; Wilkinson, K.A. Neuronal SUMOylation: Mechanisms, physiology, and roles in neuronal dysfunction. Physiol. Rev. 2014, 94, 1249–1285. [Google Scholar] [CrossRef] [PubMed]
- Stabell, M.; Saether, T.; Rohr, A.K.; Gabrielsen, O.S.; Myklebost, O. Methylation-dependent SUMOylation of the architectural transcription factor HMGA2. Biochem. Biophys. Res. Commun. 2021, 552, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Eifler, K.; Vertegaal, A.C.O. SUMOylation-Mediated Regulation of Cell Cycle Progression and Cancer. Trends Biochem. Sci. 2015, 40, 779–793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seeler, J.S.; Dejean, A. SUMO and the robustness of cancer. Nat. Rev. Cancer 2017, 17, 184–197. [Google Scholar] [CrossRef] [PubMed]
- de The, H.; Pandolfi, P.P.; Chen, Z. Acute Promyelocytic Leukemia: A Paradigm for Oncoprotein-Targeted Cure. Cancer Cell 2017, 32, 552–560. [Google Scholar] [CrossRef] [Green Version]
- Han, Z.J.; Feng, Y.H.; Gu, B.H.; Li, Y.M.; Chen, H. The post-translational modification, SUMOylation, and cancer (Review). Int. J. Oncol. 2018, 52, 1081–1094. [Google Scholar] [CrossRef] [Green Version]
- Kroonen, J.S.; Vertegaal, A.C.O. Targeting SUMO Signaling to Wrestle Cancer. Trends Cancer 2021, 7, 496–510. [Google Scholar] [CrossRef]
- Kukkula, A.; Ojala, V.K.; Mendez, L.M.; Sistonen, L.; Elenius, K.; Sundvall, M. Therapeutic Potential of Targeting the SUMO Pathway in Cancer. Cancers 2021, 13, 4402. [Google Scholar] [CrossRef]
- Gonzalez-Prieto, R.; Cuijpers, S.A.; Kumar, R.; Hendriks, I.A.; Vertegaal, A.C. c-Myc is targeted to the proteasome for degradation in a SUMOylation-dependent manner, regulated by PIAS1, SENP7 and RNF4. Cell Cycle 2015, 14, 1859–1872. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.X.; Chen, Y.; Su, Y.; Wang, X.; Chauhan, K.M.; Liang, J.; Daniel, C.J.; Sears, R.C.; Dai, M.S. SUMO protease SENP1 deSUMOylates and stabilizes c-Myc. Proc. Natl. Acad. Sci. USA 2018, 115, 10983–10988. [Google Scholar] [CrossRef] [Green Version]
- Hoellein, A.; Fallahi, M.; Schoeffmann, S.; Steidle, S.; Schaub, F.X.; Rudelius, M.; Laitinen, I.; Nilsson, L.; Goga, A.; Peschel, C.; et al. Myc-induced SUMOylation is a therapeutic vulnerability for B-cell lymphoma. Blood 2014, 124, 2081–2090. [Google Scholar] [CrossRef] [Green Version]
- Kessler, J.D.; Kahle, K.T.; Sun, T.; Meerbrey, K.L.; Schlabach, M.R.; Schmitt, E.M.; Skinner, S.O.; Xu, Q.; Li, M.Z.; Hartman, Z.C.; et al. A SUMOylation-dependent transcriptional subprogram is required for Myc-driven tumorigenesis. Science 2012, 335, 348–353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, B.; Sun, Y.; Huang, J. Overexpression of SKI oncoprotein leads to p53 degradation through regulation of MDM2 protein sumoylation. J. Biol. Chem. 2012, 287, 14621–14630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pfeiffer, A.; Luijsterburg, M.S.; Acs, K.; Wiegant, W.W.; Helfricht, A.; Herzog, L.K.; Minoia, M.; Bottcher, C.; Salomons, F.A.; van Attikum, H.; et al. Ataxin-3 consolidates the MDC1-dependent DNA double-strand break response by counteracting the SUMO-targeted ubiquitin ligase RNF4. EMBO J. 2017, 36, 1066–1083. [Google Scholar] [CrossRef] [PubMed]
- Xie, M.; Yu, J.; Ge, S.; Huang, J.; Fan, X. SUMOylation homeostasis in tumorigenesis. Cancer Lett. 2020, 469, 301–309. [Google Scholar] [CrossRef]
- Garvin, A.J.; Densham, R.M.; Blair-Reid, S.A.; Pratt, K.M.; Stone, H.R.; Weekes, D.; Lawrence, K.J.; Morris, J.R. The deSUMOylase SENP7 promotes chromatin relaxation for homologous recombination DNA repair. EMBO Rep. 2013, 14, 975–983. [Google Scholar] [CrossRef]
- Guervilly, J.H.; Takedachi, A.; Naim, V.; Scaglione, S.; Chawhan, C.; Lovera, Y.; Despras, E.; Kuraoka, I.; Kannouche, P.; Rosselli, F.; et al. The SLX4 complex is a SUMO E3 ligase that impacts on replication stress outcome and genome stability. Mol. Cell 2015, 57, 123–137. [Google Scholar] [CrossRef] [Green Version]
- Tokarz, P.; Wozniak, K. SENP Proteases as Potential Targets for Cancer Therapy. Cancers 2021, 13, 2059. [Google Scholar] [CrossRef]
- Wei, B.; Huang, C.; Liu, B.; Wang, Y.; Xia, N.; Fan, Q.; Chen, G.Q.; Cheng, J. Mitotic Phosphorylation of SENP3 Regulates DeSUMOylation of Chromosome-Associated Proteins and Chromosome Stability. Cancer Res. 2018, 78, 2171–2178. [Google Scholar] [CrossRef] [Green Version]
- Long, X.; Zhao, B.; Lu, W.; Chen, X.; Yang, X.; Huang, J.; Zhang, Y.; An, S.; Qin, Y.; Xing, Z.; et al. The Critical Roles of the SUMO-Specific Protease SENP3 in Human Diseases and Clinical Implications. Front. Physiol. 2020, 11, 558220. [Google Scholar] [CrossRef]
- Adorisio, S.; Fierabracci, A.; Muscari, I.; Liberati, A.M.; Ayroldi, E.; Migliorati, G.; Thuy, T.T.; Riccardi, C.; Delfino, D.V. SUMO proteins: Guardians of immune system. J. Autoimmun. 2017, 84, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Xie, H.; Gu, Y.; Wang, W.; Wang, X.; Ye, X.; Xin, C.; Lu, M.; Reddy, B.A.; Shu, P. Silencing of SENP2 in Multiple Myeloma Induces Bortezomib Resistance by Activating NF-kappaB Through the Modulation of IkappaBalpha Sumoylation. Sci. Rep. 2020, 10, 766. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Yu, H.; Zheng, X.; Peng, R.; Wang, Q.; Zhou, Y.; Wang, R.; Wang, J.; Qu, B.; Shen, N.; et al. SENP7 Potentiates cGAS Activation by Relieving SUMO-Mediated Inhibition of Cytosolic DNA Sensing. PLoS Pathog. 2017, 13, e1006156. [Google Scholar] [CrossRef] [PubMed]
- Barry, R.; John, S.W.; Liccardi, G.; Tenev, T.; Jaco, I.; Chen, C.H.; Choi, J.; Kasperkiewicz, P.; Fernandes-Alnemri, T.; Alnemri, E.; et al. SUMO-mediated regulation of NLRP3 modulates inflammasome activity. Nat. Commun. 2018, 9, 3001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Jiao, F.; Hong, J.; Yang, F.; Wang, L.; Gong, Z. SENP7 knockdown inhibited pyroptosis and NF-kappaB/NLRP3 inflammasome pathway activation in Raw 264.7 cells. Sci. Rep. 2020, 10, 16265. [Google Scholar] [CrossRef]
- Mattoscio, D.; Medda, A.; Chiocca, S. Recent Highlights: Onco Viral Exploitation of the SUMO System. Curr. Issues Mol. Biol. 2020, 35, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Ryu, H.Y.; Hochstrasser, M. Histone sumoylation and chromatin dynamics. Nucleic Acids Res. 2021, 49, 6043–6052. [Google Scholar] [CrossRef]
- Cossec, J.C.; Theurillat, I.; Chica, C.; Bua Aguin, S.; Gaume, X.; Andrieux, A.; Iturbide, A.; Jouvion, G.; Li, H.; Bossis, G.; et al. SUMO Safeguards Somatic and Pluripotent Cell Identities by Enforcing Distinct Chromatin States. Cell Stem Cell 2018, 23, 742–757.e8. [Google Scholar] [CrossRef] [Green Version]
- Theurillat, I.; Hendriks, I.A.; Cossec, J.C.; Andrieux, A.; Nielsen, M.L.; Dejean, A. Extensive SUMO Modification of Repressive Chromatin Factors Distinguishes Pluripotent from Somatic Cells. Cell Rep. 2020, 32, 108146. [Google Scholar] [CrossRef]
- Juarez-Vicente, F.; Luna-Pelaez, N.; Garcia-Dominguez, M. The Sumo protease Senp7 is required for proper neuronal differentiation. Biochim. Biophys. Acta 2016, 1863, 1490–1498. [Google Scholar] [CrossRef]
- Zaidan, N.Z.; Walker, K.J.; Brown, J.E.; Schaffer, L.V.; Scalf, M.; Shortreed, M.R.; Iyer, G.; Smith, L.M.; Sridharan, R. Compartmentalization of HP1 Proteins in Pluripotency Acquisition and Maintenance. Stem Cell Rep. 2018, 10, 627–641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindenmann, U.; Brand, M.; Gall, F.; Frasson, D.; Hunziker, L.; Kroslakova, I.; Sievers, M.; Riedl, R. Discovery of a Class of Potent and Selective Non-competitive Sentrin-Specific Protease 1 Inhibitors. ChemMedChem 2020, 15, 675–679. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Bian, Z.; Liu, B.; Zhang, Y.; Cao, Y.; Cui, K.; Sun, S.; Li, J.; Zhang, J.; Wang, X.; et al. Long noncoding RNA MCM3AP-AS1 enhances cell proliferation and metastasis in colorectal cancer by regulating miR-193a-5p/SENP1. Cancer Med. 2021, 10, 2470–2481. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.H.; Chang, C.C.; Lee, T.H.; Luo, M.; Huang, P.; Liao, P.H.; Wei, S.; Li, F.A.; Chen, R.H.; Zhou, X.Z.; et al. SENP1 deSUMOylates and regulates Pin1 protein activity and cellular function. Cancer Res. 2013, 73, 3951–3962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.; Tang, Y.; Guo, W.; Du, Y.; Wang, Y.; Li, P.; Zang, W.; Yin, X.; Wang, H.; Chu, H.; et al. Up-regulation of microRNA-138 induce radiosensitization in lung cancer cells. Tumour Biol. 2014, 35, 6557–6565. [Google Scholar] [CrossRef]
- Jacques, C.; Baris, O.; Prunier-Mirebeau, D.; Savagner, F.; Rodien, P.; Rohmer, V.; Franc, B.; Guyetant, S.; Malthiery, Y.; Reynier, P. Two-step differential expression analysis reveals a new set of genes involved in thyroid oncocytic tumors. J. Clin. Endocrinol. Metab. 2005, 90, 2314–2320. [Google Scholar] [CrossRef] [Green Version]
- Bouchard, D.M.; Matunis, M.J. A cellular and bioinformatics analysis of the SENP1 SUMO isopeptidase in pancreatic cancer. J. Gastrointest. Oncol. 2019, 10, 821–830. [Google Scholar] [CrossRef]
- Cheng, J.; Perkins, N.D.; Yeh, E.T. Differential regulation of c-Jun-dependent transcription by SUMO-specific proteases. J. Biol. Chem. 2005, 280, 14492–14498. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Yan, S.; Ding, J.; Yu, T.T.; Cheng, S.Y. DeSUMOylation of Gli1 by SENP1 Attenuates Sonic Hedgehog Signaling. Mol. Cell. Biol. 2017, 37, e00579-16. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Jin, J.; Zhang, J.; Wang, L.; Cao, J. Depletion of SENP1 suppresses the proliferation and invasion of triple-negative breast cancer cells. Oncol. Rep. 2016, 36, 2071–2078. [Google Scholar] [CrossRef] [Green Version]
- Xiang-Ming, Y.; Zhi-Qiang, X.; Ting, Z.; Jian, W.; Jian, P.; Li-Qun, Y.; Ming-Cui, F.; Hong-Liang, X.; Xu, C.; Yun, Z. SENP1 regulates cell migration and invasion in neuroblastoma. Biotechnol. Appl. Biochem. 2016, 63, 435–440. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, K.M.; Chen, Y.; Chen, Y.; Liu, A.T.; Sun, X.X.; Dai, M.S. The SUMO-specific protease SENP1 deSUMOylates p53 and regulates its activity. J. Cell. Biochem. 2021, 122, 189–197. [Google Scholar] [CrossRef] [PubMed]
- Tao, Y.; Li, R.; Shen, C.; Li, J.; Zhang, Q.; Ma, Z.; Wang, F.; Wang, Z. SENP1 is a crucial promotor for hepatocellular carcinoma through deSUMOylation of UBE2T. Aging 2020, 12, 1563–1576. [Google Scholar] [CrossRef] [PubMed]
- Patra, K.C.; Wang, Q.; Bhaskar, P.T.; Miller, L.; Wang, Z.; Wheaton, W.; Chandel, N.; Laakso, M.; Muller, W.J.; Allen, E.L.; et al. Hexokinase 2 is required for tumor initiation and maintenance and its systemic deletion is therapeutic in mouse models of cancer. Cancer Cell 2013, 24, 213–228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeWaal, D.; Nogueira, V.; Terry, A.R.; Patra, K.C.; Jeon, S.M.; Guzman, G.; Au, J.; Long, C.P.; Antoniewicz, M.R.; Hay, N. Hexokinase-2 depletion inhibits glycolysis and induces oxidative phosphorylation in hepatocellular carcinoma and sensitizes to metformin. Nat. Commun. 2018, 9, 446. [Google Scholar] [CrossRef] [PubMed]
- Nogueira, V.; Patra, K.C.; Hay, N. Selective eradication of cancer displaying hyperactive Akt by exploiting the metabolic consequences of Akt activation. eLife 2018, 7, e32213. [Google Scholar] [CrossRef]
- Shangguan, X.; He, J.; Ma, Z.; Zhang, W.; Ji, Y.; Shen, K.; Yue, Z.; Li, W.; Xin, Z.; Zheng, Q.; et al. SUMOylation controls the binding of hexokinase 2 to mitochondria and protects against prostate cancer tumorigenesis. Nat. Commun. 2021, 12, 1812. [Google Scholar] [CrossRef]
- Chen, M.C.; Nhan, D.C.; Hsu, C.H.; Wang, T.F.; Li, C.C.; Ho, T.J.; Mahalakshmi, B.; Chen, M.C.; Yang, L.Y.; Huang, C.Y. SENP1 participates in Irinotecan resistance in human colon cancer cells. J. Cell. Biochem. 2021, 122, 1277–1294. [Google Scholar] [CrossRef]
- Fang, G.; Chen, T.; Mao, R.; Huang, X.; Ji, L. Circular RNA circ_0089153 acts as a competing endogenous RNA to regulate colorectal cancer development by the miR-198/SUMO-specific peptidase 1 (SENP1) axis. Bioengineered 2021, 12, 5664–5678. [Google Scholar] [CrossRef]
- Gao, Q.; Wang, S.; Zhang, Z. E3 ubiquitin ligase SMURF2 prevents colorectal cancer by reducing the stability of the YY1 protein and inhibiting the SENP1/c-myc axis. Gene Ther. 2021; Epub ahead of print. [Google Scholar] [CrossRef]
- Xu, Y.; Li, J.; Zuo, Y.; Deng, J.; Wang, L.S.; Chen, G.Q. SUMO-specific protease 1 regulates the in vitro and in vivo growth of colon cancer cells with the upregulated expression of CDK inhibitors. Cancer Lett. 2011, 309, 78–84. [Google Scholar] [CrossRef]
- Zhou, G.Q.; Han, F.; Shi, Z.L.; Yu, L.; Li, X.F.; Yu, C.; Shen, C.L.; Wan, D.W.; Zhu, X.G.; Li, R.; et al. miR-133a-3p Targets SUMO-Specific Protease 1 to Inhibit Cell Proliferation and Cell Cycle Progress in Colorectal Cancer. Oncol. Res. 2018, 26, 795–800. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Sun, H.Y.; Xiao, F.J.; Wang, H.; Yang, Y.; Wang, L.; Gao, C.J.; Guo, Z.K.; Wu, C.T.; Wang, L.S. SENP1 inhibition induces apoptosis and growth arrest of multiple myeloma cells through modulation of NF-kappaB signaling. Biochem. Biophys. Res. Commun. 2015, 460, 409–415. [Google Scholar] [CrossRef] [PubMed]
- Ao, Q.; Su, W.; Guo, S.; Cai, L.; Huang, L. SENP1 desensitizes hypoxic ovarian cancer cells to cisplatin by up-regulating HIF-1alpha. Sci. Rep. 2015, 5, 16396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.; Lei, H.; Zhang, J.; Chen, X.; Tang, C.; Wang, W.; Xu, H.; Xiao, W.; Gu, W.; Wu, Y. Momordin Ic, a new natural SENP1 inhibitor, inhibits prostate cancer cell proliferation. Oncotarget 2016, 7, 58995–59005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, M.; Zhang, D.; Zhang, E.; Xu, D.; Liu, Z.; Qiu, J.; Fan, Y.; Shen, B. SENP2 suppresses epithelial-mesenchymal transition of bladder cancer cells through deSUMOylation of TGF-betaRI. Mol. Carcinog. 2017, 56, 2332–2341. [Google Scholar] [CrossRef]
- Gao, X.; Wu, Y.; Qiao, L.; Feng, X. SENP2 suppresses NF-kappaB activation and sensitizes breast cancer cells to doxorubicin. Eur. J. Pharmacol. 2019, 854, 179–186. [Google Scholar] [CrossRef]
- Hu, X.Y.; Liu, Z.; Zhang, K.L.; Feng, J.; Liu, X.F.; Wang, L.Y.; Wang, Z.W. SUMO-specific protease 2-mediated deSUMOylation is required for NDRG2 stabilization in gastric cancer cells. Cancer Biomark. 2017, 21, 195–201. [Google Scholar] [CrossRef]
- Tang, X.; Liu, B.; Zhang, C.; Tang, W.; Liang, S.; Xiao, Y.; Deng, R.; Li, Z. SENP2 Reduces Hepatocellular Carcinoma Stemness and Improves Sorafenib Sensitivity Through Inactivating the AKT/GSK3beta/CTNNB1 Pathway. Front. Oncol. 2021, 11, 773045. [Google Scholar] [CrossRef]
- Erazo, T.; Espinosa-Gil, S.; Dieguez-Martinez, N.; Gomez, N.; Lizcano, J.M. SUMOylation Is Required for ERK5 Nuclear Translocation and ERK5-Mediated Cancer Cell Proliferation. Int. J. Mol. Sci. 2020, 21, 2203. [Google Scholar] [CrossRef] [Green Version]
- Xu, R.; Yu, S.; Zhu, D.; Huang, X.; Xu, Y.; Lao, Y.; Tian, Y.; Zhang, J.; Tang, Z.; Zhang, Z.; et al. hCINAP regulates the DNA-damage response and mediates the resistance of acute myelocytic leukemia cells to therapy. Nat. Commun. 2019, 10, 3812. [Google Scholar] [CrossRef]
- Graves, J.D.; Lee, Y.J.; Liu, K.; Li, G.; Lin, F.T.; Lin, W.C. E2F1 sumoylation as a protective cellular mechanism in oxidative stress response. Proc. Natl. Acad. Sci. USA 2020, 117, 14958–14969. [Google Scholar] [CrossRef] [PubMed]
- Xiao, M.; Bian, Q.; Lao, Y.; Yi, J.; Sun, X.; Sun, X.; Yang, J. SENP3 loss promotes M2 macrophage polarization and breast cancer progression. Mol. Oncol. 2022, 16, 1026–1044. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Huang, C.; Sun, X.; Xiang, B.; Wang, M.; Yeh, E.T.; Chen, Y.; Li, H.; Shi, G.; Cang, H.; et al. SENP3-mediated de-conjugation of SUMO2/3 from promyelocytic leukemia is correlated with accelerated cell proliferation under mild oxidative stress. J. Biol. Chem. 2010, 285, 12906–12915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Z.; Teng, X.L.; Zhang, T.; Yu, X.; Ding, R.; Yi, J.; Deng, L.; Wang, Z.; Zou, Q. SENP3 senses oxidative stress to facilitate STING-dependent dendritic cell antitumor function. Mol. Cell 2021, 81, 940–952.e5. [Google Scholar] [CrossRef] [PubMed]
- Ren, Y.H.; Liu, K.J.; Wang, M.; Yu, Y.N.; Yang, K.; Chen, Q.; Yu, B.; Wang, W.; Li, Q.W.; Wang, J.; et al. De-SUMOylation of FOXC2 by SENP3 promotes the epithelial-mesenchymal transition in gastric cancer cells. Oncotarget 2014, 5, 7093–7104. [Google Scholar] [CrossRef] [Green Version]
- Wang, P.; Xue, N.; Zhang, C.; Shan, S.; Jiang, Z.; Wu, W.; Liu, X. Inhibition of SUMO2/3 antagonizes isoflurane-induced cancer-promoting effect in hepatocellular carcinoma Hep3B cells. Oncol. Lett. 2021, 21, 274. [Google Scholar] [CrossRef]
- Zhao, Y.; Yang, B.; Chen, D.; Zhou, X.; Wang, M.; Jiang, J.; Wei, L.; Chen, Z. Combined identification of ARID1A, CSMD1, and SENP3 as effective prognostic biomarkers for hepatocellular carcinoma. Aging 2021, 13, 4696–4712. [Google Scholar] [CrossRef]
- Zhou, Z.; Wang, M.; Li, J.; Xiao, M.; Chin, Y.E.; Cheng, J.; Yeh, E.T.; Yang, J.; Yi, J. SUMOylation and SENP3 regulate STAT3 activation in head and neck cancer. Oncogene 2016, 35, 5826–5838. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Z.; Xu, J.; Bao, X.; Shi, J.; Liu, B.; Chen, Y.; Li, J. Nuclear Nrf2 Activity in Laryngeal Carcinoma is Regulated by SENP3 After Cisplatin-Induced Reactive Oxygen Species Stress. J. Cancer 2019, 10, 3427–3434. [Google Scholar] [CrossRef] [Green Version]
- Sun, Z.; Hu, S.; Luo, Q.; Ye, D.; Hu, D.; Chen, F. Overexpression of SENP3 in oral squamous cell carcinoma and its association with differentiation. Oncol. Rep. 2013, 29, 1701–1706. [Google Scholar] [CrossRef] [Green Version]
- Yang, P.; Liu, Y.; Qi, Y.C.; Lian, Z.H. High SENP3 Expression Promotes Cell Migration, Invasion, and Proliferation by Modulating DNA Methylation of E-Cadherin in Osteosarcoma. Technol. Cancer Res. Treat. 2020, 19, 1533033820956988. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Su, M.; Jin, Y.; Xi, Q.; Deng, Y.; Chen, J.; Wang, W.; Chen, Y.; Chen, L.; Shi, N.; et al. Upregulation of SENP3/SMT3IP1 promotes epithelial ovarian cancer progression and forecasts poor prognosis. Tumour Biol. 2017, 39, 1010428317694543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cashman, R.; Cohen, H.; Ben-Hamo, R.; Zilberberg, A.; Efroni, S. SENP5 mediates breast cancer invasion via a TGFbetaRI SUMOylation cascade. Oncotarget 2014, 5, 1071–1082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, Z.L.; Pei, H.; Xu, Y.H.; Yu, J.; Deng, T. The SUMO-specific protease SENP5 controls DNA damage response and promotes tumorigenesis in hepatocellular carcinoma. Eur. Rev. Med. Pharmacol. Sci. 2016, 20, 3566–3573. [Google Scholar] [PubMed]
- Zhang, C.Y.; Jiang, Z.M.; Ma, X.F.; Li, Y.; Liu, X.Z.; Li, L.L.; Wu, W.H.; Wang, T. Saikosaponin-d Inhibits the Hepatoma Cells and Enhances Chemosensitivity Through SENP5-Dependent Inhibition of Gli1 SUMOylation Under Hypoxia. Front. Pharmacol. 2019, 10, 1039. [Google Scholar] [CrossRef]
- Cheng, Y.; Guo, X.; Gong, Y.; Ding, X.; Yu, Y. Sentrin/small ubiquitin-like modifier-specific protease 5 protects oral cancer cells from oxidative stress-induced apoptosis. Mol. Med. Rep. 2015, 12, 2009–2014. [Google Scholar] [CrossRef] [Green Version]
- Ding, X.; Sun, J.; Wang, L.; Li, G.; Shen, Y.; Zhou, X.; Chen, W. Overexpression of SENP5 in oral squamous cell carcinoma and its association with differentiation. Oncol. Rep. 2008, 20, 1041–1045. [Google Scholar]
- Wang, K.; Zhang, X.C. Inhibition of SENP5 suppresses cell growth and promotes apoptosis in osteosarcoma cells. Exp. Ther. Med. 2014, 7, 1691–1695. [Google Scholar] [CrossRef] [Green Version]
- Shi, L.; Huang, Y.; Huang, X.; Zhou, W.; Wei, J.; Deng, D.; Lai, Y. Analyzing the key gene expression and prognostics values for acute myeloid leukemia. Transl. Cancer Res. 2020, 9, 7284–7298. [Google Scholar] [CrossRef]
- Schick, M.; Zhang, L.; Maurer, S.; Maurer, H.C.; Isaakaidis, K.; Schneider, L.; Patra, U.; Schunck, K.; Rohleder, E.; Hofstetter, J.; et al. Genetic alterations of the SUMO isopeptidase SENP6 drive lymphomagenesis and genetic instability in diffuse large B-cell lymphoma. Nat. Commun. 2022, 13, 281. [Google Scholar] [CrossRef]
- Karami, S.; Lin, F.M.; Kumar, S.; Bahnassy, S.; Thangavel, H.; Quttina, M.; Li, Y.; Ren, J.; Bawa-Khalfe, T. Novel SUMO-Protease SENP7S Regulates beta-catenin Signaling and Mammary Epithelial Cell Transformation. Sci. Rep. 2017, 7, 46477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gallardo-Chamizo, F.; Lara-Ureña, N.; Correa-Vázquez, J.F.; Reyes, J.C.; Gauthier, B.R.; García-Domínguez, M. SENP7 overexpression protects cancer cells from oxygen and glucose deprivation and associated with poor prognosis in colon cancer. Genes Dis. 2022; in press. [Google Scholar] [CrossRef]
- Wu, Z.; Huang, H.; Han, Q.; Hu, Z.; Teng, X.L.; Ding, R.; Ye, Y.; Yu, X.; Zhao, R.; Wang, Z.; et al. SENP7 senses oxidative stress to sustain metabolic fitness and antitumor functions of CD8+ T cells. J. Clin. Investig. 2022, 132, e155224. [Google Scholar] [CrossRef] [PubMed]
- Ji, P.; Liang, S.; Li, P.; Xie, C.; Li, J.; Zhang, K.; Zheng, X.; Feng, M.; Li, Q.; Jiao, H.; et al. Speckle-type POZ protein suppresses hepatocellular carcinoma cell migration and invasion via ubiquitin-dependent proteolysis of SUMO1/sentrin specific peptidase 7. Biochem. Biophys. Res. Commun. 2018, 502, 30–42. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Ren, S.; Bitler, B.G.; Aird, K.M.; Tu, Z.; Skordalakes, E.; Zhu, Y.; Yan, J.; Sun, Y.; Zhang, R. SPOP E3 Ubiquitin Ligase Adaptor Promotes Cellular Senescence by Degrading the SENP7 deSUMOylase. Cell Rep. 2015, 13, 1183–1193. [Google Scholar] [CrossRef] [Green Version]
- Xianjun, F.; Xirui, X.; Jie, T.; Huiwen, M.; Shaojun, Z.; Qiaoyun, L.; Yunxin, L.; Xuqun, S. Momordin Ic induces G0/1 phase arrest and apoptosis in colon cancer cells by suppressing SENP1/c-MYC signaling pathway. J. Pharmacol. Sci. 2021, 146, 249–258. [Google Scholar] [CrossRef]
- Taghvaei, S.; Sabouni, F.; Minuchehr, Z. Evidence of Omics, Immune Infiltration, and Pharmacogenomic for SENP1 in the Pan-Cancer Cohort. Front. Pharmacol. 2021, 12, 700454. [Google Scholar] [CrossRef]
- Wu, T.; Donohoe, M.E. Yy1 regulates Senp1 contributing to AMPA receptor GluR1 expression following neuronal depolarization. J. Biomed. Sci. 2019, 26, 79. [Google Scholar] [CrossRef] [Green Version]
- Guo, C.; Hildick, K.L.; Luo, J.; Dearden, L.; Wilkinson, K.A.; Henley, J.M. SENP3-mediated deSUMOylation of dynamin-related protein 1 promotes cell death following ischaemia. EMBO J. 2013, 32, 1514–1528. [Google Scholar] [CrossRef]
- Maison, C.; Romeo, K.; Bailly, D.; Dubarry, M.; Quivy, J.P.; Almouzni, G. The SUMO protease SENP7 is a critical component to ensure HP1 enrichment at pericentric heterochromatin. Nat. Struct. Mol. Biol. 2012, 19, 458–460. [Google Scholar] [CrossRef]
- Huang, C.; Han, Y.; Wang, Y.; Sun, X.; Yan, S.; Yeh, E.T.; Chen, Y.; Cang, H.; Li, H.; Shi, G.; et al. SENP3 is responsible for HIF-1 transactivation under mild oxidative stress via p300 de-SUMOylation. EMBO J. 2009, 28, 2748–2762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.Y.; Ju, M.K.; Jeon, H.M.; Lee, Y.J.; Kim, C.H.; Park, H.G.; Han, S.I.; Kang, H.S. Reactive oxygen species induce epithelialmesenchymal transition, glycolytic switch, and mitochondrial repression through the Dlx2/Snail signaling pathways in MCF7 cells. Mol. Med. Rep. 2019, 20, 2339–2346. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Yang, J.; Yang, K.; Cang, H.; Huang, X.Z.; Li, H.; Yi, J. The biphasic redox sensing of SENP3 accounts for the HIF-1 transcriptional activity shift by oxidative stress. Acta Pharmacol. Sin. 2012, 33, 953–963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suhail, A.; Rizvi, Z.A.; Mujagond, P.; Ali, S.A.; Gaur, P.; Singh, M.; Ahuja, V.; Awasthi, A.; Srikanth, C.V. DeSUMOylase SENP7-Mediated Epithelial Signaling Triggers Intestinal Inflammation via Expansion of Gamma-Delta T Cells. Cell Rep. 2019, 29, 3522–3538.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, L.; Yeh, E.T. Characterization of a family of nucleolar SUMO-specific proteases with preference for SUMO-2 or SUMO-3. J. Biol. Chem. 2006, 281, 15869–15877. [Google Scholar] [CrossRef] [Green Version]
- Di Bacco, A.; Ouyang, J.; Lee, H.Y.; Catic, A.; Ploegh, H.; Gill, G. The SUMO-specific protease SENP5 is required for cell division. Mol. Cell. Biol. 2006, 26, 4489–4498. [Google Scholar] [CrossRef] [Green Version]
- Zunino, R.; Schauss, A.; Rippstein, P.; Andrade-Navarro, M.; McBride, H.M. The SUMO protease SENP5 is required to maintain mitochondrial morphology and function. J. Cell Sci. 2007, 120, 1178–1188. [Google Scholar] [CrossRef] [Green Version]
- Stehmeier, P.; Muller, S. Regulation of p53 family members by the ubiquitin-like SUMO system. DNA Repair 2009, 8, 491–498. [Google Scholar] [CrossRef]
- Bischof, O.; Schwamborn, K.; Martin, N.; Werner, A.; Sustmann, C.; Grosschedl, R.; Dejean, A. The E3 SUMO ligase PIASy is a regulator of cellular senescence and apoptosis. Mol. Cell 2006, 22, 783–794. [Google Scholar] [CrossRef]
- Hemmings, B.A.; Restuccia, D.F. PI3K-PKB/Akt pathway. Cold Spring Harb. Perspect. Biol. 2012, 4, a011189. [Google Scholar] [CrossRef] [Green Version]
- Li, R.; Wei, J.; Jiang, C.; Liu, D.; Deng, L.; Zhang, K.; Wang, P. Akt SUMOylation regulates cell proliferation and tumorigenesis. Cancer Res. 2013, 73, 5742–5753. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Chen, Y.; Wang, S.; Hu, N.; Cao, Z.; Wang, W.; Tong, T.; Zhang, X. PIASxalpha ligase enhances SUMO1 modification of PTEN protein as a SUMO E3 ligase. J. Biol. Chem. 2014, 289, 3217–3230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rabellino, A.; Carter, B.; Konstantinidou, G.; Wu, S.Y.; Rimessi, A.; Byers, L.A.; Heymach, J.V.; Girard, L.; Chiang, C.M.; Teruya-Feldstein, J.; et al. The SUMO E3-ligase PIAS1 regulates the tumor suppressor PML and its oncogenic counterpart PML-RARA. Cancer Res. 2012, 72, 2275–2284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chanda, A.; Chan, A.; Deng, L.; Kornaga, E.N.; Enwere, E.K.; Morris, D.G.; Bonni, S. Identification of the SUMO E3 ligase PIAS1 as a potential survival biomarker in breast cancer. PLoS ONE 2017, 12, e0177639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chanda, A.; Ikeuchi, Y.; Karve, K.; Sarkar, A.; Chandhoke, A.S.; Deng, L.; Bonni, A.; Bonni, S. PIAS1 and TIF1gamma collaborate to promote SnoN SUMOylation and suppression of epithelial-mesenchymal transition. Cell Death Differ. 2021, 28, 267–282. [Google Scholar] [CrossRef]
- Dadakhujaev, S.; Salazar-Arcila, C.; Netherton, S.J.; Chandhoke, A.S.; Singla, A.K.; Jirik, F.R.; Bonni, S. A novel role for the SUMO E3 ligase PIAS1 in cancer metastasis. Oncoscience 2014, 1, 229–240. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.; Tahk, S.; Yee, K.M.; Yang, R.; Yang, Y.; Mackie, R.; Hsu, C.; Chernishof, V.; O’Brien, N.; Jin, Y.; et al. PIAS1 regulates breast tumorigenesis through selective epigenetic gene silencing. PLoS ONE 2014, 9, e89464. [Google Scholar] [CrossRef] [Green Version]
- Coppola, D.; Parikh, V.; Boulware, D.; Blanck, G. Substantially reduced expression of PIAS1 is associated with colon cancer development. J. Cancer Res. Clin. Oncol. 2009, 135, 1287–1291. [Google Scholar] [CrossRef]
- Xiao, Y.; Huang, W.; Huang, H.; Wang, L.; Wang, M.; Zhang, T.; Fang, X.; Xia, X. miR-182-5p and miR-96-5p Target PIAS1 and Mediate the Negative Feedback Regulatory Loop between PIAS1 and STAT3 in Endometrial Cancer. DNA Cell Biol. 2021, 40, 618–628. [Google Scholar] [CrossRef]
- Wang, X.; Wang, L.; Xu, Y.; Zhang, G.; Wu, Y.; Chen, P. PIAS1 inhibited the metastasis of gastric cancer cell by epithelial-mesenchymal transition regulation within the inflammatory microenvironment. Oncol. Lett. 2018, 15, 3828–3837. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Meng, Y. SUMOylation Regulator-Related Molecules Can Be Used as Prognostic Biomarkers for Glioblastoma. Front. Cell Dev. Biol. 2021, 9, 658856. [Google Scholar] [CrossRef] [PubMed]
- Constanzo, J.D.; Tang, K.J.; Rindhe, S.; Melegari, M.; Liu, H.; Tang, X.; Rodriguez-Canales, J.; Wistuba, I.; Scaglioni, P.P. PIAS1-FAK Interaction Promotes the Survival and Progression of Non-Small Cell Lung Cancer. Neoplasia 2016, 18, 282–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rabellino, A.; Melegari, M.; Tompkins, V.S.; Chen, W.; Van Ness, B.G.; Teruya-Feldstein, J.; Conacci-Sorrell, M.; Janz, S.; Scaglioni, P.P. PIAS1 Promotes Lymphomagenesis through MYC Upregulation. Cell Rep. 2016, 15, 2266–2278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, G.; Xu, Y.; Ruan, N.; Li, J.; Lv, Q.; Zhang, Q.; Chen, Y.; Wang, Q.; Xia, Q.; Li, Q. Genetic alteration and clinical significance of SUMOylation regulators in multiple cancer types. J. Cancer 2020, 11, 6823–6833. [Google Scholar] [CrossRef]
- Driscoll, J.J.; Pelluru, D.; Lefkimmiatis, K.; Fulciniti, M.; Prabhala, R.H.; Greipp, P.R.; Barlogie, B.; Tai, Y.T.; Anderson, K.C.; Shaughnessy, J.D., Jr.; et al. The sumoylation pathway is dysregulated in multiple myeloma and is associated with adverse patient outcome. Blood 2010, 115, 2827–2834. [Google Scholar] [CrossRef] [Green Version]
- Adamson, A.L.; Kenney, S. Epstein-barr virus immediate-early protein BZLF1 is SUMO-1 modified and disrupts promyelocytic leukemia bodies. J. Virol. 2001, 75, 2388–2399. [Google Scholar] [CrossRef] [Green Version]
- Saiada, F.; Zhang, K.; Li, R. PIAS1 potentiates the anti-EBV activity of SAMHD1 through SUMOylation. Cell Biosci. 2021, 11, 127. [Google Scholar] [CrossRef]
- Yoon, C.J.; Chang, M.S.; Kim, D.H.; Kim, W.; Koo, B.K.; Yun, S.C.; Kim, S.H.; Kim, Y.S.; Woo, J.H. Epstein-Barr virus-encoded miR-BART5-5p upregulates PD-L1 through PIAS3/pSTAT3 modulation, worsening clinical outcomes of PD-L1-positive gastric carcinomas. Gastric Cancer 2020, 23, 780–795. [Google Scholar] [CrossRef]
- Meng, Y.; Li, X. Expression and Prognosis Analysis of SUMOylation Regulators in Oral Squamous Cell Carcinoma Based on High-Throughput Sequencing. Front Genet. 2021, 12, 671392. [Google Scholar] [CrossRef]
- Gross, M.; Liu, B.; Tan, J.; French, F.S.; Carey, M.; Shuai, K. Distinct effects of PIAS proteins on androgen-mediated gene activation in prostate cancer cells. Oncogene 2001, 20, 3880–3887. [Google Scholar] [CrossRef] [Green Version]
- Puhr, M.; Hoefer, J.; Eigentler, A.; Dietrich, D.; van Leenders, G.; Uhl, B.; Hoogland, M.; Handle, F.; Schlick, B.; Neuwirt, H.; et al. PIAS1 is a determinant of poor survival and acts as a positive feedback regulator of AR signaling through enhanced AR stabilization in prostate cancer. Oncogene 2016, 35, 2322–2332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, N.; Liu, S.; Qin, T.; Liu, X.; Watanabe, N.; Mayo, K.H.; Li, J.; Li, X. SUMO3 modification by PIAS1 modulates androgen receptor cellular distribution and stability. Cell Commun. Signal. 2019, 17, 153. [Google Scholar] [CrossRef] [Green Version]
- Tuccilli, C.; Baldini, E.; Sorrenti, S.; Di Gioia, C.; Bosco, D.; Ascoli, V.; Mian, C.; Barollo, S.; Rendina, R.; Coccaro, C.; et al. Papillary Thyroid Cancer Is Characterized by Altered Expression of Genes Involved in the Sumoylation Process. J. Biol. Regul. Homeost. Agents 2015, 29, 655–662. [Google Scholar] [PubMed]
- Wu, R.; Fang, J.; Liu, M.; Jun, A.; Liu, J.; Chen, W.; Li, J.; Ma, G.; Zhang, Z.; Zhang, B.; et al. SUMOylation of the transcription factor ZFHX3 at Lys-2806 requires SAE1, UBC9, and PIAS2 and enhances its stability and function in cell proliferation. J. Biol. Chem. 2020, 295, 6741–6753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, S.; Si, J.; Liu, T.; DeWille, J.W. PIASy represses CCAAT/enhancer-binding protein delta (C/EBPdelta) transcriptional activity by sequestering C/EBPdelta to the nuclear periphery. J. Biol. Chem. 2008, 283, 20137–20148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Ni, J.; Yi, S.; Song, D.; Ding, M. Protein inhibitor of activated STAT xalpha depresses cyclin D and cyclin D kinase, and contributes to the inhibition of osteosarcoma cell progression. Mol. Med. Rep. 2016, 13, 1645–1652. [Google Scholar] [CrossRef] [Green Version]
- Chandhoke, A.S.; Chanda, A.; Karve, K.; Deng, L.; Bonni, S. The PIAS3-Smurf2 sumoylation pathway suppresses breast cancer organoid invasiveness. Oncotarget 2017, 8, 21001–21014. [Google Scholar] [CrossRef] [Green Version]
- Jiang, M.; Zhang, W.; Zhang, R.; Liu, P.; Ye, Y.; Yu, W.; Guo, X.; Yu, J. Cancer exosome-derived miR-9 and miR-181a promote the development of early-stage MDSCs via interfering with SOCS3 and PIAS3 respectively in breast cancer. Oncogene 2020, 39, 4681–4694. [Google Scholar] [CrossRef]
- Knittle, A.M.; Helkkula, M.; Johnson, M.S.; Sundvall, M.; Elenius, K. SUMOylation regulates nuclear accumulation and signaling activity of the soluble intracellular domain of the ErbB4 receptor tyrosine kinase. J. Biol. Chem. 2017, 292, 19890–19904. [Google Scholar] [CrossRef] [Green Version]
- Park, Y.Y.; Kim, K.; Kim, S.B.; Hennessy, B.T.; Kim, S.M.; Park, E.S.; Lim, J.Y.; Li, J.; Lu, Y.; Gonzalez-Angulo, A.M.; et al. Reconstruction of nuclear receptor network reveals that NR2E3 is a novel upstream regulator of ESR1 in breast cancer. EMBO Mol. Med. 2012, 4, 52–67. [Google Scholar] [CrossRef]
- Yang, S.F.; Hou, M.F.; Chen, F.M.; Ou-Yang, F.; Wu, Y.C.; Chai, C.Y.; Yeh, Y.T. Prognostic value of protein inhibitor of activated STAT3 in breast cancer patients receiving hormone therapy. BMC Cancer 2016, 16, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qu, D.; Yang, Y.; Huang, X. miR-199a-5p promotes proliferation and metastasis and epithelial-mesenchymal transition through targeting PIAS3 in cervical carcinoma. J. Cell. Biochem. 2019, 120, 13562–13572. [Google Scholar] [CrossRef] [PubMed]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, X.; Feng, J.; Zhu, Z.; Yao, L.; Ma, S.; Hao, B.; Zhang, G. A positive feedback loop between miR-181b and STAT3 that affects Warburg effect in colon cancer via regulating PIAS3 expression. J. Cell. Mol. Med. 2018, 22, 5040–5049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polimeno, L.; Francavilla, A.; Piscitelli, D.; Fiore, M.G.; Polimeno, R.; Topi, S.; Haxhirexha, K.; Ballini, A.; Daniele, A.; Santacroce, L. The role of PIAS3, p-STAT3 and ALR in colorectal cancer: New translational molecular features for an old disease. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 10496–10511. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Banerjee, S. Differential PIAS3 expression in human malignancy. Oncol. Rep. 2004, 11, 1319–1324. [Google Scholar] [CrossRef]
- Jiao, J.; Zhang, R.; Li, Z.; Yin, Y.; Fang, X.; Ding, X.; Cai, Y.; Yang, S.; Mu, H.; Zong, D.; et al. Nuclear Smad6 promotes gliomagenesis by negatively regulating PIAS3-mediated STAT3 inhibition. Nat. Commun. 2018, 9, 2504. [Google Scholar] [CrossRef]
- Zhang, C.; Mukherjee, S.; Tucker-Burden, C.; Ross, J.L.; Chau, M.J.; Kong, J.; Brat, D.J. TRIM8 regulates stemness in glioblastoma through PIAS3-STAT3. Mol. Oncol. 2017, 11, 280–294. [Google Scholar] [CrossRef] [Green Version]
- Sun, H.; Li, Y.; Quan, X.; Chen, N.; Jin, X.; Jin, W.; Jin, Y.; Shen, X. PIAS3/SOCS1-STAT3 axis responses to oxidative stress in hepatocellular cancer cells. Am. J. Transl. Res. 2021, 13, 12395–12409. [Google Scholar]
- Hoefer, J.; Schafer, G.; Klocker, H.; Erb, H.H.; Mills, I.G.; Hengst, L.; Puhr, M.; Culig, Z. PIAS1 is increased in human prostate cancer and enhances proliferation through inhibition of p21. Am. J. Pathol. 2012, 180, 2097–2107. [Google Scholar] [CrossRef] [Green Version]
- Tseng, J.C.; Huang, S.H.; Lin, C.Y.; Wang, B.J.; Huang, S.F.; Shen, Y.Y.; Chuu, C.P. ROR2 suppresses metastasis of prostate cancer via regulation of miR-199a-5p-PIAS3-AKT2 signaling axis. Cell Death Dis. 2020, 11, 376. [Google Scholar] [CrossRef] [PubMed]
- Van Nguyen, T.; Angkasekwinai, P.; Dou, H.; Lin, F.M.; Lu, L.S.; Cheng, J.; Chin, Y.E.; Dong, C.; Yeh, E.T. SUMO-specific protease 1 is critical for early lymphoid development through regulation of STAT5 activation. Mol. Cell 2012, 45, 210–221. [Google Scholar] [CrossRef] [PubMed]
- Citro, S.; Jaffray, E.; Hay, R.T.; Seiser, C.; Chiocca, S. A role for paralog-specific sumoylation in histone deacetylase 1 stability. J. Mol. Cell. Biol. 2013, 5, 416–427. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Ollila, S.; Wong, I.P.L.; Vallenius, T.; Palvimo, J.J.; Vaahtomeri, K.; Makela, T.P. SUMOylation of AMPKalpha1 by PIAS4 specifically regulates mTORC1 signalling. Nat. Commun. 2015, 6, 8979. [Google Scholar] [CrossRef] [PubMed]
- Kang, X.; Li, J.; Zou, Y.; Yi, J.; Zhang, H.; Cao, M.; Yeh, E.T.; Cheng, J. PIASy stimulates HIF1alpha SUMOylation and negatively regulates HIF1alpha activity in response to hypoxia. Oncogene 2010, 29, 5568–5578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, B.; Zhu, Y.; Xu, W.; Zhou, Q.; Tan, L.; Zhu, L.; Chen, H.; Feng, L.; Hou, T.; Wang, X.; et al. Hypoxia Stimulates SUMOylation-Dependent Stabilization of KDM5B. Front. Cell Dev. Biol. 2021, 9, 741736. [Google Scholar] [CrossRef]
- Liu, Q.; Zhou, B.; Liao, R.; Zhou, X.; Yan, X. PIAS4, upregulated in hepatocellular carcinoma, promotes tumorigenicity and metastasis. J. Cell. Biochem. 2020, 121, 3372–3381. [Google Scholar] [CrossRef]
- Chen, B.; Luo, J.; Zhou, Y.; Xin, X.; Cai, R.; Ling, C. PIASy antagonizes Ras-driven NSCLC survival by promoting GATA2 SUMOylation. J. Cancer 2018, 9, 1689–1697. [Google Scholar] [CrossRef] [Green Version]
- Hung, P.F.; Hong, T.M.; Chang, C.C.; Hung, C.L.; Hsu, Y.L.; Chang, Y.L.; Wu, C.T.; Chang, G.C.; Chan, N.L.; Yu, S.L.; et al. Hypoxia-induced Slug SUMOylation enhances lung cancer metastasis. J. Exp. Clin. Cancer Res. 2019, 38, 5. [Google Scholar] [CrossRef]
- Sun, L.; Li, H.; Chen, J.; Iwasaki, Y.; Kubota, T.; Matsuoka, M.; Shen, A.; Chen, Q.; Xu, Y. PIASy mediates hypoxia-induced SIRT1 transcriptional repression and epithelial-to-mesenchymal transition in ovarian cancer cells. J. Cell Sci. 2013, 126, 3939–3947. [Google Scholar] [CrossRef] [Green Version]
- Chien, W.; Lee, K.L.; Ding, L.W.; Wuensche, P.; Kato, H.; Doan, N.B.; Poellinger, L.; Said, J.W.; Koeffler, H.P. PIAS4 is an activator of hypoxia signalling via VHL suppression during growth of pancreatic cancer cells. Br. J. Cancer 2013, 109, 1795–1804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanyal, S.; Mondal, P.; Sen, S.; Sengupta Bandyopadhyay, S.; Das, C. SUMO E3 ligase CBX4 regulates hTERT-mediated transcription of CDH1 and promotes breast cancer cell migration and invasion. Biochem. J. 2020, 477, 3803–3818. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Du, J.; Wang, Y.; Shi, H.; Jiang, Q.; Wang, Y.; Zhang, H.; Wei, Y.; Xue, W.; Pu, Z.; et al. MicroRNA-497-5p Induces Cell Cycle Arrest Of Cervical Cancer Cells In S Phase By Targeting CBX4. Onco Targets Ther. 2019, 12, 10535–10545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, J.; Yang, T.; Li, L. LncRNA FOXP4-AS1 Is Involved in Cervical Cancer Progression via Regulating miR-136-5p/CBX4 Axis. Onco Targets Ther. 2020, 13, 2347–2355. [Google Scholar] [CrossRef] [Green Version]
- Wei, W.; Zhao, W.; Zhang, Y. CBX4 Provides an Alternate Mode of Colon Cancer Development via Potential Influences on Circadian Rhythm and Immune Infiltration. Front. Cell Dev. Biol. 2021, 9, 669254. [Google Scholar] [CrossRef]
- Pan, Y.; Li, Q.; Cao, Z.; Zhao, S. The SUMO E3 ligase CBX4 is identified as a poor prognostic marker of gastric cancer through multipronged OMIC analyses. Genes Dis. 2021, 8, 827–837. [Google Scholar] [CrossRef]
- Li, J.; Xu, Y.; Long, X.D.; Wang, W.; Jiao, H.K.; Mei, Z.; Yin, Q.Q.; Ma, L.N.; Zhou, A.W.; Wang, L.S.; et al. Cbx4 governs HIF-1alpha to potentiate angiogenesis of hepatocellular carcinoma by its SUMO E3 ligase activity. Cancer Cell 2014, 25, 118–131. [Google Scholar] [CrossRef] [Green Version]
- Hu, C.; Zhang, Q.; Tang, Q.; Zhou, H.; Liu, W.; Huang, J.; Liu, Y.; Wang, Q.; Zhang, J.; Zhou, M.; et al. CBX4 promotes the proliferation and metastasis via regulating BMI-1 in lung cancer. J. Cell. Mol. Med. 2020, 24, 618–631. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Qin, G.; Liang, X.; Wang, W.; Wang, Z.; Liao, D.; Zhong, L.; Zhang, R.; Zeng, Y.X.; Wu, Y.; et al. Targeting the CK1alpha/CBX4 axis for metastasis in osteosarcoma. Nat. Commun. 2020, 11, 1141. [Google Scholar] [CrossRef] [Green Version]
- Choi, H.J.; Park, J.H.; Park, J.H.; Lee, K.B.; Oh, S.M. Pc2-mediated SUMOylation of WWOX is essential for its suppression of DU145 prostate tumorigenesis. FEBS Lett. 2015, 589, 3977–3988. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Z.; Qiu, K.; Huang, W. Long Non-Coding RNA (lncRNA) RAMS11 Promotes Metastatis and Cell Growth of Prostate Cancer by CBX4 Complex Binding to Top2alpha. Cancer Manag. Res. 2021, 13, 913–923. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Luo, Q.; Kang, J.; Wei, Q.; Yang, Y.; Yang, D.; Liu, X.; Liu, T.; Yi, P. YTHDF1 Aggravates the Progression of Cervical Cancer Through m(6)A-Mediated Up-Regulation of RANBP2. Front. Oncol. 2021, 11, 650383. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Liu, Y.; Wang, B.; Lan, H.; Liu, Y.; Chen, F.; Zhang, J.; Luo, J. Sumoylation in p27kip1 via RanBP2 promotes cancer cell growth in cholangiocarcinoma cell line QBC939. BMC Mol. Biol. 2017, 18, 23. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Chen, X.; Xiao, M.; Zhu, Y.; Gong, R.; Liu, J.; Zeng, Q.; Xu, C.; Chen, X.; Wang, F.; et al. RANBP2 Activates O-GlcNAcylation through Inducing CEBPalpha-Dependent OGA Downregulation to Promote Hepatocellular Carcinoma Malignant Phenotypes. Cancers 2021, 13, 3475. [Google Scholar] [CrossRef]
- Horio, Y.; Osada, H.; Shimizu, J.; Ogawa, S.; Hida, T.; Sekido, Y. Relationship of mRNA expressions of RanBP2 and topoisomerase II isoforms to cytotoxicity of amrubicin in human lung cancer cell lines. Cancer Chemother. Pharmacol. 2010, 66, 237–243. [Google Scholar] [CrossRef]
- Renner, O.; Fominaya, J.; Alonso, S.; Blanco-Aparicio, C.; Leal, J.F.; Carnero, A. Mst1, RanBP2 and eIF4G are new markers for in vivo PI3K activation in murine and human prostate. Carcinogenesis 2007, 28, 1418–1425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, Y.; Wu, H.; Xu, Y.; Zhang, Z.; Liu, T.; Lin, X.; Feng, X.H. Zinc finger protein 451 is a novel Smad corepressor in transforming growth factor-beta signaling. J. Biol. Chem. 2014, 289, 2072–2083. [Google Scholar] [CrossRef] [Green Version]
- Zeng, W.; Gu, S.; Yu, Y.; Feng, Y.; Xiao, M.; Feng, X.H. ZNF451 stabilizes TWIST2 through SUMOylation and promotes epithelial-mesenchymal transition. Am. J. Cancer Res. 2021, 11, 898–915. [Google Scholar]
- Zhang, Z.; Chen, H.; Lu, Y.; Feng, T.; Sun, W. LncRNA BC032020 suppresses the survival of human pancreatic ductal adenocarcinoma cells by targeting ZNF451. Int. J. Oncol. 2018, 52, 1224–1234. [Google Scholar] [CrossRef] [Green Version]
- Karvonen, U.; Jaaskelainen, T.; Rytinki, M.; Kaikkonen, S.; Palvimo, J.J. ZNF451 is a novel PML body- and SUMO-associated transcriptional coregulator. J. Mol. Biol. 2008, 382, 585–600. [Google Scholar] [CrossRef]
- Kakizuka, A.; Miller, W.H., Jr.; Umesono, K.; Warrell, R.P., Jr.; Frankel, S.R.; Murty, V.V.; Dmitrovsky, E.; Evans, R.M. Chromosomal translocation t(15;17) in human acute promyelocytic leukemia fuses RAR alpha with a novel putative transcription factor, PML. Cell 1991, 66, 663–674. [Google Scholar] [CrossRef]
- Song, W.; Wang, Z.; Gu, X.; Wang, A.; Chen, X.; Miao, H.; Chu, J.; Tian, Y. TRIM11 promotes proliferation and glycolysis of breast cancer cells via targeting AKT/GLUT1 pathway. Onco Targets Ther. 2019, 12, 4975–4984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, J.; Luo, Y.; Tian, Z.; Liao, X.; Cui, Q.; Yang, Q.; Wu, G. TRIM11 promotes breast cancer cell proliferation by stabilizing estrogen receptor alpha. Neoplasia 2020, 22, 343–351. [Google Scholar] [CrossRef]
- Zhao, T.T.; Jin, F.; Li, J.G.; Xu, Y.Y.; Dong, H.T.; Liu, Q.; Xing, P.; Zhu, G.L.; Xu, H.; Yin, S.C.; et al. TRIM32 promotes proliferation and confers chemoresistance to breast cancer cells through activation of the NF-kappaB pathway. J. Cancer 2018, 9, 1349–1356. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Wu, Z.; Wang, L.; Wang, Q.; Sun, X.; Niu, S. Knockdown of TRIM11 suppresses cell progression and apoptosis of cervical cancer cells via PI3K/AKT pathway. Am. J. Transl. Res. 2021, 13, 10328–10340. [Google Scholar]
- Liu, S.; Tian, Y.; Zheng, Y.; Cheng, Y.; Zhang, D.; Jiang, J.; Li, S. TRIM27 acts as an oncogene and regulates cell proliferation and metastasis in non-small cell lung cancer through SIX3-beta-catenin signaling. Aging 2020, 12, 25564–25580. [Google Scholar] [CrossRef]
- Su, X.; Wang, B.; Wang, Y.; Wang, B. Inhibition of TRIM32 Induced by miR-519d Increases the Sensitivity of Colorectal Cancer Cells to Cisplatin. Onco Targets Ther. 2020, 13, 277–289. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Feng, Y.; Ji, D.; Wang, Q.; Qian, W.; Wang, S.; Zhang, Z.; Ji, B.; Zhang, C.; Sun, Y.; et al. TRIM27 functions as an oncogene by activating epithelial-mesenchymal transition and p-AKT in colorectal cancer. Int. J. Oncol. 2018, 53, 620–632. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Zhang, B.; Wei, M.; Xu, Z.; Kong, W.; Deng, K.; Xu, X.; Zhang, L.; Zetahao, X.; Yan, L. TRIM22 inhibits endometrial cancer progression through the NOD2/NFkappaB signaling pathway and confers a favorable prognosis. Int. J. Oncol. 2020, 56, 1225–1239. [Google Scholar] [CrossRef] [Green Version]
- Ma, L.; Yao, N.; Chen, P.; Zhuang, Z. TRIM27 promotes the development of esophagus cancer via regulating PTEN/AKT signaling pathway. Cancer Cell Int. 2019, 19, 283. [Google Scholar] [CrossRef]
- Lan, Q.; Tan, X.; He, P.; Li, W.; Tian, S.; Dong, W. TRIM11 Promotes Proliferation, Migration, Invasion and EMT of Gastric Cancer by Activating beta-Catenin Signaling. Onco Targets Ther. 2021, 14, 1429–1440. [Google Scholar] [CrossRef]
- Luo, N.; Wang, Z. TRIM11 stimulates the proliferation of gastric cancer through targeting CPEB3/EGFR axis. J. BUON 2020, 25, 2097–2104. [Google Scholar] [PubMed]
- Wang, C.; Xu, J.; Fu, H.; Zhang, Y.; Zhang, X.; Yang, D.; Zhu, Z.; Wei, Z.; Hu, Z.; Yan, R.; et al. TRIM32 promotes cell proliferation and invasion by activating beta-catenin signalling in gastric cancer. J. Cell. Mol. Med. 2018, 22, 5020–5028. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Fang, Y.; Liu, T. TRIM32 Promotes the Growth of Gastric Cancer Cells through Enhancing AKT Activity and Glucose Transportation. Biomed. Res. Int. 2020, 2020, 4027627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, Y.; Liu, Z.; Cao, Y.; Guo, H.; Jiang, B.; Deng, J.; Xiong, J. Downregulation of TRIM27 suppresses gastric cancer cell proliferation via inhibition of the Hippo-BIRC5 pathway. Pathol. Res. Pract. 2020, 216, 153048. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Gao, W.; Yuan, B.; Zhang, S.; Wang, K.; Du, T. TRIM22 inhibits the proliferation of gastric cancer cells through the Smad2 protein. Cell Death Discov. 2021, 7, 234. [Google Scholar] [CrossRef]
- Herquel, B.; Ouararhni, K.; Khetchoumian, K.; Ignat, M.; Teletin, M.; Mark, M.; Bechade, G.; Van Dorsselaer, A.; Sanglier-Cianferani, S.; Hamiche, A.; et al. Transcription cofactors TRIM24, TRIM28, and TRIM33 associate to form regulatory complexes that suppress murine hepatocellular carcinoma. Proc. Natl. Acad. Sci. USA 2011, 108, 8212–8217. [Google Scholar] [CrossRef] [Green Version]
- Du, Y.; Zhang, W.; Du, B.; Zang, S.; Wang, X.; Mao, X.; Hu, Z. TRIM32 overexpression improves chemoresistance through regulation of mitochondrial function in non-small-cell lung cancers. Onco Targets Ther. 2018, 11, 7841–7852. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Zhou, X.M.; Yang, F.F.; Miao, Y.; Yin, Y.; Hu, X.J.; Hou, G.; Wang, Q.Y.; Kang, J. TRIM22 confers poor prognosis and promotes epithelial-mesenchymal transition through regulation of AKT/GSK3beta/beta-catenin signaling in non-small cell lung cancer. Oncotarget 2017, 8, 62069–62080. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.L.; Lu, S.C.; Sun, C.; Jin, W.G.; Fan, Y.W.; Shu, Y.S.; Shi, H.C.; Min, L.F. Tripartite motif protein 11 (TRIM11), an oncogene for human lung cancer via the DUSP6-mediated ERK1/2 signaling pathway. Cancer Biol. Ther. 2021, 22, 324–332. [Google Scholar] [CrossRef]
- Wang, Y.; Liang, H.X.; Zhang, C.M.; Zou, M.; Zou, B.B.; Wei, W.; Hu, W. FOXO3/TRIM22 axis abated the antitumor effect of gemcitabine in non-small cell lung cancer via autophagy induction. Transl. Cancer Res. 2020, 9, 937–948. [Google Scholar] [CrossRef]
- Yin, H.; Li, Z.; Chen, J.; Hu, X. Expression and the potential functions of TRIM32 in lung cancer tumorigenesis. J. Cell. Biochem. 2019, 120, 5232–5243. [Google Scholar] [CrossRef]
- Chen, Y.; Sun, J.; Ma, J. Proliferation and invasion of ovarian cancer cells are suppressed by knockdown of TRIM11. Oncol. Lett. 2017, 14, 2125–2130. [Google Scholar] [CrossRef] [Green Version]
- Jiang, J.; Xie, C.; Liu, Y.; Shi, Q.; Chen, Y. Up-regulation of miR-383-5p suppresses proliferation and enhances chemosensitivity in ovarian cancer cells by targeting TRIM27. Biomed. Pharmacother. 2019, 109, 595–601. [Google Scholar] [CrossRef]
- Song, Z.; Guo, Q.; Wang, H.; Gao, L.; Wang, S.; Liu, D.; Liu, J.; Qi, Y.; Lin, B. miR-5193, regulated by FUT1, suppresses proliferation and migration of ovarian cancer cells by targeting TRIM11. Pathol. Res. Pract. 2020, 216, 153148. [Google Scholar] [CrossRef]
- Zhang, B.; Yan, Y.Y.; Gu, Y.Q.; Teng, F.; Lin, X.; Zhou, X.L.; Che, J.X.; Dong, X.W.; Zhou, L.X.; Lin, N.M. Inhibition of TRIM32 by ibr-7 treatment sensitizes pancreatic cancer cells to gemcitabine via mTOR/p70S6K pathway. J. Cell. Mol. Med. 2022, 26, 515–526. [Google Scholar] [CrossRef]
- Pan, Y.; Zhang, R.; Chen, H.; Chen, W.; Wu, K.; Lv, J. Expression of Tripartite Motif-Containing Proteactiin 11 (TRIM11) is Associated with the Progression of Human Prostate Cancer and is Downregulated by MicroRNA-5193. Med. Sci. Monit. 2019, 25, 98–106. [Google Scholar] [CrossRef]
- Xiao, C.; Zhang, W.; Hua, M.; Chen, H.; Yang, B.; Wang, Y.; Yang, Q. TRIM27 interacts with Ikappabalpha to promote the growth of human renal cancer cells through regulating the NF-kappaB pathway. BMC Cancer 2021, 21, 841. [Google Scholar] [CrossRef]
- Tang, J.; Tian, Z.; Liao, X.; Wu, G. SOX13/TRIM11/YAP axis promotes the proliferation, migration and chemoresistance of anaplastic thyroid cancer. Int. J. Biol. Sci. 2021, 17, 417–429. [Google Scholar] [CrossRef]
- Kamitani, T.; Nguyen, H.P.; Kito, K.; Fukuda-Kamitani, T.; Yeh, E.T. Covalent modification of PML by the sentrin family of ubiquitin-like proteins. J. Biol. Chem. 1998, 273, 3117–3120. [Google Scholar] [CrossRef] [Green Version]
- de The, H.; Chen, Z. Acute promyelocytic leukaemia: Novel insights into the mechanisms of cure. Nat. Rev. Cancer 2010, 10, 775–783. [Google Scholar] [CrossRef]
- Zhu, J.; Zhou, J.; Peres, L.; Riaucoux, F.; Honore, N.; Kogan, S.; de The, H. A sumoylation site in PML/RARA is essential for leukemic transformation. Cancer Cell 2005, 7, 143–153. [Google Scholar] [CrossRef] [Green Version]
- Lallemand-Breitenbach, V.; Jeanne, M.; Benhenda, S.; Nasr, R.; Lei, M.; Peres, L.; Zhou, J.; Zhu, J.; Raught, B.; de The, H. Arsenic degrades PML or PML-RARalpha through a SUMO-triggered RNF4/ubiquitin-mediated pathway. Nat. Cell Biol. 2008, 10, 547–555. [Google Scholar] [CrossRef]
- de The, H.; Le Bras, M.; Lallemand-Breitenbach, V. The cell biology of disease: Acute promyelocytic leukemia, arsenic, and PML bodies. J. Cell Biol. 2012, 198, 11–21. [Google Scholar] [CrossRef] [Green Version]
- Kadare, G.; Toutant, M.; Formstecher, E.; Corvol, J.C.; Carnaud, M.; Boutterin, M.C.; Girault, J.A. PIAS1-mediated sumoylation of focal adhesion kinase activates its autophosphorylation. J. Biol. Chem. 2003, 278, 47434–47440. [Google Scholar] [CrossRef] [Green Version]
- Sulzmaier, F.J.; Jean, C.; Schlaepfer, D.D. FAK in cancer: Mechanistic findings and clinical applications. Nat. Rev. Cancer 2014, 14, 598–610. [Google Scholar] [CrossRef] [Green Version]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef] [Green Version]
- Li, X.X.; Shi, L.; Zhou, X.J.; Wu, J.; Xia, T.S.; Zhou, W.B.; Sun, X.; Zhu, L.; Wei, J.F.; Ding, Q. The role of c-Myc-RBM38 loop in the growth suppression in breast cancer. J. Exp. Clin. Cancer Res. 2017, 36, 49. [Google Scholar] [CrossRef]
- Sentis, S.; Le Romancer, M.; Bianchin, C.; Rostan, M.C.; Corbo, L. Sumoylation of the estrogen receptor alpha hinge region regulates its transcriptional activity. Mol. Endocrinol. 2005, 19, 2671–2684. [Google Scholar] [CrossRef]
- Kim, J.H.; Jang, J.W.; Lee, Y.S.; Lee, J.W.; Chi, X.Z.; Li, Y.H.; Kim, M.K.; Kim, D.M.; Choi, B.S.; Kim, J.; et al. RUNX family members are covalently modified and regulated by PIAS1-mediated sumoylation. Oncogenesis 2014, 3, e101. [Google Scholar] [CrossRef] [Green Version]
- Nishida, T.; Yasuda, H. PIAS1 and PIASxalpha function as SUMO-E3 ligases toward androgen receptor and repress androgen receptor-dependent transcription. J. Biol. Chem. 2002, 277, 41311–41317. [Google Scholar] [CrossRef] [Green Version]
- Liao, Z.; Lutz, J.; Nevalainen, M.T. Transcription factor Stat5a/b as a therapeutic target protein for prostate cancer. Int. J. Biochem. Cell Biol. 2010, 42, 186–192. [Google Scholar] [CrossRef] [Green Version]
- Myatt, S.S.; Lam, E.W. The emerging roles of forkhead box (Fox) proteins in cancer. Nat. Rev. Cancer 2007, 7, 847–859. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, N.; John, R.; Chand, V.; Nag, A. Oncogenic Human Papillomavirus 16E7 modulates SUMOylation of FoxM1b. Int. J. Biochem. Cell Biol. 2015, 58, 28–36. [Google Scholar] [CrossRef]
- Myatt, S.S.; Kongsema, M.; Man, C.W.; Kelly, D.J.; Gomes, A.R.; Khongkow, P.; Karunarathna, U.; Zona, S.; Langer, J.K.; Dunsby, C.W.; et al. SUMOylation inhibits FOXM1 activity and delays mitotic transition. Oncogene 2014, 33, 4316–4329. [Google Scholar] [CrossRef] [Green Version]
- zur Hausen, H. Papillomaviruses in the causation of human cancers—A brief historical account. Virology 2009, 384, 260–265. [Google Scholar] [CrossRef] [Green Version]
- Thompson, M.P.; Kurzrock, R. Epstein-Barr virus and cancer. Clin. Cancer Res. 2004, 10, 803–821. [Google Scholar] [CrossRef] [Green Version]
- Tsurumi, T.; Fujita, M.; Kudoh, A. Latent and lytic Epstein-Barr virus replication strategies. Rev. Med. Virol. 2005, 15, 3–15. [Google Scholar] [CrossRef]
- Robinson, N.J.; Miyagi, M.; Scarborough, J.A.; Scott, J.G.; Taylor, D.J.; Schiemann, W.P. SLX4IP promotes RAP1 SUMOylation by PIAS1 to coordinate telomere maintenance through NF-kappaB and Notch signaling. Sci. Signal. 2021, 14, eabe9613. [Google Scholar] [CrossRef]
- Parsons, M.J.; Brancaccio, M.; Sethi, S.; Maywood, E.S.; Satija, R.; Edwards, J.K.; Jagannath, A.; Couch, Y.; Finelli, M.J.; Smyllie, N.J.; et al. The Regulatory Factor ZFHX3 Modifies Circadian Function in SCN via an AT Motif-Driven Axis. Cell 2015, 162, 607–621. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; Fu, X.; Li, J.; Xing, C.; Martin, D.W.; Zhang, H.H.; Chen, Z.; Dong, J.T. Heterozygous deletion of Atbf1 by the Cre-loxP system in mice causes preweaning mortality. Genesis 2012, 50, 819–827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, X.; Xing, C.; Fu, X.; Li, J.; Zhang, B.; Frierson, H.F., Jr.; Dong, J.T. Additive Effect of Zfhx3/Atbf1 and Pten Deletion on Mouse Prostatic Tumorigenesis. J. Genet. Genom. 2015, 42, 373–382. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; Li, J.; Dong, F.N.; Dong, J.T. Characterization of nuclear localization and SUMOylation of the ATBF1 transcription factor in epithelial cells. PLoS ONE 2014, 9, e92746. [Google Scholar] [CrossRef]
- Junttila, T.T.; Sundvall, M.; Lundin, M.; Lundin, J.; Tanner, M.; Harkonen, P.; Joensuu, H.; Isola, J.; Elenius, K. Cleavable ErbB4 isoform in estrogen receptor-regulated growth of breast cancer cells. Cancer Res. 2005, 65, 1384–1393. [Google Scholar] [CrossRef] [Green Version]
- Maatta, J.A.; Sundvall, M.; Junttila, T.T.; Peri, L.; Laine, V.J.; Isola, J.; Egeblad, M.; Elenius, K. Proteolytic cleavage and phosphorylation of a tumor-associated ErbB4 isoform promote ligand-independent survival and cancer cell growth. Mol. Biol. Cell 2006, 17, 67–79. [Google Scholar] [CrossRef] [Green Version]
- Ni, C.Y.; Murphy, M.P.; Golde, T.E.; Carpenter, G. γ-Secretase cleavage and nuclear localization of ErbB-4 receptor tyrosine kinase. Science 2001, 294, 2179–2181. [Google Scholar] [CrossRef]
- Wang, L.; Zhang, J.; Banerjee, S.; Barnes, L.; Barnes, L.; Sajja, V.; Liu, Y.; Guo, B.; Du, Y.; Agarwal, M.K.; et al. Sumoylation of vimentin354 is associated with PIAS3 inhibition of glioma cell migration. Oncotarget 2010, 1, 620–627. [Google Scholar] [CrossRef] [Green Version]
- Ropero, S.; Esteller, M. The role of histone deacetylases (HDACs) in human cancer. Mol. Oncol. 2007, 1, 19–25. [Google Scholar] [CrossRef]
- Cai, Q.; Verma, S.C.; Kumar, P.; Ma, M.; Robertson, E.S. Hypoxia inactivates the VHL tumor suppressor through PIASy-mediated SUMO modification. PLoS ONE 2010, 5, e9720. [Google Scholar] [CrossRef] [Green Version]
- Aqeilan, R.I.; Kuroki, T.; Pekarsky, Y.; Albagha, O.; Trapasso, F.; Baffa, R.; Huebner, K.; Edmonds, P.; Croce, C.M. Loss of WWOX expression in gastric carcinoma. Clin. Cancer Res. 2004, 10, 3053–3058. [Google Scholar] [CrossRef] [Green Version]
- Donati, V.; Fontanini, G.; Dell’Omodarme, M.; Prati, M.C.; Nuti, S.; Lucchi, M.; Mussi, A.; Fabbri, M.; Basolo, F.; Croce, C.M.; et al. WWOX expression in different histologic types and subtypes of non-small cell lung cancer. Clin. Cancer Res. 2007, 13, 884–891. [Google Scholar] [CrossRef] [Green Version]
- Guler, G.; Uner, A.; Guler, N.; Han, S.Y.; Iliopoulos, D.; Hauck, W.W.; McCue, P.; Huebner, K. The fragile genes FHIT and WWOX are inactivated coordinately in invasive breast carcinoma. Cancer 2004, 100, 1605–1614. [Google Scholar] [CrossRef]
- Nunez, M.I.; Rosen, D.G.; Ludes-Meyers, J.H.; Abba, M.C.; Kil, H.; Page, R.; Klein-Szanto, A.J.; Godwin, A.K.; Liu, J.; Mills, G.B.; et al. WWOX protein expression varies among ovarian carcinoma histotypes and correlates with less favorable outcome. BMC Cancer 2005, 5, 64. [Google Scholar] [CrossRef] [Green Version]
- Park, S.W.; Ludes-Meyers, J.; Zimonjic, D.B.; Durkin, M.E.; Popescu, N.C.; Aldaz, C.M. Frequent downregulation and loss of WWOX gene expression in human hepatocellular carcinoma. Br. J. Cancer 2004, 91, 753–759. [Google Scholar] [CrossRef] [Green Version]
- Packham, S.; Warsito, D.; Lin, Y.; Sadi, S.; Karlsson, R.; Sehat, B.; Larsson, O. Nuclear translocation of IGF-1R via p150(Glued) and an importin-beta/RanBP2-dependent pathway in cancer cells. Oncogene 2015, 34, 2227–2238. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.; Rohan, T. Role of the insulin-like growth factor family in cancer development and progression. J. Natl. Cancer Inst. 2000, 92, 1472–1489. [Google Scholar] [CrossRef]
- Deng, H.; Lin, Y.; Badin, M.; Vasilcanu, D.; Stromberg, T.; Jernberg-Wiklund, H.; Sehat, B.; Larsson, O. Over-accumulation of nuclear IGF-1 receptor in tumor cells requires elevated expression of the receptor and the SUMO-conjugating enzyme Ubc9. Biochem. Biophys. Res. Commun. 2011, 404, 667–671. [Google Scholar] [CrossRef]
- Aleksic, T.; Chitnis, M.M.; Perestenko, O.V.; Gao, S.; Thomas, P.H.; Turner, G.D.; Protheroe, A.S.; Howarth, M.; Macaulay, V.M. Type 1 insulin-like growth factor receptor translocates to the nucleus of human tumor cells. Cancer Res. 2010, 70, 6412–6419. [Google Scholar] [CrossRef] [Green Version]
- van Gaal, J.C.; Roeffen, M.H.; Flucke, U.E.; van der Laak, J.A.; van der Heijden, G.; de Bont, E.S.; Suurmeijer, A.J.; Versleijen-Jonkers, Y.M.; van der Graaf, W.T. Simultaneous targeting of insulin-like growth factor-1 receptor and anaplastic lymphoma kinase in embryonal and alveolar rhabdomyosarcoma: A rational choice. Eur. J. Cancer 2013, 49, 3462–3470. [Google Scholar] [CrossRef]
- Burger, A.M.; Kona, F.; Amemiya, Y.; Gao, Y.; Bacopulos, S.; Seth, A.K. Role of the BCA2 ubiquitin E3 ligase in hormone responsive breast cancer. Open Cancer J. 2010, 3, 116–123. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Nie, Z.; Chen, W.; Zhou, Z.; Kong, Q.; Seth, A.K.; Liu, R.; Chen, C. RNF115/BCA2 E3 ubiquitin ligase promotes breast cancer cell proliferation through targeting p21Waf1/Cip1 for ubiquitin-mediated degradation. Neoplasia 2013, 15, 1028–1035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narita, R.; Kitaura, H.; Torii, A.; Tashiro, E.; Miyazawa, M.; Ariga, H.; Iguchi-Ariga, S.M. Rabring7 degrades c-Myc through complex formation with MM-1. PLoS ONE 2012, 7, e41891. [Google Scholar] [CrossRef] [PubMed]
- Colomer-Lluch, M.; Serra-Moreno, R. BCA2/Rabring7 Interferes with HIV-1 Proviral Transcription by Enhancing the SUMOylation of IkappaBalpha. J. Virol. 2017, 91, e02098-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
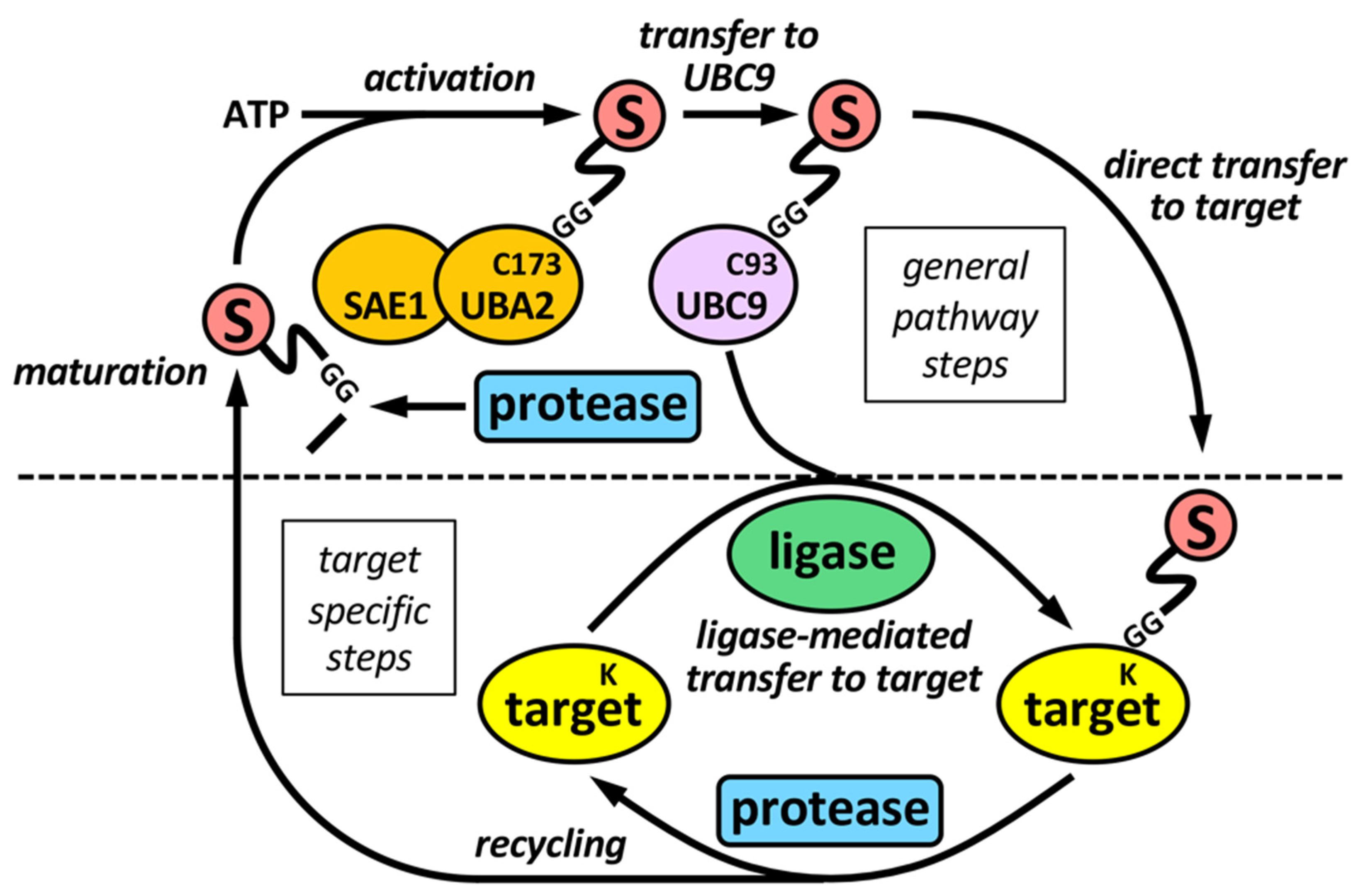
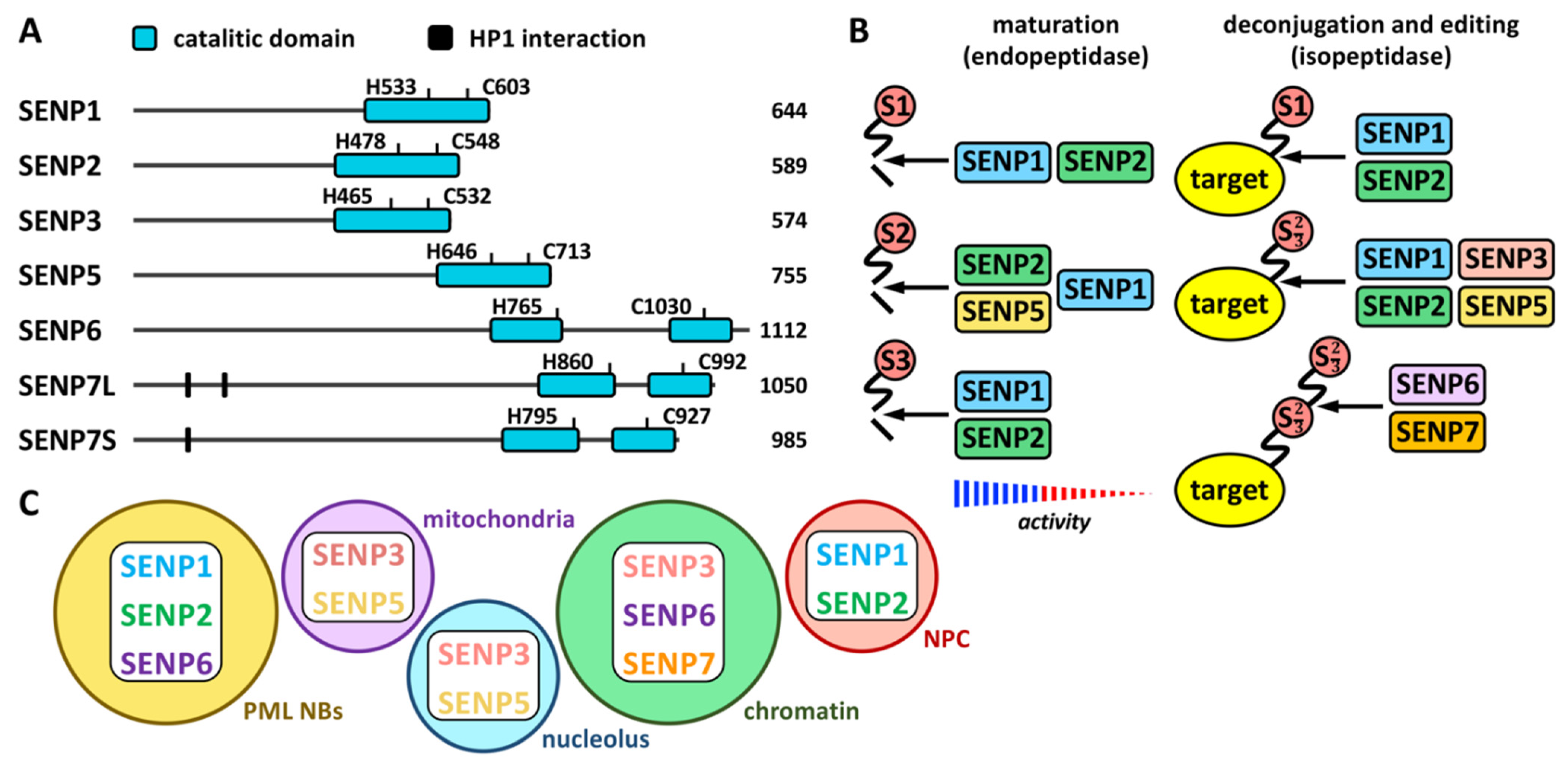
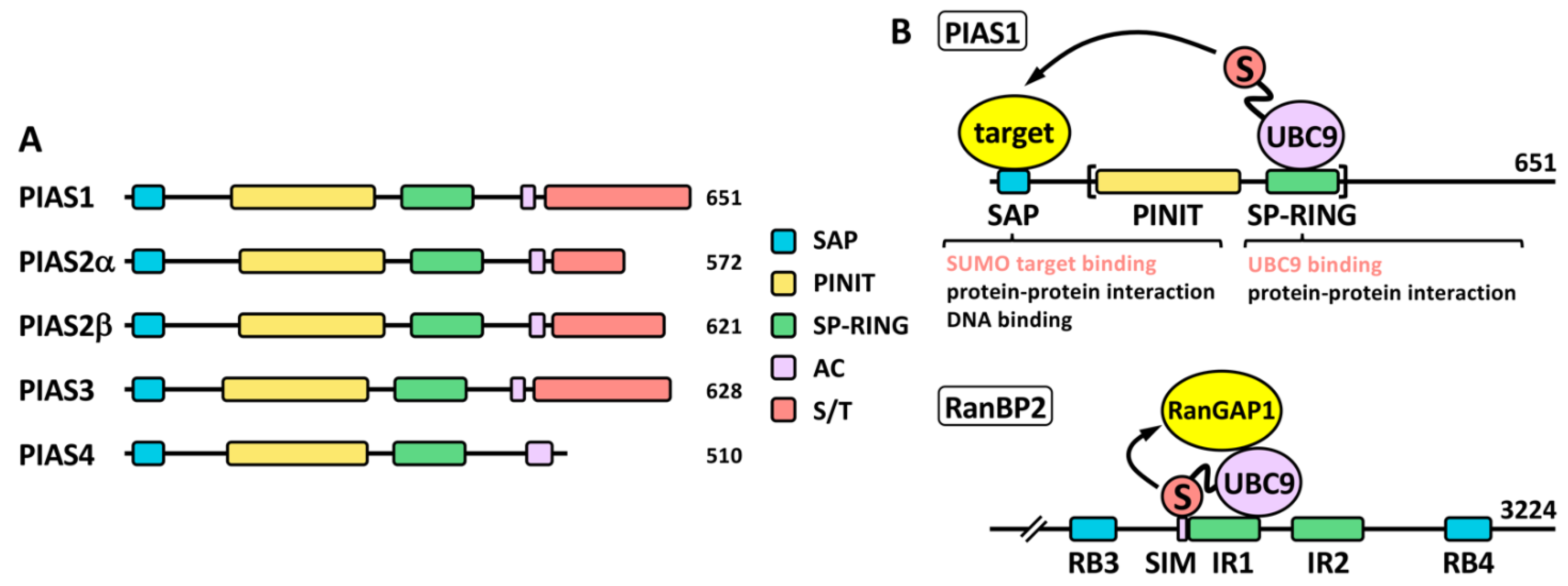
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lara-Ureña, N.; Jafari, V.; García-Domínguez, M. Cancer-Associated Dysregulation of Sumo Regulators: Proteases and Ligases. Int. J. Mol. Sci. 2022, 23, 8012. https://doi.org/10.3390/ijms23148012
Lara-Ureña N, Jafari V, García-Domínguez M. Cancer-Associated Dysregulation of Sumo Regulators: Proteases and Ligases. International Journal of Molecular Sciences. 2022; 23(14):8012. https://doi.org/10.3390/ijms23148012
Chicago/Turabian StyleLara-Ureña, Nieves, Vahid Jafari, and Mario García-Domínguez. 2022. "Cancer-Associated Dysregulation of Sumo Regulators: Proteases and Ligases" International Journal of Molecular Sciences 23, no. 14: 8012. https://doi.org/10.3390/ijms23148012