
Comparative Transcriptome Analysis Reveals Coordinated Transcriptional Regulation of Central and Secondary Metabolism in the Trichomes of Cannabis Cultivars

 , ,
, ,
Abstract
:1. Introduction
2. Results
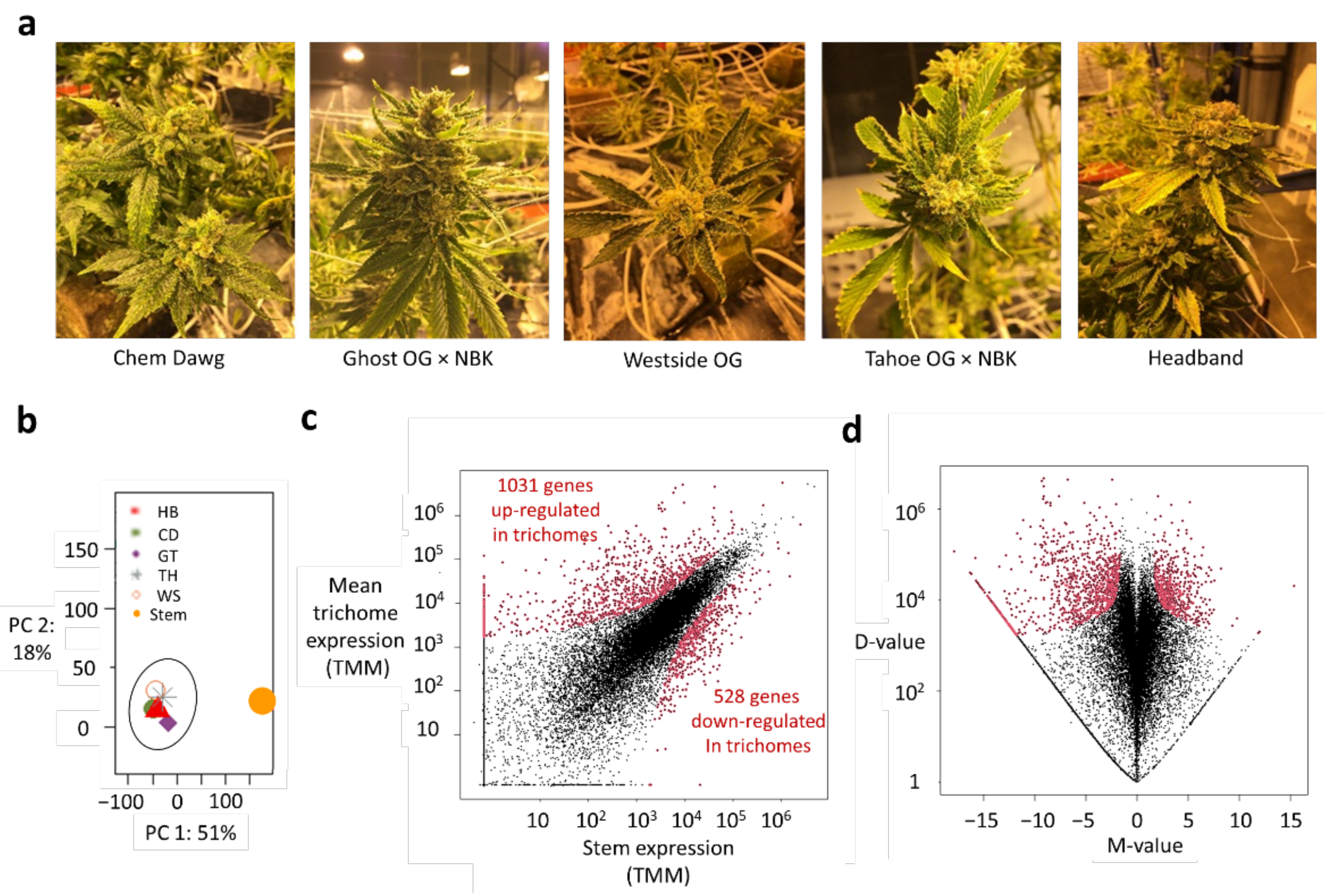
2.1. RNA-seq-Based Approach for Deciphering Conserved Expressions of Metabolic Enzymes
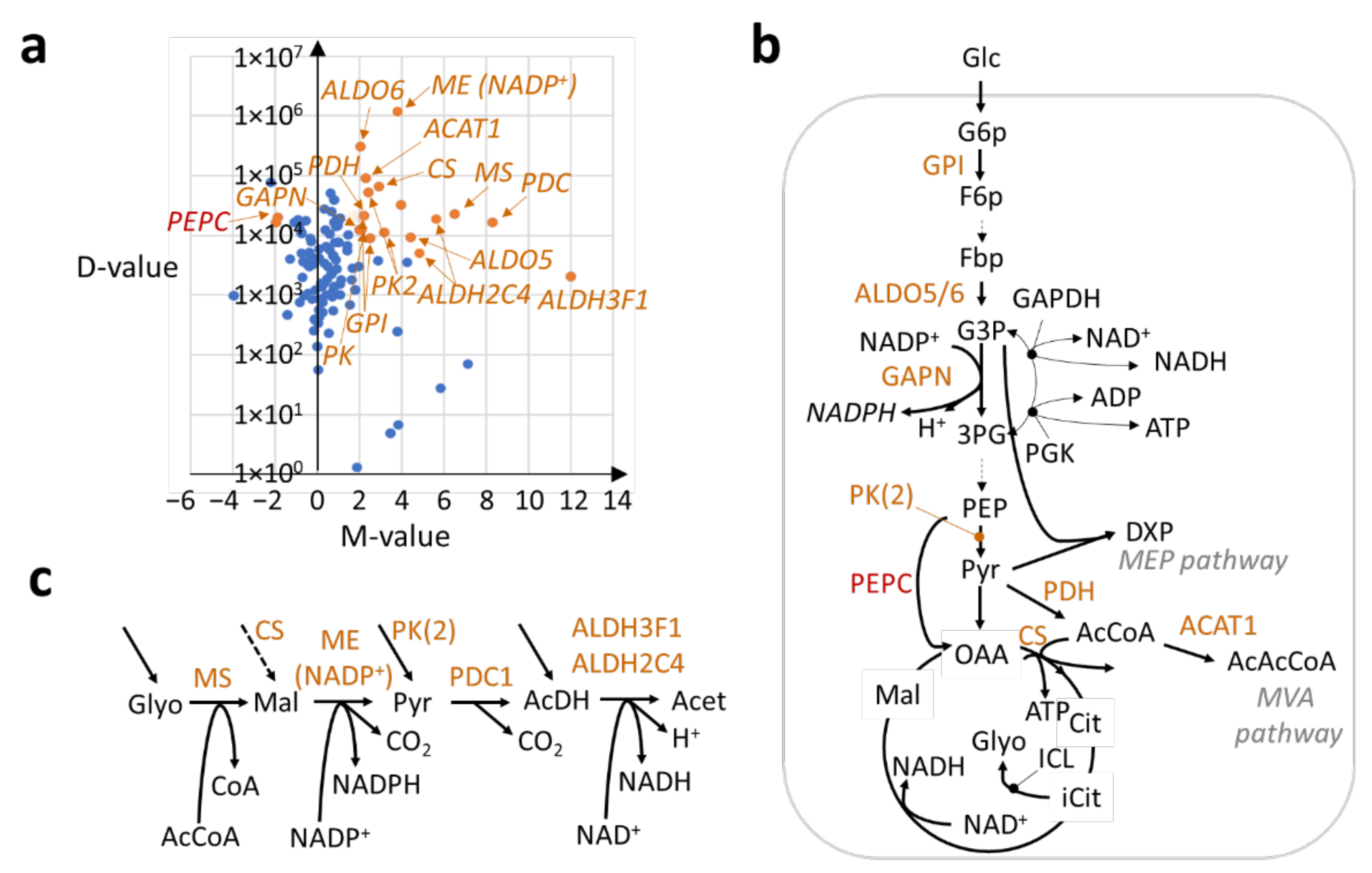
2.2. Transcriptional Regulation of the Central Metabolism in Trichomes for Enhancing Carbon Feedstock Production
2.3. An Uncovered Pathway for Meeting NAD(P)H Demand in Trichomes
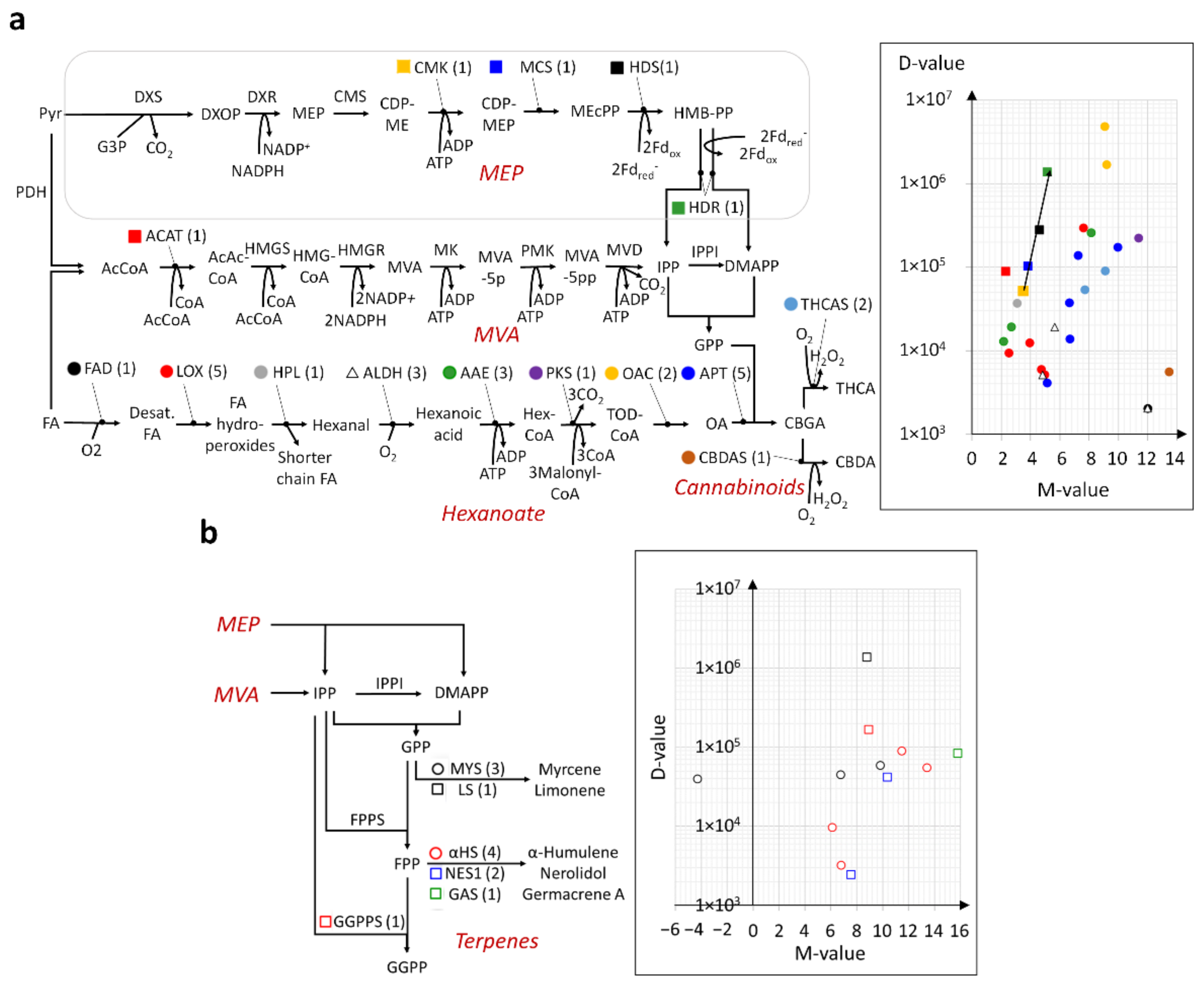
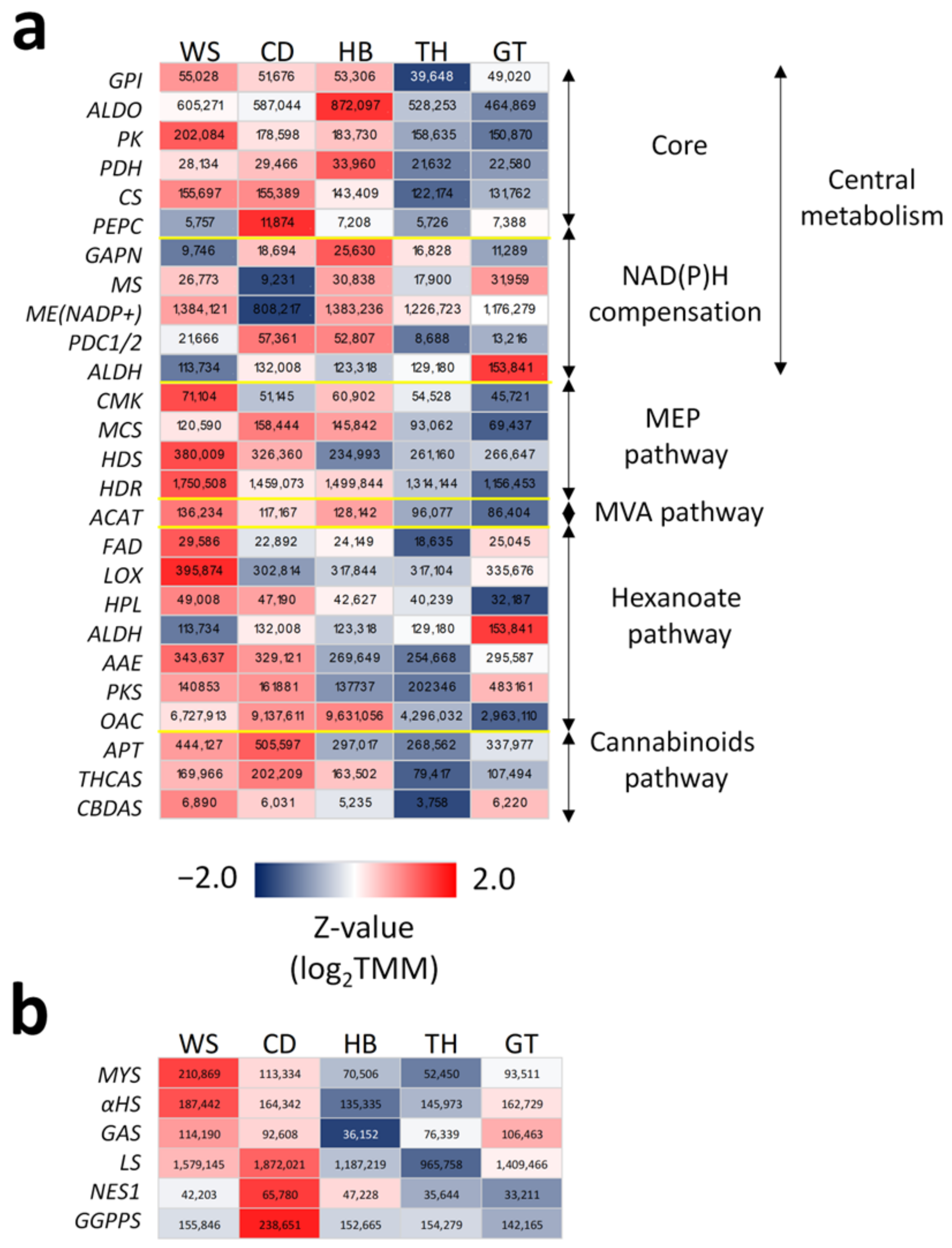
2.4. Modular and Calibrated Regulation of MEP and MVA Pathways
2.5. En Bloc and Multi-Loci Up-Regulation of the Hexanoate Pathway and Cannabinoid Biosynthesis
2.6. Metabolic Profiles of Cultivars
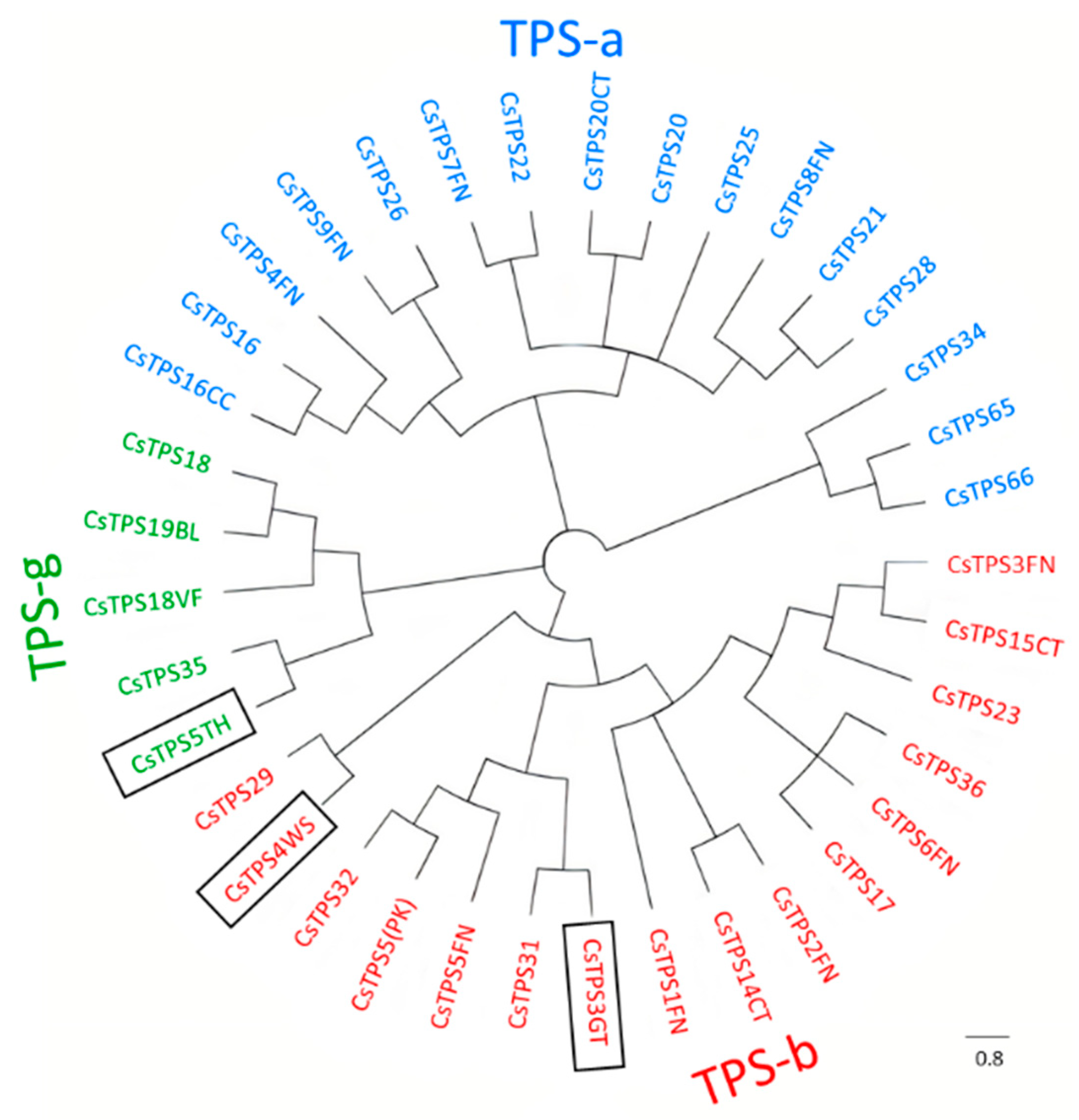
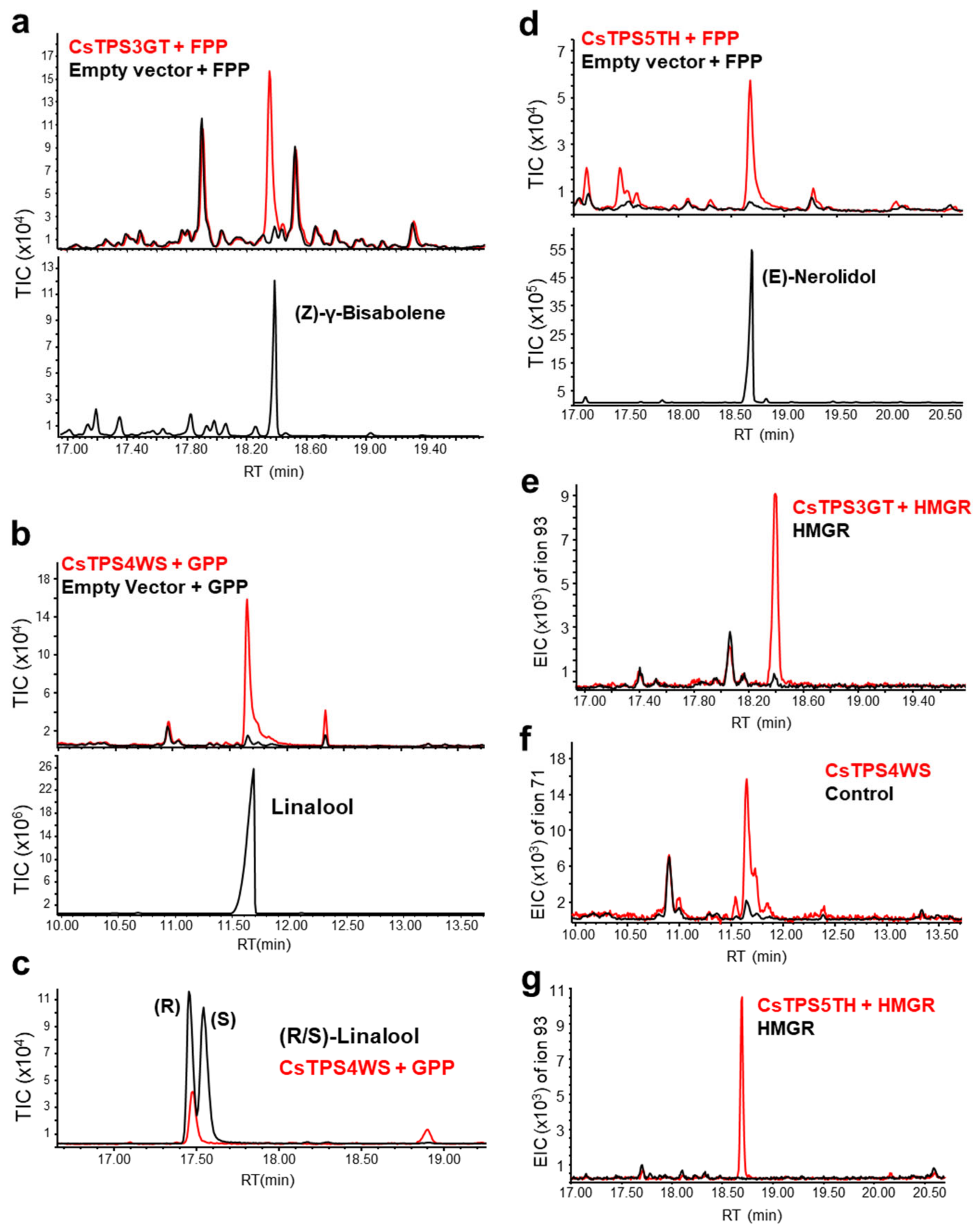
2.7. Functional Characterization of New TPSs
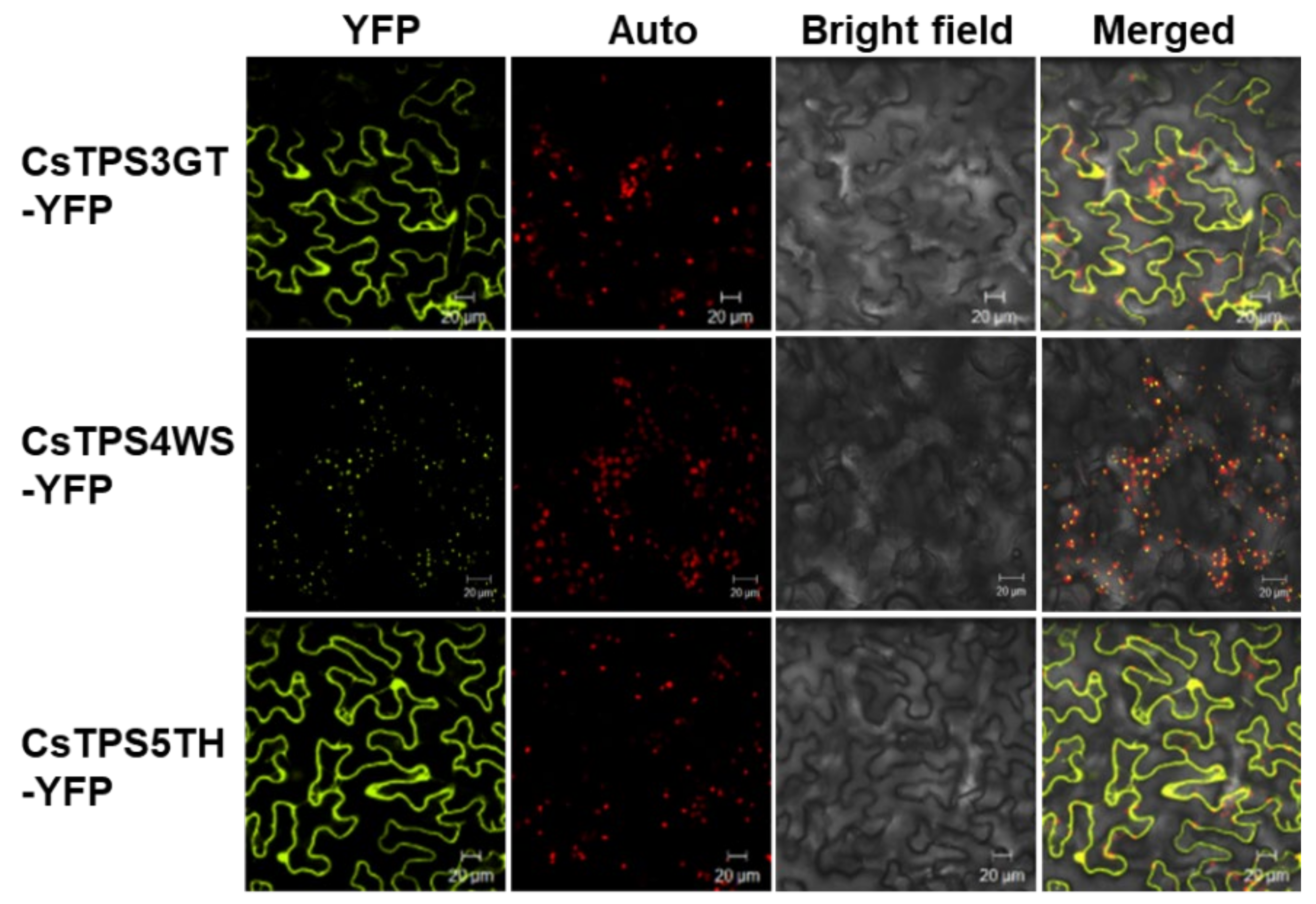
2.8. In Vitro and In Vivo Identification of CsTPSs
3. Discussion
4. Materials and Methods
4.1. Plant Material, Trichome Isolation, and RNA Isolation
4.2. RNA Library, RNA-seq, Pre-Processing, and Quality Control
4.3. Reference Genome Mapping and GC-Bias Correction
4.4. Gene-Level Expression, Normalization, and Quality Control
4.5. Differential Expression Analysis
4.6. Hierarchical Clustering and Venn Diagram Analysis of DEG
4.7. Gene Mapping to Metabolic Pathways
4.8. Quantitative Real-Time PCR (qRT-PCR)
4.9. Phylogenetic Tree and Clustal Analysis
4.10. Subcellular Localization of TPSs
4.11. In Vitro and In Vivo TPS Assays
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Izzo, A.A.; Borrelli, F.; Capasso, R.; Di Marzo, V.; Mechoulam, R. Non-psychotropic plant cannabinoids: New therapeutic opportunities from an ancient herb. Trends Pharmacol. Sci. 2009, 30, 515–527. [Google Scholar] [CrossRef]
- Namdar, D.; Anis, O.; Poulin, P.; Koltai, H. Chronological Review and Rational and Future Prospects of Cannabis-Based Drug Development. Molecules 2020, 25, 4821. [Google Scholar] [CrossRef]
- Small, E.; Beckstead, H.D.; Chan, A. The Evolution of Cannabinoid Phenotypes in Cannabis. Econ. Bot. 1975, 29, 219–232. [Google Scholar] [CrossRef]
- Gülck, T.; Møller, B.L. Phytocannabinoids: Origins and Biosynthesis. Trends Plant Sci. 2020, 25, 985–1004. [Google Scholar] [CrossRef] [PubMed]
- Fellermeier, M.; Eisenreich, W.; Bacher, A.; Zenk, M.H. Biosynthesis of cannabinoids. Eur. J. Biochem. 2001, 268, 1596–1604. [Google Scholar] [CrossRef] [PubMed]
- Rodziewicz, P.; Loroch, S.; Marczak, Ł.; Sickmann, A.; Kayser, O. Cannabinoid synthases and osmoprotective metabolites accumulate in the exudates of Cannabis sativa L. glandular trichomes. Plant Sci. 2019, 284, 108–116. [Google Scholar] [CrossRef] [PubMed]
- Van Bakel, H.; Stout, J.M.; Cote, A.G.; Tallon, C.M.; Sharpe, A.G.; Hughes, T.R.; Page, J.E. The draft genome and transcriptome of Cannabis sativa. Genome Biol. 2011, 12, R102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanuš, L.O.; Meyer, S.M.; Muñoz, E.; Taglialatela-Scafati, O.; Appendino, G. Phytocannabinoids: A unified critical inventory. Nat. Prod. Rep. 2016, 33, 1357–1392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- ElSohly, M.A.; Radwan, M.M.; Gul, W.; Chandra, S.; Galal, A. Phytochemistry of Cannabis sativa L. In Phytocannabinoids: Unraveling the Complex Chemistry and Pharmacology of Cannabis sativa; Kinghorn, A.D., Falk, H., Gibbons, S., Kobayashi, J.I., Eds.; Springer International Publishing: Cham, Switzerland, 2017; pp. 1–36. [Google Scholar]
- ElSohly, M.A.; Slade, D. Chemical constituents of marijuana: The complex mixture of natural cannabinoids. Life Sci. 2005, 78, 539–548. [Google Scholar] [CrossRef] [PubMed]
- Sirikantaramas, S.; Morimoto, S.; Shoyama, Y.; Ishikawa, Y.; Wada, Y.; Shoyama, Y.; Taura, F. The gene controlling marijuana psychoactivity: Molecular cloning and heterologous expression of δ1-tetrahydrocannabinolic acid synthase from Cannabis sativa L. J. Biol. Chem. 2004, 279, 39767–39774. [Google Scholar] [CrossRef] [Green Version]
- Taura, F.; Sirikantaramas, S.; Shoyama, Y.; Yoshikai, K.; Shoyama, Y.; Morimoto, S. Cannabidiolic-acid synthase, the chemotype-determining enzyme in the fiber-type Cannabis sativa. FEBS Lett. 2007, 581, 2929–2934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taura, F.; Tanaka, S.; Taguchi, C.; Fukamizu, T.; Tanaka, H.; Shoyama, Y.; Morimoto, S. Characterization of olivetol synthase, a polyketide synthase putatively involved in cannabinoid biosynthetic pathway. FEBS Lett. 2009, 583, 2061–2066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Welling, M.T.; Shapter, T.; Rose, T.J.; Liu, L.; Stanger, R.; King, G.J. A Belated Green Revolution for Cannabis: Virtual Genetic Resources to Fast-Track Cultivar Development. Front. Plant Sci. 2016, 7, 1113. [Google Scholar] [CrossRef]
- Booth, J.K.; Bohlmann, J. Terpenes in Cannabis sativa—From plant genome to humans. Plant Sci. 2019, 284, 67–72. [Google Scholar] [CrossRef] [PubMed]
- Russo, E.B. Taming THC: Potential cannabis synergy and phytocannabinoid-terpenoid entourage effects. Br. J. Pharmacol. 2011, 163, 1344–1364. [Google Scholar] [CrossRef]
- Russo, E.B. The Case for the Entourage Effect and Conventional Breeding of Clinical Cannabis: No “Strain,” No Gain. Front. Plant Sci. 2019, 9, 1969. [Google Scholar] [CrossRef]
- Gertsch, J.; Leonti, M.; Raduner, S.; Racz, I.; Chen, J.-Z.; Xie, X.-Q.; Altmann, K.-H.; Karsak, M.; Zimmer, A. Beta-caryophyllene is a dietary cannabinoid. Proc. Natl. Acad. Sci. USA 2008, 105, 9099–9104. [Google Scholar] [CrossRef] [Green Version]
- Guirimand, G.; Guihur, A.; Perello, C.; Phillips, M.; Mahroug, S.; Oudin, A.; Dugé de Bernonville, T.; Besseau, S.; Lanoue, A.; Giglioli-Guivarc’h, N.; et al. Cellular and Subcellular Compartmentation of the 2C-Methyl-D-Erythritol 4-Phosphate Pathway in the Madagascar Periwinkle. Plants 2020, 9, 462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vranová, E.; Coman, D.; Gruissem, W. Network Analysis of the MVA and MEP Pathways for Isoprenoid Synthesis. Annu. Rev. Plant Biol. 2013, 64, 665–700. [Google Scholar] [CrossRef] [PubMed]
- Thabet, I.; Guirimand, G.; Guihur, A.; Lanoue, A.; Courdavault, V.; Papon, N.; Bouzid, S.; Giglioli-Guivarc’h, N.; Simkin, A.J.; Clastre, M. Characterization and subcellular localization of geranylgeranyl diphosphate synthase from Catharanthus roseus. Mol. Biol. Rep. 2012, 39, 3235–3243. [Google Scholar] [CrossRef]
- Okada, K.; Saito, T.; Nakagawa, T.; Kawamukai, M.; Kamiya, Y. Five geranylgeranyl diphosphate synthases expressed in different organs are localized into three subcellular compartments in Arabidopsis. Plant Physiol. 2000, 122, 1045–1056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pu, X.; Dong, X.; Li, Q.; Chen, Z.; Liu, L. An update on the function and regulation of methylerythritol phosphate and mevalonate pathways and their evolutionary dynamics. J. Integr. Plant Biol. 2021, 63, 1211–1226. [Google Scholar] [CrossRef] [PubMed]
- Shiba, Y.; Paradise, E.M.; Kirby, J.; Ro, D.K.; Keasling, J.D. Engineering of the pyruvate dehydrogenase bypass in Saccharomyces cerevisiae for high-level production of isoprenoids. Metab. Eng. 2007, 9, 160–168. [Google Scholar] [CrossRef] [PubMed]
- Brocker, C.; Vasiliou, M.; Carpenter, S.; Carpenter, C.; Zhang, Y.; Wang, X.; Kotchoni, S.O.; Wood, A.J.; Kirch, H.-H.; Kopečný, D.; et al. Aldehyde dehydrogenase (ALDH) superfamily in plants: Gene nomenclature and comparative genomics. Planta 2013, 237, 189–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, L.; Chang, W.-C.; Xiao, Y.; Liu, H.-W.; Liu, P. Methylerythritol phosphate pathway of isoprenoid biosynthesis. Annu. Rev. Biochem. 2013, 82, 497–530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodríguez-Concepción, M. Early Steps in Isoprenoid Biosynthesis: Multilevel Regulation of the Supply of Common Precursors in Plant Cells. Phytochem. Rev. 2006, 5, 1–15. [Google Scholar] [CrossRef]
- Booth, J.K.; Page, J.E.; Bohlmann, J. Terpene synthases from Cannabis sativa. PLoS ONE 2017, 12, e0173911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Booth, J.K.; Yuen, M.M.S.; Jancsik, S.; Madilao, L.L.; Page, J.E.; Bohlmann, J. Terpene Synthases and Terpene Variation in Cannabis sativa. Plant Physiol. 2020, 184, 130–147. [Google Scholar] [CrossRef]
- Allen, K.D.; McKernan, K.; Pauli, C.; Roe, J.; Torres, A.; Gaudino, R. Genomic characterization of the complete terpene synthase gene family from Cannabis sativa. PLoS ONE 2019, 14, e0222363. [Google Scholar]
- Zager, J.J.; Lange, I.; Srividya, N.; Smith, A.; Lange, B.M. Gene Networks Underlying Cannabinoid and Terpenoid Accumulation in Cannabis. J. Plant Physiol. 2019, 180, 1877–1897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, D.M.; Aubourg, S.; Schouwey, M.B.; Daviet, L.; Schalk, M.; Toub, O.; Lund, S.T.; Bohlmann, J. Functional Annotation, Genome Organization and Phylogeny of the Grapevine (Vitis vinifera) Terpene Synthase Gene Family Based on Genome Assembly, FLcDNA Cloning, and Enzyme Assays. BMC Plant Biol. 2010, 10, 226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, Y.; Khan, F.; Rastogi, S.; Shasany, A.K. Genome-wide detection of terpene synthase genes in holy basil (Ocimum sanctum L.). PLoS ONE 2018, 13, e0207097. [Google Scholar] [CrossRef] [PubMed]
- Dhandapani, S.; Jin, J.; Sridhar, V.; Sarojam, R.; Chua, N.-H.; Jang, I.-C. Integrated metabolome and transcriptome analysis of Magnolia champaca identifies biosynthetic pathways for floral volatile organic compounds. BMC Genom. 2017, 18, 463. [Google Scholar] [CrossRef] [PubMed]
- Pichersky, E.; Raguso, R.A.; Lewinsohn, E.; Croteau, R. Floral Scent Production in Clarkia (Onagraceae) (I. Localization and Developmental Modulation of Monoterpene Emission and Linalool Synthase Activity). J. Plant Physiol. 1994, 106, 1533. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Quan, S.; Xiao, H. Towards efficient terpenoid biosynthesis: Manipulating IPP and DMAPP supply. Bioresour. Bioprocess. 2019, 6, 6. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Sun, J.; Yang, Q.; Yang, J. Metabolic Engineering Escherichia coli for the Production of Lycopene. Molecules 2020, 25, 3136. [Google Scholar] [CrossRef] [PubMed]
- Coussement, P.; Bauwens, D.; Maertens, J.; De Mey, M. Direct Combinatorial Pathway Optimization. ACS Synth. Biol. 2017, 6, 224–232. [Google Scholar] [CrossRef]
- Zhao, J.; Li, Q.; Sun, T.; Zhu, X.; Xu, H.; Tang, J.; Zhang, X.; Ma, Y. Engineering central metabolic modules of Escherichia coli for improving β-carotene production. Metab. Eng. 2013, 17, 42–50. [Google Scholar] [CrossRef] [PubMed]
- Rasheed, S.; Bashir, K.; Kim, J.-M.; Ando, M.; Tanaka, M.; Seki, M. The modulation of acetic acid pathway genes in Arabidopsis improves survival under drought stress. Sci. Rep. 2018, 8, 7831. [Google Scholar] [CrossRef]
- Rivoal, J.; Thind, S.; Pradet, A.; Ricard, B. Differential Induction of Pyruvate Decarboxylase Subunits and Transcripts in Anoxic Rice Seedlings. Plant Physiol. 1997, 114, 1021–1029. [Google Scholar] [CrossRef] [Green Version]
- Fu, X.; Yang, H.; Pangestu, F.; Nikolau, B.J. Failure to Maintain Acetate Homeostasis by Acetate-Activating Enzymes Impacts Plant Development. Plant Physiol. 2020, 182, 1256–1271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, S.R.; Lange, I.; Srividya, N.; Lange, B.M. Bioenergetics of Monoterpenoid Essential Oil Biosynthesis in Nonphotosynthetic Glandular Trichomes. Plant Physiol. 2017, 175, 681–695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Livingston, S.J.; Quilichini, T.D.; Booth, J.K.; Wong, D.C.J.; Rensing, K.H.; Laflamme-Yonkman, J.; Castellarin, S.D.; Bohlmann, J.; Page, J.E.; Samuels, A.L. Cannabis glandular trichomes alter morphology and metabolite content during flower maturation. Plant J. 2020, 101, 37–56. [Google Scholar] [CrossRef] [PubMed]
- Shlomi, T.; Benyamini, T.; Gottlieb, E.; Sharan, R.; Ruppin, E. Genome-Scale Metabolic Modeling Elucidates the Role of Proliferative Adaptation in Causing the Warburg Effect. PLoS Comput. Biol. 2011, 7, e1002018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeo, H.C.; Hong, J.; Lakshmanan, M.; Lee, D.-Y. Enzyme capacity-based genome scale modelling of CHO cells. Metab. Eng. 2020, 60, 138–147. [Google Scholar] [CrossRef] [PubMed]
- Booth, J. Terpene and Isoprenoid Biosynthesis in Cannabis Sativa; The University of British Columbia: Vancouver, BC, Canada, 2020. [Google Scholar]
- Braich, S.; Baillie, R.C.; Jewell, L.S.; Spangenberg, G.C.; Cogan, N.O.I. Generation of a Comprehensive Transcriptome Atlas and Transcriptome Dynamics in Medicinal Cannabis. Sci. Rep. 2019, 9, 16583. [Google Scholar] [CrossRef] [PubMed]
- Aizpurua-Olaizola, O.; Soydaner, U.; Ozturk, E.; Schibano, D.; Simsir, Y.; Navarro, P.; Etxebarria, N.; Usobiaga, A. Evolution of the Cannabinoid and Terpene Content during the Growth of Cannabis sativa Plants from Different Chemotypes. J. Nat. Prod. 2016, 79, 324–331. [Google Scholar] [CrossRef] [PubMed]
- Whited, G.M.; Feher, F.J.; Benko, D.A.; Cervin, M.A.; Chotani, G.K.; McAuliffe, J.C.; LaDuca, R.J.; Ben-Shoshan, E.A.; Sanford, K.J. TECHNOLOGY UPDATE: Development of a gas-phase bioprocess for isoprene-monomer production using metabolic pathway engineering. Ind. Biotechnol. 2010, 6, 152–163. [Google Scholar] [CrossRef]
- Rude, M.A.; Schirmer, A. New microbial fuels: A biotech perspective. Curr. Opin. Microbiol. 2009, 12, 274–281. [Google Scholar] [CrossRef] [PubMed]
- Wille, A.; Zimmermann, P.; Vranová, E.; Fürholz, A.; Laule, O.; Bleuler, S.; Hennig, L.; Prelic, A.; von Rohr, P.; Thiele, L.; et al. Sparse graphical Gaussian modeling of the isoprenoid gene network in Arabidopsis thaliana. Genome Biol. 2004, 5, R92. [Google Scholar] [CrossRef] [Green Version]
- Estévez, J.M.; Cantero, A.; Reindl, A.; Reichler, S.; León, P. 1-Deoxy-d-xylulose-5-phosphate Synthase, a Limiting Enzyme for Plastidic Isoprenoid Biosynthesis in Plants*. J. Biol. Chem. 2001, 276, 22901–22909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hemmerlin, A.; Harwood, J.L.; Bach, T.J. A raison d’être for two distinct pathways in the early steps of plant isoprenoid biosynthesis? Prog. Lipid Res. 2012, 51, 95–148. [Google Scholar] [CrossRef] [PubMed]
- Pechous, S.W.; Whitaker, B.D. Cloning and functional expression of an (E,E)-α-farnesene synthase cDNA from peel tissue of apple fruit. Planta 2004, 219, 84–94. [Google Scholar] [CrossRef] [Green Version]
- Baser, K.H.C.; Özek, T.; Konakchiev, A. Enantiomeric Distribution of Linalool, Linalyl Acetate and Camphor in Bulgarian Lavender Oil. J. Essent. Oil Res. 2005, 17, 135–136. [Google Scholar] [CrossRef]
- Padrayuttawat, A.; Yoshizawa, T.; Tamura, H.; Tokunaga, T. Optical Isomers and Odor Thresholds of Volatile Constituents in Citrus sudachi. Food Sci. Technol. Int. Tokyo 1997, 3, 402–408. [Google Scholar] [CrossRef] [Green Version]
- Jin, J.; Panicker, D.; Wang, Q.; Kim, M.J.; Liu, J.; Yin, J.-L.; Wong, L.; Jang, I.-C.; Chua, N.-H.; Sarojam, R. Next generation sequencing unravels the biosynthetic ability of Spearmint (Mentha spicata) peltate glandular trichomes through comparative transcriptomics. BMC Plant Biol. 2014, 14, 292. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.J.; Jin, J.; Zheng, J.; Wong, L.; Chua, N.-H.; Jang, I.-C. Comparative Transcriptomics Unravel Biochemical Specialization of Leaf Tissues of Stevia for Diterpenoid Production. Plant Physiol. 2015, 169, 2462–2480. [Google Scholar] [CrossRef] [Green Version]
- Jin, D.; Dai, K.; Xie, Z.; Chen, J. Secondary Metabolites Profiled in Cannabis Inflorescences, Leaves, Stem Barks, and Roots for Medicinal Purposes. Sci. Rep. 2020, 10, 3309. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. v0.11.9. 2010. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc (accessed on 16 September 2020).
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 2017, 14, 417–419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soneson, C.; Love, M.I.; Robinson, M.D. Differential analyses for RNA-seq: Transcript-level estimates improve gene-level inferences. F1000Research 2015, 4, 1521. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; Oshlack, A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 2010, 11, R25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tarazona, S.; Furió-Tarí, P.; Turrà, D.; Pietro, A.D.; Nueda, M.J.; Ferrer, A.; Conesa, A. Data quality aware analysis of differential expression in RNA-seq with NOISeq R/Bioc package. Nucleic Acids Res. 2015, 43, e140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heberle, H.; Meirelles, G.V.; da Silva, F.R.; Telles, G.P.; Minghim, R. InteractiVenn: A web-based tool for the analysis of sets through Venn diagrams. BMC Bioinform. 2015, 16, 169. [Google Scholar] [CrossRef] [PubMed]
- Götz, S.; García-Gómez, J.M.; Terol, J.; Williams, T.D.; Nagaraj, S.H.; Nueda, M.J.; Robles, M.; Talón, M.; Dopazo, J.; Conesa, A. High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res. 2008, 36, 3420–3435. [Google Scholar] [CrossRef] [PubMed]
- Walker, N.J. A Technique Whose Time Has Come. Science 2002, 296, 557. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Garreton, V.; Chua, N.-H. The AIP2 E3 ligase acts as a novel negative regulator of ABA signaling by promoting ABI3 degradation. Genes Dev. 2005, 19, 1532–1543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Höfgen, R.; Willmitzer, L. Storage of competent cells for Agrobacterium transformation. Nucleic Acids Res. 1988, 16, 9877. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tissue | Cultivar | Raw Reads (mil) | Reads after QC (mil) | Mapped Reads (mil) | % Raw Reads after QC | % Raw Reads after Further Mapping |
|---|---|---|---|---|---|---|
| Stem | HB | 248 | 228 | 200 | 92% | 88% |
| Trichome | CD | 273 | 261 | 227 | 96% | 87% |
| Trichome | GT | 256 | 249 | 215 | 97% | 86% |
| Trichome | HB | 260 | 251 | 215 | 97% | 86% |
| Trichome | TH | 273 | 262 | 229 | 96% | 87% |
| Trichome | WS | 277 | 267 | 235 | 96% | 88% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yeo, H.C.; Reddy, V.A.; Mun, B.-G.; Leong, S.H.; Dhandapani, S.; Rajani, S.; Jang, I.-C. Comparative Transcriptome Analysis Reveals Coordinated Transcriptional Regulation of Central and Secondary Metabolism in the Trichomes of Cannabis Cultivars. Int. J. Mol. Sci. 2022, 23, 8310. https://doi.org/10.3390/ijms23158310
Yeo HC, Reddy VA, Mun B-G, Leong SH, Dhandapani S, Rajani S, Jang I-C. Comparative Transcriptome Analysis Reveals Coordinated Transcriptional Regulation of Central and Secondary Metabolism in the Trichomes of Cannabis Cultivars. International Journal of Molecular Sciences. 2022; 23(15):8310. https://doi.org/10.3390/ijms23158310
Chicago/Turabian StyleYeo, Hock Chuan, Vaishnavi Amarr Reddy, Bong-Gyu Mun, Sing Hui Leong, Savitha Dhandapani, Sarojam Rajani, and In-Cheol Jang. 2022. "Comparative Transcriptome Analysis Reveals Coordinated Transcriptional Regulation of Central and Secondary Metabolism in the Trichomes of Cannabis Cultivars" International Journal of Molecular Sciences 23, no. 15: 8310. https://doi.org/10.3390/ijms23158310
APA StyleYeo, H. C., Reddy, V. A., Mun, B.-G., Leong, S. H., Dhandapani, S., Rajani, S., & Jang, I.-C. (2022). Comparative Transcriptome Analysis Reveals Coordinated Transcriptional Regulation of Central and Secondary Metabolism in the Trichomes of Cannabis Cultivars. International Journal of Molecular Sciences, 23(15), 8310. https://doi.org/10.3390/ijms23158310