
Targeting Fibronectin to Overcome Remyelination Failure in Multiple Sclerosis: The Need for Brain- and Lesion-Targeted Drug Delivery



Abstract
:1. Introduction
2. Multiple Sclerosis: An Unmet Need for a Remyelination-Based Therapy to Stop Disease Progression
3. Remyelination Failure in MS: Perturbed ECM Remodeling in White Matter Lesions
4. Fibronectin: Multifaceted Roles in the CNS and in the Pathogenesis of MS
4.1. Fibronectin and Its Role in BBB Functioning
4.2. Fibronectin as a Vasculogenic Regulator in MS
4.3. Fibronectin (Aggregates) as a Remyelination Inhibitor in MS
5. Promoting Remyelination in MS: Therapeutic Strategies to Overcome Fn-Mediated Inhibition of Remyelination Failure

{kind=link}
{kind=link}
{kind=link}
| Strategy | Method | Mechanism of Action | Reference |
|---|---|---|---|
| Prevent Fn expression | Prevent TG2 expression or activity | Mediates Fn expression and deposition | [161,225] |
| Prevent Fn aggregation | Modulate Fn splicing | Induces conformational changes in Fn to increase cell surface binding | [198] |
| Prevent Fn aggregation | Prevent TLR3 signaling (astrocytes) | Prevents the release of Fn fibrils from the cell surface | [198] |
| Prevent Fn aggregation | Modulate HSP90β activity | Contributes to the unfolding of Fn to facilitate Fn fibrillogenesis | [223] |
| Degrade Fn aggregates | Increase MMP7 expression and activity | Cleaves Fn, including Fn aggregates | [211] |
| Bypass Fn aggregates | Treat with ganglioside GD1a | Overcomes the Fn-mediated inhibition of OPC maturation via a PKA-mediated signaling pathway | [18] |
| Bypass Fn aggregates | Treat with PDE inhibitors | Prolongs cAMP levels, thereby potentially activating PKA, and enhances CNS remyelination | [194,195,196,197] |
6. MS Therapeutics: Drug Delivery Vehicles for Delivery to the Brain
6.1. Lipid-Based Nanoparticles as Drug Delivery Vehicle for RRMS Treatment
| Treatment | Administration Means | Administration Time Point | Outcome Measure | Reference |
|---|---|---|---|---|
| Experimental MS Models | ||||
| 99mTc-DTPA-loaded liposomes in EAE | Intravenous | At induction of disease | Biodistribution of liposomes | [258] |
| MOG40–55-loaded liposomes in EAE | Intraperitoneal | At induction of disease | Preventive and preclinical treatment effects on EAE development | [267] |
| MBP-loaded liposomes in EAE | Subcutaneous | At disease onset for 6 days | Effect of different MBP isoforms on EAE progression | [275] |
| Prednisolone-loaded liposomes in EAE | Intravenous | At peak of disease | Effect on EAE progression, BBB permeability, and drug biodistribution | [261] |
| (Methyl)prednisolone-loaded liposomes in EAE | Intravenous | At peak of disease | Effect on EAE progression and macrophage functioning | [262] |
| Methylprednisolone-loaded liposomes in EAE | Intravenous | Prophylactic, at disease onset, and disease peak | Brain-targeted effect on EAE symptoms | [264] |
| MOG-loaded PLGA particles in EAE | Intravenous/subcutaneous | Prophylactic | Effect on EAE development | [268] |
| MOG-anti-Fas-PD-L1-Fc-CD47-Fc-TGFβ-loaded PLGA particles in EAE | Intravenous | At disease onset and disease peak | Modulation of auto-reactive T cells in EAE and disease progression | [269] |
| MOG-IL10-loaded PLGA particles in EAE | Subcutaneous | Prophylactic, at disease onset, and disease peak | Effect of ‘inverse vaccination’ on EAE progression | [270] |
| PLP-coupled PLGA particles in EAE | Intravenous | At disease onset | Treatment of EAE and nanoparticle uptake in vitro by antigen-presenting cells | [272] |
| PHCCC-loaded PLGA particles in EAE | Subcutaneous | From induction of disease, every 3 or 5 days | Effect on DC activation and EAE disease progression | [273] |
| miR-219a-5p liposomes, PLGA particles, and extracellular vesicles in EAE | Intranasal | 2 and 8 days post-induction of disease (before symptom onset) | Effect on remyelination in EAE | [274] |
| Curcumin-loaded HPPS in EAE | Intravenous | 8, 10, 12, and 14 days post-induction of disease | Restriction of immune cell infiltration of the brain in EAE by modulation of monocytes | [259] |
| PLP-coupled PLGA particles in relapsing–remitting EAE | Intravenous | At disease onset, disease peak, and disease remission | Prevention and treatment of relapsing EAE disease | [271] |
| (Methyl)prednisolone-loaded liposomes in chronic relapsing EAE | Intravenous | At first peak of disease | Effect on disease progression, their effect on relapse risk, and macrophage CNS infiltration | [263] |
| Dimethyl-fumarate-loaded solid lipid nanoparticles in cuprizone | Oral | Daily cuprizone and nanoparticles for 30 days | Effect on remyelination | [266] |
| LIF-loaded PLGA particles in focal demyelination | Intralesional | 8 days post-lysolecithin lesioning | Effect on OPC differentiation in vitro and remyelination in vivo | [279] |
| MS Patients | ||||
| MBP-loaded liposomes | Subcutaneous | Once a week for 6 weeks | Safety profile of CD206-targeted liposomal MBP treatment in RRMS and SPMS patients | [286] |
| MBP-loaded liposomes | Subcutaneous | Once a week for 6 weeks | Serum cytokine analysis and Th1/Th2 ratio in RRMS and SPMS patients | [278] |
6.2. Polymer-Based Nanoparticles as a Drug Delivery Vehicle for RRMS Treatment
6.3. Drug Delivery Vehicles for Treatment of Progressive MS
7. Progressive MS Treatment: Considerations for Designing a Brain-Targeted Drug Delivery System
7.1. Receptor-Mediated Transcytosis (RMT)
7.2. Adsorptive-Mediated Transport (AMT)
7.3. Focused Ultrasound
7.4. Intranasal Drug Delivery
8. MS Therapeutics: Considerations for Intracellular Delivery of Therapeutic Agents
9. Active Targeting to MS Lesions: Considerations for Controlled Drug Delivery to Overcome Fibronectin-Mediated Inhibition of Remyelination
9.1. Active Targeting to Fn Aggregates
9.2. Active Targeting of Cells
10. Concluding Remarks and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Oh, J.; Vidal-Jordana, A.; Montalban, X. Multiple Sclerosis. Curr. Opin. Neurol. 2018, 31, 752–759. [Google Scholar] [CrossRef] [PubMed]
- Montalban, X.; Tintoré, M.; Swanton, J.; Barkhof, F.; Fazekas, F.; Filippi, M.; Frederiksen, J.; Kappos, L.; Palace, J.; Polman, C.; et al. MRI Criteria for MS in Patients with Clinically Isolated Syndromes. Neurology 2010, 74, 427–434. [Google Scholar] [CrossRef] [PubMed]
- Mahad, D.H.; Trapp, B.D.; Lassmann, H. Pathological Mechanisms in Progressive Multiple Sclerosis. Lancet Neurol. 2015, 14, 183–193. [Google Scholar] [CrossRef]
- Lublin, F.D.; Reingold, S.C. Defining the Clinical Course of Multiple Sclerosis: Results of an International Survey. Neurology 1996, 46, 907–911. [Google Scholar] [CrossRef] [PubMed]
- Gourraud, P.-A.; Harbo, H.F.; Hauser, S.L.; Baranzini, S.E. The Genetics of Multiple Sclerosis: An up-to-Date Review. Immunol. Rev. 2012, 248, 87–103. [Google Scholar] [CrossRef] [Green Version]
- Ascherio, A.; Munger, K.L. Environmental Risk Factors for Multiple Sclerosis. Part I: The Role of Infection. Ann. Neurol. 2007, 61, 288–299. [Google Scholar] [CrossRef] [PubMed]
- Bjornevik, K.; Cortese, M.; Healy, B.C.; Kuhle, J.; Mina, M.J.; Leng, Y.; Elledge, S.J.; Niebuhr, D.W.; Scher, A.I.; Munger, K.L.; et al. Longitudinal Analysis Reveals High Prevalence of Epstein-Barr Virus Associated with Multiple Sclerosis. Science 2022, 375, 296–301. [Google Scholar] [CrossRef] [PubMed]
- Amato, M.P.; Derfuss, T.; Hemmer, B.; Liblau, R.; Montalban, X.; Soelberg Sørensen, P.; Miller, D.H.; Alfredsson, L.; Aloisi, F.; Ascherio, A.; et al. Environmental Modifiable Risk Factors for Multiple Sclerosis: Report from the 2016 ECTRIMS Focused Workshop. Mult. Scler. J. 2018, 24, 590–603. [Google Scholar] [CrossRef] [Green Version]
- Trapp, B.D.; Nave, K.-A. Multiple Sclerosis: An Immune or Neurodegenerative Disorder? Annu. Rev. Neurosci. 2008, 31, 247–269. [Google Scholar] [CrossRef] [PubMed]
- Fünfschilling, U.; Supplie, L.M.; Mahad, D.; Boretius, S.; Saab, A.S.; Edgar, J.; Brinkmann, B.G.; Kassmann, C.M.; Tzvetanova, I.D.; Möbius, W.; et al. Glycolytic Oligodendrocytes Maintain Myelin and Long-Term Axonal Integrity. Nature 2012, 485, 517–521. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.; Morrison, B.M.; Li, Y.; Lengacher, S.; Farah, M.H.; Hoffman, P.N.; Liu, Y.; Tsingalia, A.; Jin, L.; Zhang, P.W.; et al. Oligodendroglia Metabolically Support Axons and Contribute to Neurodegeneration. Nature 2012, 487, 443–448. [Google Scholar] [CrossRef] [PubMed]
- Irvine, K.A.; Blakemore, W.F. Remyelination Protects Axons from Demyelination-Associated Axon Degeneration. Brain 2008, 131, 1464–1477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franklin, R.J.M.; Frisén, J.; Lyons, D.A. Revisiting Remyelination: Towards a Consensus on the Regeneration of CNS Myelin. Semin. Cell Dev. Biol. 2020, 116, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Heß, K.; Starost, L.; Kieran, N.W.; Thomas, C.; Vincenten, M.C.J.; Antel, J.; Martino, G.; Huitinga, I.; Healy, L.; Kuhlmann, T. Lesion Stage-Dependent Causes for Impaired Remyelination in MS. Acta Neuropathol. 2020, 140, 359–375. [Google Scholar] [CrossRef] [PubMed]
- de Jong, J.M.; Wang, P.; Oomkens, M.; Baron, W. Remodeling of the Interstitial Extracellular Matrix in White Matter Multiple Sclerosis Lesions: Implications for Remyelination (Failure). J. Neurosci. Res. 2020, 98, 1370–1397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghorbani, S.; Yong, V.W. The Extracellular Matrix as Modifier of Neuroinflammation and Remyelination in Multiple Sclerosis. Brain 2021, 144, 1958–1973. [Google Scholar] [CrossRef]
- Stoffels, J.M.J.; de Jonge, J.C.; Stancic, M.; Nomden, A.; van Strien, M.E.; Ma, D.; Šišková, Z.; Maier, O.; ffrench-Constant, C.; Franklin, R.J.M.; et al. Fibronectin Aggregation in Multiple Sclerosis Lesions Impairs Remyelination. Brain 2013, 136, 116–131. [Google Scholar] [CrossRef] [Green Version]
- Qin, J.; Sikkema, A.H.; van der Bij, K.; de Jonge, J.C.; Klappe, K.; Nies, V.; Jonker, J.W.; Kok, J.W.; Hoekstra, D.; Baron, W. GD1a Overcomes Inhibition of Myelination by Fibronectin via Activation of Protein Kinase A: Implications for Multiple Sclerosis. J. Neurosci. 2017, 37, 9925–9938. [Google Scholar] [CrossRef]
- Hohlfeld, R.; Wekerle, H. Autoimmune Concepts of Multiple Sclerosis as a Basis for Selective Immunotherapy: From Pipe Dreams to (Therapeutic) Pipelines. Proc. Natl. Acad. Sci. USA 2004, 101, 14599–14606. [Google Scholar] [CrossRef] [Green Version]
- Lucchinetti, C.; Brück, W.; Parisi, J.; Scheithauer, B.; Rodriguez, M.; Lassmann, H. Heterogeneity of Multiple Sclerosis Lesions: Implications for the Pathogenesis of Demyelination. Ann. Neurol. 2000, 47, 707–717. [Google Scholar] [CrossRef]
- Qin, Y.; Duquette, P.; Zhang, Y.; Talbot, P.; Poole, R.; Antel, J. Clonal Expansion and Somatic Hypermutation of V(H) Genes of B Cells from Cerebrospinal Fluid in Multiple Sclerosis. J. Clin. Investig. 1998, 102, 1045–1050. [Google Scholar] [CrossRef] [PubMed]
- Coles, A.J. Alemtuzumab Therapy for Multiple Sclerosis. Neurotherapeutics 2013, 10, 29–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bermel, R.A.; Bakshi, R. The Measurement and Clinical Relevance of Brain Atrophy in Multiple Sclerosis. Lancet Neurol. 2006, 5, 158–170. [Google Scholar] [PubMed]
- Franklin, R.J.M.; ffrench-Constant, C. Regenerating CNS Myelin—From Mechanisms to Experimental Medicines. Nat. Rev. Neurosci. 2017, 18, 753–769. [Google Scholar] [CrossRef]
- Kuhlmann, T.; Ludwin, S.; Prat, A.; Antel, J.; Brück, W.; Lassmann, H. An Updated Histological Classification System for Multiple Sclerosis Lesions. Acta Neuropathol. 2017, 133, 13–24. [Google Scholar] [CrossRef]
- Luchetti, S.; Fransen, N.L.; van Eden, C.G.; Ramaglia, V.; Mason, M.; Huitinga, I. Progressive Multiple Sclerosis Patients Show Substantial Lesion Activity that Correlates with Clinical Disease Severity and Sex: A Retrospective Autopsy Cohort Analysis. Acta Neuropathol. 2018, 135, 511–528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kutzelnigg, A.; Lucchinetti, C.F.; Stadelmann, C.; Brück, W.; Rauschka, H.; Bergmann, M.; Schmidbauer, M.; Parisi, J.E.; Lassmann, H. Cortical Demyelination and Diffuse White Matter Injury in Multiple Sclerosis. Brain 2005, 128, 2705–2712. [Google Scholar] [CrossRef]
- Ferguson, B.; Matyszak, M.K.; Esiri, M.M.; Perry, V.H. Axonal Damage in Acute Multiple Sclerosis Lesions. Brain 1997, 120, 393–399. [Google Scholar] [CrossRef] [PubMed]
- Trapp, B.D.; Peterson, J.; Ransohoff, R.M.; Rudick, R.; Mörk, S.; Bö, L. Axonal Transection in the Lesions of Multiple Sclerosis. N. Engl. J. Med. 1998, 338, 278–285. [Google Scholar] [CrossRef]
- Peterson, J.W.; Bö, L.; Mörk, S.; Chang, A.; Trapp, B.D. Transected Neurites, Apoptotic Neurons, and Reduced Inflammation in Cortical Multiple Sclerosis Lesions. Ann. Neurol. 2001, 50, 389–400. [Google Scholar] [CrossRef]
- Calabrese, M.; Agosta, F.; Rinaldi, F.; Mattisi, I.; Grossi, P.; Favaretto, A.; Atzori, M.; Bernardi, V.; Barachino, L.; Rinaldi, L.; et al. Cortical Lesions and Atrophy Associated with Cognitive Impairment in Relapsing-Remitting Multiple Sclerosis. Arch. Neurol. 2009, 66, 1144–1150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nielsen, A.S.; Kinkel, R.P.; Madigan, N.; Tinelli, E.; Benner, T.; Mainero, C. Contribution of Cortical Lesion Subtypes at 7T MRI to Physical and Cognitive Performance in MS. Neurology 2013, 81, 641–649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiaravalloti, N.D.; DeLuca, J. Cognitive Impairment in Multiple Sclerosis. Lancet. Neurol. 2008, 7, 1139–1151. [Google Scholar] [CrossRef]
- Kouvatsou, Z.; Masoura, E.; Kimiskidis, V. Working Memory Deficits in Multiple Sclerosis: An Overview of the Findings. Front. Psychol. 2022, 13, 866885. [Google Scholar] [CrossRef]
- Santiago, O.; Guàrdia, J.; Casado, V.; Carmona, O.; Arbizu, T. Specificity of Frontal Dysfunctions in Relapsing–Remitting Multiple Sclerosis. Arch. Clin. Neuropsychol. 2007, 22, 623–629. [Google Scholar] [CrossRef] [Green Version]
- Engl, C.; Tiemann, L.; Grahl, S.; Bussas, M.; Schmidt, P.; Pongratz, V.; Berthele, A.; Beer, A.; Gaser, C.; Kirschke, J.S.; et al. Cognitive Impairment in Early MS: Contribution of White Matter Lesions, Deep Grey Matter Atrophy, and Cortical Atrophy. J. Neurol. 2020, 267, 2307. [Google Scholar] [CrossRef] [PubMed]
- Cunniffe, N.; Coles, A. Promoting Remyelination in Multiple Sclerosis. J. Neurol. 2021, 268, 30–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bramow, S.; Frischer, J.M.; Lassmann, H.; Koch-Henriksen, N.; Lucchinetti, C.F.; Sørensen, P.S.; Laursen, H. Demyelination versus Remyelination in Progressive Multiple Sclerosis. Brain 2010, 133, 2983–2998. [Google Scholar] [CrossRef] [Green Version]
- Goldschmidt, T.; Antel, J.; König, F.B.; Brück, W.; Kuhlmann, T. Remyelination Capacity of the MS Brain Decreases with Disease Chronicity. Neurology 2009, 72, 1914–1921. [Google Scholar] [CrossRef]
- Rodgers, J.M.; Robinson, A.P.; Miller, S.D. Strategies for Protecting Oligodendrocytes and Enhancing Remyelination in Multiple Sclerosis. Discov. Med. 2013, 16, 53–63. [Google Scholar]
- Duncan, I.D.; Radcliff, A.B.; Heidari, M.; Kidd, G.; August, B.K.; Wierenga, L.A. The Adult Oligodendrocyte Can Participate in Remyelination. Proc. Natl. Acad. Sci. USA 2018, 115, e11807–e11816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeung, M.S.Y.; Djelloul, M.; Steiner, E.; Bernard, S.; Salehpour, M.; Possnert, G.; Brundin, L.; Frisén, J. Dynamics of Oligodendrocyte Generation in Multiple Sclerosis. Nature 2019, 566, 538–542. [Google Scholar] [CrossRef] [PubMed]
- Neely, S.A.; Williamson, J.M.; Klingseisen, A.; Zoupi, L.; Early, J.J.; Williams, A.; Lyons, D.A. New Oligodendrocytes Exhibit More Abundant and Accurate Myelin Regeneration than Those That Survive Demyelination. Nat. Neurosci. 2022, 25, 415–420. [Google Scholar] [CrossRef]
- Rivera, F.J.; Steffenhagen, C.; Kremer, D.; Kandasamy, M.; Sandner, B.; Couillard-Despres, S.; Weidner, N.; Küry, P.; Aigner, L. Deciphering the Oligodendrogenic Program of Neural Progenitors: Cell Intrinsic and Extrinsic Regulators. Stem Cells Dev. 2010, 19, 595–606. [Google Scholar] [CrossRef] [PubMed]
- Wolswijk, G. Chronic Stage Multiple Sclerosis Lesions Contain a Relatively Quiescent Population of Oligodendrocyte Precursor Cells. J. Neurosci. 1998, 18, 601–609. [Google Scholar] [CrossRef] [Green Version]
- Kuhlmann, T.; Miron, V.; Cuo, Q.; Wegner, C.; Antel, J.; Brück, W. Differentiation Block of Oligodendroglial Progenitor Cells as a Cause for Remyelination Failure in Chronic Multiple Sclerosis. Brain 2008, 131, 1749–1758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boyd, A.; Zhang, H.; Williams, A. Insufficient OPC Migration into Demyelinated Lesions Is a Cause of Poor Remyelination in MS and Mouse Models. Acta Neuropathol. 2013, 125, 841–859. [Google Scholar] [CrossRef] [Green Version]
- Plemel, J.R.; Liu, W.Q.; Yong, V.W. Remyelination Therapies: A New Direction and Challenge in Multiple Sclerosis. Nat. Rev. Drug Discov. 2017, 16, 617–634. [Google Scholar] [CrossRef]
- Galloway, D.A.; Gowing, E.; Setayeshgar, S.; Kothary, R. Inhibitory Milieu at the Multiple Sclerosis Lesion Site and the Challenges for Remyelination. Glia 2020, 68, 859–877. [Google Scholar] [CrossRef]
- Quraishe, S.; Forbes, L.H.; Andrews, M.R. The Extracellular Environment of the CNS: Influence on Plasticity, Sprouting, and Axonal Regeneration after Spinal Cord Injury. Neural Plast. 2018, 2018, 2952386. [Google Scholar] [CrossRef] [Green Version]
- Câmara, J.; ffrench-Constant, C. Lessons from Oligodendrocyte Biology on Promoting Repair in Multiple Sclerosis. J. Neurol. 2007, 254, I15–I22. [Google Scholar] [CrossRef]
- McTigue, D.M.; Tripathi, R.B. The Life, Death and Replacement of Oligodendrocytes in the Adult Central Nervous System. J Neurochem. 2008, 107, 1–19. [Google Scholar] [CrossRef]
- Sim, F.J.; Zhao, C.; Penderis, J.; Franklin, R.J.M. The Age-Related Decrease in CNS Remyelination Efficiency Is Attributable to an Impairment of Both Oligodendrocyte Progenitor Recruitment and Differentiation. J. Neurosci. 2002, 22, 2451–2459. [Google Scholar] [CrossRef] [PubMed]
- Neumann, B.; Segel, M.; Chalut, K.J.; Franklin, R.J.M. Remyelination and Ageing: Reversing the Ravages of Time. Mult. Scler. J. 2019, 25, 1835–1841. [Google Scholar] [CrossRef] [Green Version]
- Alroughani, R.A.; Akhtar, S.; Ahmed, S.F.; Al-Hashel, J.Y. Clinical Predictors of Disease Progression in Multiple Sclerosis Patients with Relapsing Onset in a Nation-Wide Cohort. Int. J. Neurosci. 2015, 125, 831–837. [Google Scholar] [CrossRef] [PubMed]
- Confavreux, C.; Vukusic, S. Age at Disability Milestones in Multiple Sclerosis. Brain 2006, 129, 595–605. [Google Scholar] [CrossRef] [Green Version]
- Shields, S.A.; Gilson, J.M.; Blakemore, W.; Franklin, R.J. Remyelination Occurs as Extensively but More Slowly in Old Rats Compared to Young Rats Following Fliotoxin-Induced CNS Demyelination. Glia 1999, 28, 77–83. [Google Scholar] [CrossRef]
- Shen, S.; Sandoval, J.; Swiss, V.A.; Li, J.; Dupree, J.; Franklin, R.J.M.; Casaccia-Bonnefil, P. Age-Dependent Epigenetic Control of Differentiation Inhibitors Is Critical for Remyelination Efficiency. Nat. Neurosci. 2008, 11, 1024–1034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neumann, B.; Baror, R.; Zhao, C.; Segel, M.; Dietmann, S.; Rawji, K.S.; Foerster, S.; McClain, C.R.; Chalut, K.; van Wijngaarden, P.; et al. Metformin Restores CNS Remyelination Capacity by Rejuvenating Aged Stem Cells. Cell Stem Cell 2019, 25, 473–485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Segel, M.; Neumann, B.; Hill, M.F.E.; Weber, I.P.; Viscomi, C.; Zhao, C.; Young, A.; Agley, C.C.; Thompson, A.J.; Gonzalez, G.A.; et al. Niche Stiffness Underlies the Ageing of Central Nervous System Progenitor Cells. Nature 2019, 573, 130–134. [Google Scholar] [CrossRef] [PubMed]
- Zobel, K.; Hansen, U.; Galla, H.J. Blood-Brain Barrier Properties in Vitro Depend on Composition and Assembly of Endogenous Extracellular Matrices. Cell Tissue Res. 2016, 365, 233–245. [Google Scholar] [CrossRef] [PubMed]
- Davis, G.E.; Senger, D.R. Endothelial Extracellular Matrix: Biosynthesis, Remodeling, and Functions during Vascular Morphogenesis and Neovessel Stabilization. Circ. Res. 2005, 97, 1093–1107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Theocharis, A.D.; Skandalis, S.S.; Gialeli, C.; Karamanos, N.K. Extracellular Matrix Structure. Adv. Drug Deliv. Rev. 2016, 97, 4–27. [Google Scholar] [CrossRef] [PubMed]
- Rozario, T.; DeSimone, D.W. The Extracellular Matrix in Development and Morphogenesis: A Dynamic View. Dev. Biol. 2010, 341, 126–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sorokin, L. The Impact of the Extracellular Matrix on Inflammation. Nat. Rev. Immunol. 2010, 10, 712–723. [Google Scholar] [CrossRef] [PubMed]
- Boyd, D.F.; Thomas, P.G. Towards Integrating Extracellular Matrix and Immunological Pathways. Cytokine 2017, 98, 79–86. [Google Scholar] [CrossRef] [PubMed]
- Schonherr, E.; Hausser, H.J. Extracellular Matrix and Cytokines: A Functional Unit. Dev. Immunol. 2000, 7, 89–101. [Google Scholar] [CrossRef]
- Wijelath, E.S.; Rahman, S.; Namekata, M.; Murray, J.; Nishimura, T.; Mostafavi-Pour, Z.; Patel, Y.; Suda, Y.; Humphries, M.J.; Sobel, M. Heparin-II Domain of Fibronectin Is a Vascular Endothelial Growth Factor-Binding Domain: Enhancement of VEGF Biological Activity by a Singular Growth Factor/Matrix Protein Synergism. Circ. Res. 2006, 99, 853–860. [Google Scholar] [CrossRef]
- Rahman, S.; Patel, Y.; Murray, J.; Patel, K.V.; Sumathipala, R.; Sobel, M.; Wijelath, E.S. Novel Hepatocyte Growth Factor (HGF) Binding Domains on Fibronectin and Vitronectin Coordinate a Distinct and Amplified Met-Integrin Induced Signalling Pathway in Endothelial Cells. BMC Cell Biol. 2005, 6, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bauer, N.G.; ffrench-Constant, C. Physical Forces in Myelination and Repair: A Question of Balance? J. Biol. 2009, 8, 78. [Google Scholar] [CrossRef] [PubMed]
- Saha, K.; Keung, A.J.; Irwin, E.F.; Li, Y.; Little, L.; Schaffer, D.V.; Healy, K.E. Substrate Modulus Directs Neural Stem Cell Behavior. Biophys. J. 2008, 95, 4426–4438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jagielska, A.; Norman, A.L.; Whyte, G.; Vliet, K.J.V.; Guck, J.; Franklin, R.J.M. Mechanical Environment Modulates Biological Properties of Oligodendrocyte Progenitor Cells. Stem Cells Dev. 2012, 21, 2905–2914. [Google Scholar] [CrossRef] [PubMed]
- Urbanski, M.M.; Kingsbury, L.; Moussouros, D.; Kassim, I.; Mehjabeen, S.; Paknejad, N.; Melendez-Vasquez, C.V. Myelinating Glia Differentiation Is Regulated by Extracellular Matrix Elasticity. Sci. Rep. 2016, 6, 33751. [Google Scholar] [CrossRef] [PubMed]
- Stoffels, J.M.J.; Hoekstra, D.; Franklin, R.J.M.; Baron, W.; Zhao, C. The EIIIA Domain from Astrocyte-Derived Fibronectin Mediates Proliferation of Oligodendrocyte Progenitor Cells Following CNS Demyelination. Glia 2015, 63, 242–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lourenço, T.; Paes De Faria, J.; Bippes, C.A.; Maia, J.; Lopes-Da-Silva, J.A.; Relvas, J.B.; Graõs, M. Modulation of Oligodendrocyte Differentiation and Maturation by Combined Biochemical and Mechanical Cues. Sci. Rep. 2016, 6, 21563. [Google Scholar] [CrossRef] [Green Version]
- Buttery, P.C.; ffrench-Constant, C. Laminin-2/Integrin Interactions Enhance Myelin Membrane Formation by Oligodendrocytes. Mol. Cell. Neurosci. 1999, 14, 199–212. [Google Scholar] [CrossRef] [PubMed]
- Colognato, H.; Galvin, J.; Wang, Z.; Relucio, J.; Nguyen, T.; Harrison, D.; Yurchenco, P.D.; ffrench-Constant, C. Identification of Dystroglycan as a Second Laminin Receptor in Oligodendrocytes, with a Role in Myelination. Development 2007, 134, 1723–1736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Urbanski, M.M.; Brendel, M.B.; Melendez-Vasquez, C.V. Acute and Chronic Demyelinated CNS Lesions Exhibit Opposite Elastic Properties. Sci. Rep. 2019, 9, 999. [Google Scholar] [CrossRef] [PubMed]
- Yi, J.-H.; Katagiri, Y.; Susarla, B.; Figge, D.; Symes, A.J.; Geller, H.M. Alterations in Sulfated Chondroitin Glycosaminoglycans Following Controlled Cortical Impact Injury in Mice. J. Comp. Neurol. 2012, 520, 3295–3313. [Google Scholar] [CrossRef] [Green Version]
- Carulli, D.; Rhodes, K.E.; Brown, D.J.; Bonnert, T.P.; Pollack, S.J.; Oliver, K.; Strata, P.; Fawcett, J.W. Composition of Perineuronal Nets in the Adult Rat Cerebellum and the Cellular Origin of Their Components. J. Comp. Neurol. 2006, 494, 559–577. [Google Scholar] [CrossRef]
- Hara, M.; Kobayakawa, K.; Ohkawa, Y.; Kumamaru, H.; Yokota, K.; Saito, T.; Kijima, K.; Yoshizaki, S.; Harimaya, K.; Nakashima, Y.; et al. Interaction of Reactive Astrocytes with Type I Collagen Induces Astrocytic Scar Formation through the Integrin-N-Cadherin Pathway after Spinal Cord Injury. Nat. Med. 2017, 23, 818–828. [Google Scholar] [CrossRef] [PubMed]
- Lau, L.W.; Keough, M.B.; Haylock-Jacobs, S.; Cua, R.; Döring, A.; Sloka, S.; Stirling, D.P.; Rivest, S.; Yong, V.W. Chondroitin Sulfate Proteoglycans in Demyelinated Lesions Impair Remyelination. Ann. Neurol. 2012, 72, 419–432. [Google Scholar] [CrossRef] [PubMed]
- Pendleton, J.C.; Shamblott, M.J.; Gary, D.S.; Belegu, V.; Hurtado, A.; Malone, M.L.; McDonald, J.W. Chondroitin Sulfate Proteoglycans Inhibit Oligodendrocyte Myelination through PTPσ. Exp. Neurol. 2013, 247, 113–121. [Google Scholar] [CrossRef]
- Hibbits, N.; Yoshino, J.; Le, T.Q.; Armstrong, R.C. Astrogliosis during Acute and Chronic Cuprizone Demyelination and Implications for Remyelination. ASN Neuro 2012, 4, 393–408. [Google Scholar] [CrossRef]
- Karimi-Abdolrezaee, S.; Schut, D.; Wang, J.; Fehlings, M.G. Chondroitinase and Growth Factors Enhance Activation and Oligodendrocyte Differentiation of Endogenous Neural Precursor Cells after Spinal Cord Injury. PLoS ONE 2012, 7, e37589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siebert, J.R.; Stelzner, D.J.; Osterhout, D.J. Chondroitinase Treatment Following Spinal Contusion Injury Increases Migration of Oligodendrocyte Progenitor Cells. Exp. Neurol. 2011, 231, 19–29. [Google Scholar] [CrossRef]
- Kumari, S.; Mak, M.; Poh, Y.; Tohme, M.; Watson, N.; Melo, M.; Janssen, E.; Dustin, M.; Geha, R.; Irvine, D.J. Cytoskeletal Tension Actively Sustains the Migratory T-Cell Synaptic Contact. EMBO J. 2020, 39, e102783. [Google Scholar] [CrossRef]
- Keough, M.B.; Rogers, J.A.; Zhang, P.; Jensen, S.K.; Stephenson, E.L.; Chen, T.; Hurlbert, M.G.; Lau, L.W.; Rawji, K.S.; Plemel, J.R.; et al. An Inhibitor of Chondroitin Sulfate Proteoglycan Synthesis Promotes Central Nervous System Remyelination. Nat. Commun. 2016, 7, 11312. [Google Scholar] [CrossRef]
- Stephenson, E.L.; Zhang, P.; Ghorbani, S.; Wang, A.; Gu, J.; Keough, M.B.; Rawji, K.S.; Silva, C.; Yong, V.W.; Ling, C.C. Targeting the Chondroitin Sulfate Proteoglycans: Evaluating Fluorinated Glucosamines and Xylosides in Screens Pertinent to Multiple Sclerosis. ACS Cent. Sci. 2019, 5, 1223–1234. [Google Scholar] [CrossRef] [Green Version]
- Pu, A.; Mishra, M.K.; Dong, Y.; Ghorbanigazar, S.; Stephenson, E.L.; Rawji, K.S.; Silva, C.; Kitagawa, H.; Sawcer, S.; Yong, V.W. The Glycosyltransferase EXTL2 Promotes Proteoglycan Deposition and Injurious Neuroinflammation Following Demyelination. J. Neuroinflamm. 2020, 17, 220. [Google Scholar] [CrossRef]
- Luo, F.; Tran, A.P.; Xin, L.; Sanapala, C.; Lang, B.T.; Silver, J.; Yang, Y. Modulation of Proteoglycan Receptor PTPσ Enhances MMP-2 Activity to Promote Recovery from Multiple Sclerosis. Nat. Commun. 2018, 9, 4126. [Google Scholar] [CrossRef]
- Pu, A.; Stephenson, E.L.; Yong, V.W. The Extracellular Matrix: Focus on Oligodendrocyte Biology and Targeting CSPGs for Remyelination Therapies. Glia 2018, 66, 1809–1825. [Google Scholar] [CrossRef] [PubMed]
- Lau, L.W.; Cua, R.; Keough, M.B.; Haylock-Jacobs, S.; Yong, V.W. Pathophysiology of the Brain Extracellular Matrix: A New Target for Remyelination. Nat. Rev. Neurosci. 2013, 14, 722–729. [Google Scholar] [CrossRef]
- Patel, R.S.; Odermatt, E.; Schwarzbauer, J.E.; Hynes, R.O. Organization of the Fibronectin Gene Provides Evidence for Exon Shuffling during Evolution. EMBO J. 1987, 6, 2565–2572. [Google Scholar] [CrossRef] [PubMed]
- ffrench-Constant, C. Alternative Splicing of Fibronectin-Many Different Proteins but Few Different Functions. Exp. Cell Res. 1995, 221, 261–271. [Google Scholar] [CrossRef]
- Pankov, R.; Yamada, K.M. Fibronectin at a Glance. J. Cell Sci. 2002, 115, 3861–3863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plow, E.F.; Haas, T.A.; Zhang, L.; Loftus, J.; Smith, J.W. Ligand Binding to Integrins. J. Biol. Chem. 2000, 275, 21785–21788. [Google Scholar] [CrossRef] [Green Version]
- Murphy, P.A.; Begum, S.; Hynes, R.O. Tumor Angiogenesis in the Absence of Fibronectin or Its Cognate Integrin Receptors. PLoS ONE 2015, 10, e0120872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hynes, R.O. Integrins: Versatility, Modulation, and Signaling in Cell Adhesion. Cell 1992, 69, 11–25. [Google Scholar] [CrossRef]
- Sastry, S.K.; Horwitz, A.F. Integrin Cytoplasmic Domains: Mediators of Cytoskeletal Linkages and Extra- and Intracellular Initiated Transmembrane Signaling. Curr. Opin. Cell Biol. 1993, 5, 819–831. [Google Scholar] [CrossRef]
- Diamond, M.S.; Springer, T.A. The Dynamic Regulation of Integrin Adhesiveness. Curr. Biol. 1994, 4, 506–517. [Google Scholar] [CrossRef]
- Wolburg, H.; Noell, S.; Mack, A.; Wolburg-Buchholz, K.; Fallier-Becker, P. Brain Endothelial Cells and the Glio-Vascular Complex. Cell Tissue Res. 2009, 335, 75–96. [Google Scholar] [CrossRef] [PubMed]
- Mathiisen, T.M.; Lehre, K.P.; Danbolt, N.C.; Ottersen, O.P. The Perivascular Astroglial Sheath Provides a Complete Covering of the Brain Microvessels: An Electron Microscopic 3D Reconstruction. Glia 2010, 58, 1094–1103. [Google Scholar] [CrossRef] [PubMed]
- Sekiguchi, R.; Yamada, K.M. Basement Membranes in Development and Disease. Curr. Top. Dev. Biol. 2018, 130, 143–191. [Google Scholar]
- Owens, T.; Bechmann, I.; Engelhardt, B. Perivascular Spaces and the Two Steps to Neuroinflammation. J. Neuropathol. Exp. Neurol. 2008, 67, 1113–1121. [Google Scholar] [CrossRef] [Green Version]
- Baeten, K.M.; Akassoglou, K. Extracellular Matrix and Matrix Receptors in Blood-Brain Barrier Formation and Stroke. Dev. Neurobiol. 2011, 71, 1018–1039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Horssen, J.; Bö, L.; Vos, C.M.P.; Virtanen, I.; de Vries, H.E. Basement Membrane Proteins in Multiple Sclerosis-Associated Inflammatory Cuffs: Potential Role in Influx and Transport of Leukocytes. J. Neuropathol. Exp. Neurol. 2005, 64, 722–729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kouwenhoven, M.; Özenci, V.; Gomes, A.; Yarilin, D.; Giedraitis, V.; Press, R.; Link, H. Multiple Sclerosis: Elevated Expression of Matrix Metalloproteinases in Blood Monocytes. J. Autoimmun. 2001, 16, 463–470. [Google Scholar] [CrossRef]
- Korn, J.; Christ, B.; Kurz, H. Neuroectodermal Origin of Brain Pericytes and Vascular Smooth Muscle Cells. J. Comp. Neurol. 2002, 442, 78–88. [Google Scholar] [CrossRef] [PubMed]
- Etchevers, H.C.; Vincent, C.; Le Douarin, N.M.; Couly, G.F. The Cephalic Neural Crest Provides Pericytes and Smooth Muscle Cells to All Blood Vessels of the Face and Forebrain. Development 2001, 128, 1059–1068. [Google Scholar] [CrossRef]
- Yamanishi, E.; Takahashi, M.; Saga, Y.; Osumi, N. Penetration and Differentiation of Cephalic Neural Crest-Derived Cells in the Developing Mouse Telencephalon. Dev. Growth Differ. 2012, 54, 785–800. [Google Scholar] [CrossRef]
- Yamamoto, S.; Muramatsu, M.; Azuma, E.; Ikutani, M.; Nagai, Y.; Sagara, H.; Koo, B.N.; Kita, S.; O’Donnell, E.; Osawa, T.; et al. A Subset of Cerebrovascular Pericytes Originates from Mature Macrophages in the Very Early Phase of Vascular Development in CNS. Sci. Rep. 2017, 7, 3855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muoio, V.; Persson, P.B.; Sendeski, M.M. The Neurovascular Unit-Concept Review. Acta Physiol. 2014, 210, 790–798. [Google Scholar] [CrossRef]
- Preston, J.E.; Joan Abbott, N.; Begley, D.J. Transcytosis of Macromolecules at the Blood-Brain Barrier. Adv. Pharmacol. 2014, 71, 1147–1163. [Google Scholar] [CrossRef]
- Tilling, T.; Engelbertz, C.; Decker, S.; Korte, D.; Hüwel, S.; Galla, H.J. Expression and Adhesive Properties of Basement Membrane Proteins in Cerebral Capillary Endothelial Cell Cultures. Cell Tissue Res. 2002, 310, 19–29. [Google Scholar] [CrossRef] [PubMed]
- Yurchenco, P.; Patton, B. Developmental and Pathogenic Mechanisms of Basement Membrane Assembly. Curr. Pharm. Des. 2009, 15, 1277–1294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yurchenco, P.D. Basement Membranes: Cell Scaffoldings and Signaling Platforms. Cold Spring Harb. Perspect. Biol. 2011, 3, a004911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tilling, T.; Korte, D.; Hoheisel, D.; Galla, H.-J. Basement Membrane Proteins Influence Brain Capillary Endothelial Barrier Function In Vitro. J. Neurochem. 2002, 71, 1151–1157. [Google Scholar] [CrossRef]
- Wolburg, H.; Neuhaus, J.; Kniesel, U.; Krauß, B.; Schmid, E.M.; Öcalan, M.; Farrell, C.; Risau, W. Modulation of Tight Junction Structure in Blood-Brain Barrier Endothelial Cells: Effects of Tissue Culture, Second Messengers and Cocultured Astrocytes. J. Cell Sci. 1994, 107, 1347–1357. [Google Scholar] [CrossRef] [PubMed]
- Canfield, S.G.; Stebbins, M.J.; Faubion, M.G.; Gastfriend, B.D.; Palecek, S.P.; Shusta, E.V. An Isogenic Neurovascular Unit Model Comprised of Human Induced Pluripotent Stem Cell-Derived Brain Microvascular Endothelial Cells, Pericytes, Astrocytes, and Neurons. Fluids Barriers CNS 2019, 16, 25. [Google Scholar] [CrossRef]
- Nakagawa, S.; Deli, M.A.; Kawaguchi, H.; Shimizudani, T.; Shimono, T.; Kittel, Á.; Tanaka, K.; Niwa, M. A New Blood-Brain Barrier Model Using Primary Rat Brain Endothelial Cells, Pericytes and Astrocytes. Neurochem. Int. 2009, 54, 253–263. [Google Scholar] [CrossRef] [Green Version]
- Hartmann, C.; Zozulya, A.; Wegener, J.; Galla, H.J. The Impact of Glia-Derived Extracellular Matrices on the Barrier Function of Cerebral Endothelial Cells: An in Vitro Study. Exp. Cell Res. 2007, 313, 1318–1325. [Google Scholar] [CrossRef]
- Wang, J.; Milner, R. Fibronectin Promotes Brain Capillary Endothelial Cell Survival and Proliferation through αvβ1 and αvβ3 Integrins via MAP Kinase Signalling. J. Neurochem. 2006, 96, 148–159. [Google Scholar] [CrossRef]
- Mettouchi, A.; Klein, S.; Guo, W.; Lopez-Lago, M.; Lemichez, E.; Westwick, J.K.; Giancotti, F.G. Integrin-Specific Activation of Rac Controls Progression through the G1 Phase of the Cell Cycle. Mol. Cell 2001, 8, 115–127. [Google Scholar] [CrossRef]
- Wang, Y.; Jin, G.; Miao, H.; Li, J.Y.S.; Usami, S.; Chien, S. Integrins Regulate VE-Cadherin and Catenins: Dependence of This Regulation on Src, but Not on Ras. Proc. Natl. Acad. Sci. USA 2006, 103, 1774–1779. [Google Scholar] [CrossRef] [Green Version]
- Alghisi, G.C.; Ponsonnet, L.; Rüegg, C. The Integrin Antagonist Cilengitide Activates AVβ3, Disrupts VE-Cadherin Localization at Cell Junctions and Enhances Permeability in Endothelial Cells. PLoS ONE 2009, 4, e4449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pulous, F.E.; Petrich, B.G. Integrin-Dependent Regulation of the Endothelial Barrier. Tissue Barriers 2019, 7, 1685844. [Google Scholar] [CrossRef] [Green Version]
- Yadav, R.; Larbi, K.Y.; Young, R.E.; Nourshargh, S. Migration of Leukocytes through the Vessel Wall and Beyond. Thromb. Haemost. 2003, 90, 598–606. [Google Scholar] [CrossRef]
- Rosenberg, G.A.; Kornfeld, M.; Estrada, E.; Kelley, R.O.; Liotta, L.A.; Stetler-Stevenson, W.G. TIMP-2 Reduces Proteolytic Opening of Blood-Brain Barrier by Type IV Collagenase. Brain Res. 1992, 576, 203–207. [Google Scholar] [CrossRef]
- Brundula, V.; Rewcastle, N.B.; Metz, L.M.; Bernard, C.C.; Yong, V.W. Targeting Leukocyte MMPs and TransmigrationMinocycline as a Potential Therapy for Multiple Sclerosis. Brain 2002, 125, 1297–1308. [Google Scholar] [CrossRef]
- Sobel, R.A.; Mitchell, M.E.; Fondren, G. Intercellular Adhesion Molecule-1 (ICAM-1) in Cellular Immune Reactions in the Human Central Nervous System. Am. J. Pathol. 1990, 136, 1309–1316. [Google Scholar] [PubMed]
- Greenwood, J.; Etienne-Manneville, S.; Adamson, P.; Couraud, P.-O. Lymphocyte Migration into the Central Nervous System: Implication of ICAM-1 Signalling at the Blood-Brain Barrier. Vascul. Pharmacol. 2002, 38, 315–322. [Google Scholar] [CrossRef]
- van der Laan, L.J.W.; Groot, C.J.A.; De Elices, M.J.; Dijkstra, C.D. Extracellular Matrix Proteins Expressed by Human Adult Astrocytes in Vivo and in Vitro: An Astrocyte Surface Protein Containing the CS1 Domain Contributes to Binding of Lymphoblasts. J. Neurosci. Res. 1997, 50, 539–548. [Google Scholar] [CrossRef]
- Guan, J.L.; Hynes, R.O. Lymphoid Cells Recognize an Alternatively Spliced Segment of Fibronectin via the Integrin Receptor α4β1. Cell 1990, 60, 53–61. [Google Scholar] [CrossRef]
- van der Laan, L.J.W.; van der Goes, A.; Wauben, M.H.M.; Ruuls, S.R.; Döpp, A.; de Groot, C.J.A.; Kuijpers, T.W.; Elices, M.J.; Dijkstra, C.D. Beneficial Effect of Modified Peptide Inhibitor of α4 Integrins on Experimental Allergic Encephalomyelitis in Lewis Rats. J. Neurosci. Res. 2002, 67, 191–199. [Google Scholar] [CrossRef]
- Kent, S.J.; Karlik, S.J.; Cannon, C.; Hines, D.K.; Yednock, T.A.; Fritz, L.C.; Horner, H.C. A Monoclonal Antibody to α4 Integrin Suppresses and Reverses Active Experimental Allergic Encephalomyelitis. J. Neuroimmunol. 1995, 58, 1–10. [Google Scholar] [CrossRef]
- Yu, Y.; Schürpf, T.; Springer, T.A. How Natalizumab Binds and Antagonizes A4 Integrins. J. Biol. Chem. 2013, 288, 32314–32325. [Google Scholar] [CrossRef] [Green Version]
- Shirani, A.; Stüve, O. Natalizumab: Perspectives from the Bench to Bedside. Cold Spring Harb. Perspect. Med. 2018, 8, a029066. [Google Scholar] [CrossRef]
- Clerico, M.; Artusi, C.A.; Di Liberto, A.; Rolla, S.; Bardina, V.; Barbero, P.; De Mercanti, S.F.; Durelli, L. Natalizumab in Multiple Sclerosis: Long-Term Management. Int. J. Mol. Sci. 2017, 18, 940. [Google Scholar] [CrossRef] [PubMed]
- Melincovici, C.S.; Boşca, A.B.; Şuşman, S.; Mărginean, M.; Mihu, C.; Istrate, M.; Moldovan, I.M.; Roman, A.L.; Mihu, C.M. Vascular Endothelial Growth Factor (VEGF)—Key Factor in Normal and Pathological Angiogenesis. Rom. J. Morphol. Embryol. 2018, 59, 455–467. [Google Scholar]
- Hao, W.; Han, J.; Chu, Y.; Huang, L.; Zhuang, Y.; Sun, J.; Li, X.; Zhao, Y.; Chen, Y.; Dai, J. Collagen/Heparin Bi-Affinity Multilayer Modified Collagen Scaffolds for Controlled bFGF Release to Improve Angiogenesis In Vivo. Macromol. Biosci. 2018, 18, e1800086. [Google Scholar] [CrossRef] [Green Version]
- Bloch, W.; Forsberg, E.; Lentini, S.; Brakebusch, C.; Martin, K.; Krell, H.W.; Weidle, U.H.; Addicks, K.; Fässler, R. β1 Integrin Is Essential for Teratoma Growth and Angiogenesis. J. Cell Biol. 1997, 139, 265–278. [Google Scholar] [CrossRef] [Green Version]
- Giancotti, F.G.; Ruoslahti, E. Integrin Signaling. Science 1999, 285, 1028–1032. [Google Scholar] [CrossRef] [PubMed]
- Ingber, D.E.; Folkman, J. Mechanochemical Switching between Growth and Differentiation during Fibroblast Growth Factor-Stimulated Angiogenesis in Vitro: Role of Extracellular Matrix. J. Cell Biol. 1989, 109, 317–330. [Google Scholar] [CrossRef] [Green Version]
- McIntosh, L.C.; Muckersie, L.; Forrester, J.V. Retinal Capillary Endothelial Cells Prefer Different Substrates for Growth and Migration. Tissue Cell 1988, 20, 193–209. [Google Scholar] [CrossRef]
- Kirkpatrick, C.J.; Kampe, M.; Rixen, H.; Fischer, E.G.; Ruchatz, D.; Mittermayer, C. In Vitro Studies on the Expansion of Endothelial Cell Monolayers on Components of the Basement Membrane. Virchows Arch. B Cell Pathol. Incl. Mol. Pathol. 1990, 58, 207–213. [Google Scholar] [CrossRef] [PubMed]
- Grant, D.S.; Kleinman, H.K. Regulation of Capillary Formation by Laminin and Other Components of the Extracellular Matrix. EXS 1997, 79, 317–333. [Google Scholar]
- Grant, D.S.; Tashiro, K.I.; Segui-Real, B.; Yamada, Y.; Martin, G.R.; Kleinman, H.K. Two Different Laminin Domains Mediate the Differentiation of Human Endothelial Cells into Capillary-like Structures in Vitro. Cell 1989, 58, 933–943. [Google Scholar] [CrossRef]
- Roscoe, W.A.; Welsh, M.E.; Carter, D.E.; Karlik, S.J. VEGF and Angiogenesis in Acute and Chronic MOG(35-55) Peptide Induced EAE. J. Neuroimmunol. 2009, 209, 6–15. [Google Scholar] [CrossRef]
- Seabrook, T.J.; Littlewood-Evans, A.; Brinkmann, V.; Pöllinger, B.; Schnell, C.; Hiestand, P.C. Angiogenesis Is Present in Experimental Autoimmune Encephalomyelitis and Pro-Angiogenic Factors Are Increased in Multiple Sclerosis Lesions. J. Neuroinflamm. 2010, 7, 95. [Google Scholar] [CrossRef] [Green Version]
- Lassmann, H. Hypoxia-like Tissue Injury as a Component of Multiple Sclerosis Lesions. J. Neurol. Sci. 2003, 206, 187–191. [Google Scholar] [CrossRef]
- Trapp, B.D.; Stys, P.K. Virtual Hypoxia and Chronic Necrosis of Demyelinated Axons in Multiple Sclerosis. Lancet Neurol. 2009, 8, 280–291. [Google Scholar] [CrossRef]
- Li, L.; Welser-Alves, J.; van der Flier, A.; Boroujerdi, A.; Hynes, R.O.; Milner, R. An Angiogenic Role for the α5β1 Integrin in Promoting Endothelial Cell Proliferation during Cerebral Hypoxia. Exp. Neurol. 2012, 237, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Boroujerdi, A.; Welser-Alves, J.V.; Milner, R. Extensive Vascular Remodeling in the Spinal Cord of Pre-Symptomatic Experimental Autoimmune Encephalomyelitis Mice; Increased Vessel Expression of Fibronectin and the α5β1 Integrin. Exp. Neurol. 2013, 250, 43–51. [Google Scholar] [CrossRef] [Green Version]
- Holley, J.E.; Newcombe, J.; Whatmore, J.L.; Gutowski, N.J. Increased Blood Vessel Density and Endothelial Cell Proliferation in Multiple Sclerosis Cerebral White Matter. Neurosci. Lett. 2010, 470, 65–70. [Google Scholar] [CrossRef] [PubMed]
- Sobel, R.A.; Mitchell, M.E. Fibronectin in Multiple Sclerosis Lesions. Am. J. Pathol. 1989, 135, 161–168. [Google Scholar] [PubMed]
- Peters, J.H.; Loredo, G.A.; Chen, G.; Maunder, R.; Hahn, T.J.; Willits, N.H.; Hynes, R.O. Plasma Levels of Fibronectin Bearing the Alternatively Spliced EIIIB Segment Are Increased after Major Trauma. J. Lab. Clin. Med. 2003, 141, 401–410. [Google Scholar] [CrossRef]
- Castellanos, M.; Leira, R.; Serena, J.; Blanco, M.; Pedraza, S.; Castillo, J.; Dávalos, A. Plasma Cellular-Fibronectin Concentration Predicts Hemorrhagic Transformation after Thrombolytic Therapy in Acute Ischemic Stroke. Stroke 2004, 35, 1671–1676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stoffels, J.M.J.; Zhao, C.; Baron, W. Fibronectin in Tissue Regeneration: Timely Disassembly of the Scaffold Is Necessary to Complete the Build. Cell. Mol. Life Sci. 2013, 70, 4243–4253. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Fancy, S.P.J.; Franklin, R.J.M.; ffrench-Constant, C. Up-Regulation of Oligodendrocyte Precursor Cell αv Integrin and Its Extracellular Ligands during Central Nervous System Remyelination. J. Neurosci. Res. 2009, 87, 3447–3455. [Google Scholar] [CrossRef]
- Espitia Pinzon, N.; Sanz-Morello, B.; Brevé, J.J.P.; Bol, J.G.J.M.; Drukarch, B.; Bauer, J.; Baron, W.; van Dam, A.M. Astrocyte-Derived Tissue Transglutaminase Affects Fibronectin Deposition, but Not Aggregation, during Cuprizone-Induced Demyelination. Sci. Rep. 2017, 7, 40995. [Google Scholar] [CrossRef]
- Paul, J.; Strickland, S.; Melchor, J.P. Fibrin Deposition Accelerates Neurovascular Damage and Neuroinflammation in Mouse Models of Alzheimer’s Disease. J. Exp. Med. 2007, 204, 1999–2008. [Google Scholar] [CrossRef] [PubMed]
- Kermode, A.G.; Thompson, A.J.; Tofts, P.; Macmanus, D.G.; Kendall, B.E.; Kingsley, D.P.E.; Moseley, I.F.; Rudge, P.; Mcdonald, W.I. Breakdown of the Blood-Brain Barrier Precedes Symptoms and Other Mri Signs of New Lesions in Multiple Sclerosis: Pathogenetic and Clinical Implications. Brain 1990, 113, 1477–1489. [Google Scholar] [CrossRef] [PubMed]
- Huber, J.D.; Egleton, R.D.; Davis, T.P. Molecular Physiology and Pathophysiology of Tight Junctions in the Blood -Brain Barrier. Trends Neurosci. 2001, 24, 719–725. [Google Scholar] [CrossRef]
- Baron, W.; Colognato, H.; ffrench-Constant, C. Integrin-Growth Factor Interactions as Regulators of Oligodendroglial Development and Function. Glia 2005, 49, 467–479. [Google Scholar] [CrossRef] [PubMed]
- Baron, W.; Shattil, S.J.; ffrench-Constant, C. The Oligodendrocyte Precursor Mitogen PDGF Stimulates Proliferation by Activation of αvβ3 Integrins. EMBO J. 2002, 21, 1957–1966. [Google Scholar] [CrossRef] [Green Version]
- Milner, R.; Edwards, G.; Streuli, C.; ffrench-Constant, C. A Role in Migration for the αvβ1 Integrin Expressed on Oligodendrocyte Precursors. J. Neurosci. 1996, 16, 7240–7252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maier, O.; van der Heide, T.; van Dam, A.M.; Baron, W.; de Vries, H.; Hoekstra, D. Alteration of the Extracellular Matrix Interferes with Raft Association of Neurofascin in Oligodendrocytes. Potential Significance for Multiple Sclerosis? Mol. Cell. Neurosci. 2005, 28, 390–401. [Google Scholar] [CrossRef] [PubMed]
- Šišková, Z.; Baron, W.; de Vries, H.; Hoekstra, D. Fibronectin Impedes “Myelin” Sheet-Directed Flow in Oligodendrocytes: A Role for a Beta 1 Integrin-Mediated PKC Signaling Pathway in Vesicular Trafficking. Mol. Cell. Neurosci. 2006, 33, 150–159. [Google Scholar] [CrossRef]
- Šišková, Z.; Yong, V.W.; Nomden, A.; van Strien, M.; Hoekstra, D.; Baron, W. Fibronectin Attenuates Process Outgrowth in Oligodendrocytes by Mislocalizing MMP-9 Activity. Mol. Cell. Neurosci. 2009, 42, 234–242. [Google Scholar] [CrossRef]
- Baron, W.; Bijlard, M.; Nomden, A.; de Jonge, J.C.; Teunissen, C.E.; Hoekstra, D. Sulfatide-Mediated Control of Extracellular Matrix-Dependent Oligodendrocyte Maturation. Glia 2014, 62, 927–942. [Google Scholar] [CrossRef] [PubMed]
- Natrajan, M.S.; Komori, M.; Kosa, P.; Johnson, K.R.; Wu, T.; Franklin, R.J.M.; Bielekova, B. Pioglitazone Regulates Myelin Phagocytosis and Multiple Sclerosis Monocytes. Ann. Clin. Transl. Neurol. 2015, 2, 1071–1084. [Google Scholar] [CrossRef] [PubMed]
- Ruckh, J.M.; Zhao, J.W.; Shadrach, J.L.; van Wijngaarden, P.; Rao, T.N.; Wagers, A.J.; Franklin, R.J.M. Rejuvenation of Regeneration in the Aging Central Nervous System. Cell Stem Cell 2012, 10, 96–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kotter, M.R.; Li, W.W.; Zhao, C.; Franklin, R.J.M. Myelin Impairs CNS Remyelination by Inhibiting Oligodendrocyte Precursor Cell Differentiation. J. Neurosci. 2006, 26, 328–332. [Google Scholar] [CrossRef] [PubMed]
- Neumann, H.; Kotter, M.R.; Franklin, R.J.M. Debris Clearance by Microglia: An Essential Link between Degeneration and Regeneration. Brain 2009, 132, 288–295. [Google Scholar] [CrossRef] [PubMed]
- Ousman, S.S.; David, S. Lysophosphatidylcholine Induces Rapid Recruitment and Activation of Macrophages in the Adult Mouse Spinal Cord. Glia 2000, 30, 92–104. [Google Scholar] [CrossRef]
- Cavone, L.; McCann, T.; Drake, L.K.; Aguzzi, E.A.; Oprişoreanu, A.M.; Pedersen, E.; Sandi, S.; Selvarajah, J.; Tsarouchas, T.M.; Wehner, D.; et al. A Unique Macrophage Subpopulation Signals Directly to Progenitor Cells to Promote Regenerative Neurogenesis in the Zebrafish Spinal Cord. Dev. Cell 2021, 56, 1617–1630. [Google Scholar] [CrossRef]
- Foote, A.K.; Blakemore, W.F. Inflammation Stimulates Remyelination in Areas of Chronic Demyelination. Brain 2005, 128, 528–539. [Google Scholar] [CrossRef] [Green Version]
- Lloyd, A.F.; Davies, C.L.; Holloway, R.K.; Labrak, Y.; Ireland, G.; Carradori, D.; Dillenburg, A.; Borger, E.; Soong, D.; Richardson, J.C.; et al. Central Nervous System Regeneration Is Driven by Microglia Necroptosis and Repopulation. Nat. Neurosci. 2019, 22, 1046–1052. [Google Scholar] [CrossRef]
- McMurran, C.E.; Jones, C.A.; Fitzgerald, D.C.; Franklin, R.J.M. CNS Remyelination and the Innate Immune System. Front. Cell Dev. Biol. 2016, 4, 39. [Google Scholar] [CrossRef]
- Milner, R.; Campbell, I.L. The Extracellular Matrix and Cytokines Regulate Microglial Integrin Expression and Activation. J. Immunol. 2003, 170, 3850–3858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milner, R.; Relvas, J.B.; Fawcett, J.; ffrench-Constant, C. Developmental Regulation of αv Integrins Produces Functional Changes in Astrocyte Behavior. Mol. Cell. Neurosci. 2001, 18, 108–118. [Google Scholar] [CrossRef] [PubMed]
- Tawil, N.J.; Wilson, P.; Carbonetto, S. Expression and Distribution of Functional Integrins in Rat CNS Glia. J. Neurosci. Res. 1994, 39, 436–447. [Google Scholar] [CrossRef]
- Sikkema, A.H.; Stoffels, J.M.J.; Wang, P.; Basedow, F.J.; Bulsink, R.; Bajramovic, J.J.; Baron, W. Fibronectin Aggregates Promote Features of a Classically and Alternatively Activated Phenotype in Macrophages. J. Neuroinflamm. 2018, 15, 218. [Google Scholar] [CrossRef] [PubMed]
- Miron, V.E.; Boyd, A.; Zhao, J.W.; Yuen, T.J.; Ruckh, J.M.; Shadrach, J.L.; van Wijngaarden, P.; Wagers, A.J.; Williams, A.; Franklin, R.J.M.; et al. M2 Microglia and Macrophages Drive Oligodendrocyte Differentiation during CNS Remyelination. Nat. Neurosci. 2013, 16, 1211–1218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peferoen, L.A.N.; Vogel, D.Y.S.; Ummenthum, K.; Breur, M.; Heijnen, P.D.A.M.; Gerritsen, W.H.; Peferoen-Baert, R.M.B.; van der Valk, P.; Dijkstra, C.D.; Amor, S. Activation Status of Human Microglia Is Dependent on Lesion Formation Stage and Remyelination in Multiple Sclerosis. J. Neuropathol. Exp. Neurol. 2015, 74, 48–63. [Google Scholar] [CrossRef] [Green Version]
- Vogel, D.Y.S.; Vereyken, E.J.F.; Glim, J.E.; Heijnen, P.D.A.M.; Moeton, M.; van der Valk, P.; Amor, S.; Teunissen, C.E.; van Horssen, J.; Dijkstra, C.D. Macrophages in Inflammatory Multiple Sclerosis Lesions Have an Intermediate Activation Status. J. Neuroinflamm. 2013, 10, 35. [Google Scholar] [CrossRef] [Green Version]
- Milner, R.; Crocker, S.J.; Hung, S.; Wang, X.; Frausto, R.F.; del Zoppo, G.J. Fibronectin- and Vitronectin-Induced Microglial Activation and Matrix Metalloproteinase-9 Expression Is Mediated by Integrins α5β1 and αvβ5. J. Immunol. 2007, 178, 8158–8167. [Google Scholar] [CrossRef] [Green Version]
- Nasu-Tada, K.; Koizumi, S.; Inoue, K. Involvement of β1 Integrin in Microglial Chemotaxis and Proliferation on Fibronectin: Different Regulations by ADP through PKA. Glia 2005, 52, 98–107. [Google Scholar] [CrossRef]
- Goos, M.; Lange, P.; Hanisch, U.K.; Prinz, M.; Scheffel, J.; Bergmann, R.; Ebert, S.; Nau, R. Fibronectin Is Elevated in the Cerebrospinal Fluid of Patients Suffering from Bacterial Meningitis and Enhances Inflammation Caused by Bacterial Products in Primary Mouse Microglial Cell Cultures. J. Neurochem. 2007, 102, 2049–2060. [Google Scholar] [CrossRef] [PubMed]
- Okamura, Y.; Watari, M.; Jerud, E.S.; Young, D.W.; Ishizaka, S.T.; Rose, J.; Chow, J.C.; Strauss, J.F. The Extra Domain A of Fibronectin Activates Toll-like Receptor 4. J. Biol. Chem. 2001, 276, 10229–10233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ribes, S.; Ebert, S.; Regen, T.; Czesnik, D.; Scheffel, J.; Zeug, A.; Bunkowski, S.; Eiffert, H.; Hanisch, U.K.; Hammerschmidt, S.; et al. Fibronectin Stimulates Escherichia Coli Phagocytosis by Microglial Cells. Glia 2010, 58, 367–376. [Google Scholar] [CrossRef] [PubMed]
- Summers, L.; Kielty, C.; Pinteaux, E. Adhesion to Fibronectin Regulates Interleukin-1 Beta Expression in Microglial Cells. Mol. Cell. Neurosci. 2009, 41, 148–155. [Google Scholar] [CrossRef] [PubMed]
- Medina-Rodríguez, E.M.; Bribián, A.; Boyd, A.; Palomo, V.; Pastor, J.; Lagares, A.; Gil, C.; Martínez, A.; Williams, A.; De Castro, F. Promoting in Vivo Remyelination with Small Molecules: A Neuroreparative Pharmacological Treatment for Multiple Sclerosis. Sci. Rep. 2017, 7, 43545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Syed, Y.A.; Baer, A.; Hofer, M.P.; González, G.A.; Rundle, J.; Myrta, S.; Huang, J.K.; Zhao, C.; Rossner, M.J.; Trotter, M.W.B.; et al. Inhibition of Phosphodiesterase-4 Promotes Oligodendrocyte Precursor Cell Differentiation and Enhances CNS Remyelination. EMBO Mol. Med. 2013, 5, 1918–1934. [Google Scholar] [CrossRef] [PubMed]
- de Santana Nunes, A.K.; Rapôso, C.; de Oliveira, W.H.; Thomé, R.; Verinaud, L.; Tovar-Moll, F.; Peixoto, C.A. Phosphodiesterase-5 Inhibition Promotes Remyelination by MCP-1/CCR-2 and MMP-9 Regulation in a Cuprizone-Induced Demyelination Model. Exp. Neurol. 2016, 275, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Bielekova, B.; Richert, N.; Howard, T.; Packer, A.N.; Blevins, G.; Ohayon, J.; McFarland, H.F.; Stürzebecher, C.S.; Martin, R. Treatment with the Phosphodiesterase Type-4 Inhibitor Rolipram Fails to Inhibit Blood--Brain Barrier Disruption in Multiple Sclerosis. Mult. Scler. 2009, 15, 1206–1214. [Google Scholar] [CrossRef] [PubMed]
- Werkman, I.; Sikkema, A.H.; Versluijs, J.B.; Qin, J.; de Boer, P.; Baron, W. TLR3 Agonists Induce Fibronectin Aggregation by Activated Astrocytes: A Role of pro-Inflammatory Cytokines and Fibronectin Splice Variants. Sci. Rep. 2020, 10, 532. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Keivenst, V.M.; O’Toole, T.E.; McDonald, J.A.; Ginsberg, M.H. Integrin Activation and Cytoskeletal Interaction Are Essential for the Assembly of a Fibronectin Matrix. Cell 1995, 83, 715–724. [Google Scholar] [CrossRef] [Green Version]
- Wierzbicka-Patynowski, I.; Schwarzbauer, J.E. The Ins and Outs of Fibronectin Matrix Assembly. J. Cell Sci. 2003, 116, 3269–3276. [Google Scholar] [CrossRef] [Green Version]
- Ohashi, T.; Erickson, H.P. Revisiting the Mystery of Fibronectin Multimers: The Fibronectin Matrix Is Composed of Fibronectin Dimers Cross-Linked by Non-Covalent Bonds. Matrix Biol. 2009, 28, 170–175. [Google Scholar] [CrossRef] [PubMed]
- Schwarzbauer, J.E.; DeSimone, D.W. Fibronectins, Their Fibrillogenesis, and in Vivo Functions. Cold Spring Harb. Perspect. Biol. 2011, 3, a005041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bazigou, E.; Xie, S.; Chen, C.; Weston, A.; Miura, N.; Sorokin, L.; Adams, R.; Muro, A.F.; Sheppard, D.; Makinen, T. Integrin-α9 Is Required for Fibronectin Matrix Assembly during Lymphatic Valve Morphogenesis. Dev. Cell 2009, 17, 175–186. [Google Scholar] [CrossRef] [PubMed]
- Astrof, S.; Crowley, D.; George, E.L.; Fukuda, T.; Sekiguchi, K.; Hanahan, D.; Hynes, R.O. Direct Test of Potential Roles of EIIIA and EIIIB Alternatively Spliced Segments of Fibronectin in Physiological and Tumor Angiogenesis. Mol. Cell. Biol. 2004, 24, 8662–8670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, M.H.; Sun, Z.; Opitz, S.L.; Schmidt, T.E.; Peters, J.H.; George, E.L. Deletion of the Alternatively Spliced Fibronectin EIIIA Domain in Mice Reduces Atherosclerosis. Blood 2004, 104, 11–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chauhan, A.K.; Moretti, F.A.; Iaconcig, A.; Baralle, F.E.; Muro, A.F. Impaired Motor Coordination in Mice Lacking the EDA Exon of the Fibronectin Gene. Behav. Brain Res. 2005, 161, 31–38. [Google Scholar] [CrossRef]
- Muro, A.F.; Chauhan, A.K.; Gajovic, S.; Iaconcig, A.; Porro, F.; Stanta, G.; Baralle, F.E. Regulated Splicing of the Fibronectin EDA Exon Is Essential for Proper Skin Wound Healing and Normal Lifespan. J. Cell Biol. 2003, 162, 149–160. [Google Scholar] [CrossRef]
- Bsibsi, M.; Bajramovic, J.J.; Vogt, M.H.J.; van Duijvenvoorden, E.; Baghat, A.; Persoon-Deen, C.; Tielen, F.; Verbeek, R.; Huitinga, I.; Ryffel, B.; et al. The Microtubule Regulator Stathmin Is an Endogenous Protein Agonist for TLR3. J. Immunol. 2010, 184, 6929–6937. [Google Scholar] [CrossRef] [Green Version]
- Liu, A.; Stadelmann, C.; Moscarello, M.; Bruck, W.; Sobel, A.; Mastronardi, F.G.; Casaccia-Bonnefil, P. Expression of Stathmin, a Developmentally Controlled Cytoskeleton- Regulating Molecule, in Demyelinating Disorders. J. Neurosci. 2005, 25, 737–747. [Google Scholar] [CrossRef] [Green Version]
- Škuljec, J.; Gudi, V.; Ulrich, R.; Frichert, K.; Yildiz, Ö.; Pul, R.; Voss, E.V.; Wissel, K.; Baumgärtner, W.; Stangel, M. Matrix Metalloproteinases and Their Tissue Inhibitors in Cuprizone-Induced Demyelination and Remyelination of Brain White and Gray Matter. J. Neuropathol. Exp. Neurol. 2011, 70, 758–769. [Google Scholar] [CrossRef]
- Wang, P.; Gorter, R.P.; de Jonge, J.C.; Nazmuddin, M.; Zhao, C.; Amor, S.; Hoekstra, D.; Baron, W. MMP7 Cleaves Remyelination-Impairing Fibronectin Aggregates and Its Expression Is Reduced in Chronic Multiple Sclerosis Lesions. Glia 2018, 66, 1625–1643. [Google Scholar] [CrossRef] [PubMed]
- Anthony, D.C.; Ferguson, B.; Matyzak, M.K.; Miller, K.M.; Esiri, M.M.; Perry, V.H. Differential Matrix Metalloproteinase Expression in Cases of Multiple Sclerosis and Stroke. Neuropathol. Appl. Neurobiol. 1997, 23, 406–415. [Google Scholar] [CrossRef]
- Cossins, J.A.; Clements, J.M.; Ford, J.; Miller, K.M.; Pigott, R.; Vos, W.; van der Valk, P.; De Groot, C.J.A. Enhanced Expression of MMP-7 and MMP-9 in Demyelinating Multiple Sclerosis Lesions. Acta Neuropathol. 1997, 94, 590–598. [Google Scholar] [CrossRef]
- Lindberg, R.L.P.; De Groot, C.J.A.; Montagne, L.; Freitag, P.; van der Valk, P.; Kappos, L.; Leppert, D. The Expression Profile of Matrix Metalloproteinases (MMPs) and Their Inhibitors (TIMPs) in Lesions and Normal Appearing White Matter of Multiple Sclerosis. Brain 2001, 124, 1743–1753. [Google Scholar] [CrossRef]
- Gorter, R.P.; Baron, W. Matrix Metalloproteinases Shape the Oligodendrocyte (Niche) during Development and upon Demyelination. Neurosci. Lett. 2020, 729, 134980. [Google Scholar] [CrossRef]
- Young, J.C.; Agashe, V.R.; Siegers, K.; Hartl, F.U. Pathways of Chaperone-Mediated Protein Folding in the Cytosol. Nat. Rev. Mol. Cell Biol. 2004, 5, 781–791. [Google Scholar] [CrossRef]
- Young, J.C. Mechanisms of the Hsp70 Chaperone System. Biochem. Cell Biol. 2010, 88, 291–300. [Google Scholar] [CrossRef] [PubMed]
- Calderwood, S.K.; Gong, J.; Murshid, A. Extracellular HSPs: The Complicated Roles of Extracellular HSPs in Immunity. Front. Immunol. 2016, 7, 159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- González-Ramos, M.; Calleros, L.; López-Ongil, S.; Raoch, V.; Griera, M.; Rodríguez-Puyol, M.; De Frutos, S.; Rodríguez-Puyol, D. HSP70 Increases Extracellular Matrix Production by Human Vascular Smooth Muscle through TGF-Β1 up-Regulation. Int. J. Biochem. Cell Biol. 2013, 45, 232–242. [Google Scholar] [CrossRef]
- Cwiklinska, H.; Mycko, M.P.; Szymanska, B.; Matysiak, M.; Selmaj, K.W. Aberrant Stress-Induced Hsp70 Expression in Immune Cells in Multiple Sclerosis. J. Neurosci. Res. 2010, 88, 3102–3110. [Google Scholar] [CrossRef] [PubMed]
- Khan, E.S.; Sankaran, S.; Llontop, L.; Del Campo, A. Exogenous Supply of Hsp47 Triggers Fibrillar Collagen Deposition in Skin Cell Cultures in Vitro. BMC Mol. Cell Biol. 2020, 21, 22. [Google Scholar] [CrossRef] [Green Version]
- Ito, S.; Nagata, K. Biology of Hsp47 (Serpin H1), a Collagen-Specific Molecular Chaperone. Semin. Cell Dev. Biol. 2017, 62, 142–151. [Google Scholar] [CrossRef] [PubMed]
- Hunter, M.C.; O’Hagan, K.L.; Kenyon, A.; Dhanani, K.C.H.; Prinsloo, E.; Edkins, A.L. Hsp90 Binds Directly to Fibronectin (FN) and Inhibition Reduces the Extracellular Fibronectin Matrix in Breast Cancer Cells. PLoS ONE 2014, 9, e86842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cid, C.; Alvarez-Cermeño, J.C.; Camafeita, E.; Salinas, M.; Alcázar, A. Antibodies Reactive to Heat Shock Protein 90 Induce Oligodendrocyte Precursor Cell Death in Culture. Implications for Demyelination in Multiple Sclerosis. FASEB J. 2004, 18, 409–411. [Google Scholar] [CrossRef] [PubMed]
- Espitia Pinzón, N.; Brevé, J.J.P.; Bol, J.G.J.M.; Drukarch, B.; Baron, W.; van Dam, A.M. Tissue Transglutaminase in Astrocytes Is Enhanced by Inflammatory Mediators and Is Involved in the Formation of Fibronectin Fibril-like Structures. J. Neuroinflamm. 2017, 14, 260. [Google Scholar] [CrossRef] [Green Version]
- Nichols, P.; Urriola, J.; Miller, S.; Bjorkman, T.; Mahady, K.; Vegh, V.; Nasrallah, F.; Winter, C. Blood-Brain Barrier Dysfunction Significantly Correlates with Serum Matrix Metalloproteinase-7 (MMP-7) Following Traumatic Brain Injury. Neuroimage Clin. 2021, 31, 102741. [Google Scholar] [CrossRef] [PubMed]
- Waubant, E.; Goodkin, D.E.; Gee, L.; Bacchetti, P.; Sloan, R.; Stewart, T.; Andersson, P.B.; Stabler, G.; Miller, K. Serum MMP-9 and TIMP-1 Levels Are Related to MRI Activity in Relapsing Multiple Sclerosis. Neurology 1999, 53, 1397–1401. [Google Scholar] [CrossRef] [PubMed]
- Lassmann, H.; van Horssen, J.; Mahad, D. Progressive Multiple Sclerosis: Pathology and Pathogenesis. Nat. Rev. Neurol. 2012, 8, 647–656. [Google Scholar] [CrossRef] [PubMed]
- Alexander, A.; Agrawal, M.; Uddin, A.; Siddique, S.; Shehata, A.M.; Shaker, M.A.; Rahman, S.A.U.; Abdul, M.I.M.; Shaker, M.A. Recent Expansions of Novel Strategies towards the Drug Targeting into the Brain. Int. J. Nanomed. 2019, 14, 5895–5909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagpal, K.; Singh, S.K.; Mishra, D.N. Drug Targeting to Brain: A Systematic Approach to Study the Factors, Parameters and Approaches for Prediction of Permeability of Drugs across BBB. Expert Opin. Drug Deliv. 2013, 10, 927–955. [Google Scholar] [CrossRef]
- Stein, E.S.; Itsekson-Hayosh, Z.; Aronovich, A.; Reisner, Y.; Bushi, D.; Pick, C.G.; Tanne, D.; Chapman, J.; Vlachos, A.; Maggio, N. Thrombin Induces Ischemic LTP (ILTP): Implications for Synaptic Plasticity in the Acute Phase of Ischemic Stroke. Sci. Rep. 2015, 5, 7912. [Google Scholar] [CrossRef] [Green Version]
- Gingrich, M.B.; Traynelis, S.F. Serine Proteases and Brain Damage-Is There a Link? Trends Neurosci. 2000, 23, 399–407. [Google Scholar] [CrossRef]
- Chong, W.; Kim, S.N.; Han, S.K.; Lee, S.Y.; Ryu, P.D. Low Non-NMDA Receptor Current Density as Possible Protection Mechanism from Neurotoxicity of Circulating Glutamate on Subfornical Organ Neurons in Rats. Korean J. Physiol. Pharmacol. 2015, 19, 177–181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abbott, N.J.; Patabendige, A.A.K.; Dolman, D.E.M.; Yusof, S.R.; Begley, D.J. Structure and Function of the Blood-Brain Barrier. Neurobiol. Dis. 2010, 37, 13–25. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.T.; Zhao, Y.Z.; Wong, H.L.; Cai, J.; Peng, L.; Tian, X.Q. Current Approaches to Enhance CNS Delivery of Drugs across the Brain Barriers. Int. J. Nanomed. 2014, 9, 2241–2257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Yang, G.; Jin, S.; Xu, L.; Zhao, C.X. Development of High-Drug-Loading Nanoparticles. Chempluschem 2020, 85, 2143–2157. [Google Scholar] [CrossRef]
- Musumeci, T.; Ventura, C.A.; Giannone, I.; Ruozi, B.; Montenegro, L.; Pignatello, R.; Puglisi, G. PLA/PLGA Nanoparticles for Sustained Release of Docetaxel. Int. J. Pharm. 2006, 325, 172–179. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, B.; Santra, K.; Pattnaik, G.; Ghosh, S. Preparation, Characterization and in-Vitro Evaluation of Sustained Release Protein-Loaded Nanoparticles Based on Biodegradable Polymers. Int. J. Nanomed. 2008, 3, 487. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.H.; Martin, D.C. Sustained Release of Dexamethasone from Hydrophilic Matrices Using PLGA Nanoparticles for Neural Drug Delivery. Biomaterials 2006, 27, 3031–3037. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, G.B.; Shinde, R.; Contag, C.H.; Zare, R.N. Sustained Release of Drugs Dispersed in Polymer Nanoparticles. Angew. Chemie 2008, 120, 7998–8000. [Google Scholar] [CrossRef]
- Su, C.W.; Chiang, C.S.; Li, W.M.; Hu, S.H.; Chen, S.Y. Multifunctional Nanocarriers for Simultaneous Encapsulation of Hydrophobic and Hydrophilic Drugs in Cancer Treatment. Nanomedicine 2014, 9, 1499–1515. [Google Scholar] [CrossRef] [PubMed]
- Zogg, H.; Singh, R.; Ro, S. Current Advances in RNA Therapeutics for Human Diseases. Int. J. Mol. Sci. 2022, 23, 2736. [Google Scholar] [CrossRef] [PubMed]
- Cheon, J.; Chan, W.; Zuhorn, I. The Future of Nanotechnology: Cross-Disciplined Progress to Improve Health and Medicine. Acc. Chem. Res. 2019, 52, 2405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitchell, M.J.; Billingsley, M.M.; Haley, R.M.; Wechsler, M.E.; Peppas, N.A.; Langer, R. Engineering Precision Nanoparticles for Drug Delivery. Nat. Rev. Drug Discov. 2021, 20, 101–124. [Google Scholar] [CrossRef] [PubMed]
- Renukuntla, J.; Vadlapudi, A.D.; Patel, A.; Boddu, S.H.S.; Mitra, A.K. Approaches for Enhancing Oral Bioavailability of Peptides and Proteins. Int. J. Pharm. 2013, 447, 75–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Odiba, A.; Ottah, V.; Ottah, C.; Anunobi, O.; Ukegbu, C.; Edeke, A.; Uroko, R.; Omeje, K. Therapeutic Nanomedicine Surmounts the Limitations of Pharmacotherapy. Open Med. 2017, 12, 271–287. [Google Scholar] [CrossRef]
- Agrahari, V.; Agrahari, V.; Mitra, A.K. Nanocarrier Fabrication and Macromolecule Drug Delivery: Challenges and Opportunities. Ther. Deliv. 2016, 7, 257–278. [Google Scholar] [CrossRef] [Green Version]
- Mahato, R.I.; Narang, A.S.; Thoma, L.; Miller, D.D. Emerging Trends in Oral Delivery of Peptide and Protein Drugs. Crit. Rev. Ther. Drug Carrier Syst. 2003, 20, 153–214. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Q.; Wei, T.; Farbiak, L.; Johnson, L.T.; Dilliard, S.A.; Siegwart, D.J. Selective ORgan Targeting (SORT) Nanoparticles for Tissue Specific MRNA Delivery and CRISPR/Cas Gene Editing. Nat. Nanotechnol. 2020, 15, 313. [Google Scholar] [CrossRef]
- Podual, K.; Doyle, F.J.; Peppas, N.A. Glucose-Sensitivity of Glucose Oxidase-Containing Cationic Copolymer Hydrogels Having Poly(Ethylene Glycol) Grafts. J. Control. Release 2000, 67, 9–17. [Google Scholar] [CrossRef]
- Marek, S.R.; Peppas, N.A. Insulin Release Dynamics from Poly(Diethylaminoethyl Methacrylate) Hydrogel Systems. AIChE J. 2013, 59, 3578–3585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Culver, H.R.; Clegg, J.R.; Peppas, N.A. Analyte-Responsive Hydrogels: Intelligent Materials for Biosensing and Drug Delivery. Acc. Chem. Res. 2017, 50, 170. [Google Scholar] [CrossRef]
- Cheng, X.; Lee, R.J. The Role of Helper Lipids in Lipid Nanoparticles (LNPs) Designed for Oligonucleotide Delivery. Adv. Drug Deliv. Rev. 2016, 99, 129–137. [Google Scholar] [CrossRef]
- Li, X.; Tsibouklis, J.; Weng, T.; Zhang, B.; Yin, G.; Feng, G.; Cui, Y.; Savina, I.N.; Mikhalovska, L.I.; Sandeman, S.R.; et al. Nano Carriers for Drug Transport across the Blood–Brain Barrier. J. Drug Target. 2017, 25, 17–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joshi, B.S.; Ortiz, D.; Zuhorn, I.S. Converting Extracellular Vesicles into Nanomedicine: Loading and Unloading of Cargo. Mater. Today Nano 2021, 16, 100148. [Google Scholar] [CrossRef]
- Aliyandi, A.; Satchell, S.; Unger, R.E.; Bartosch, B.; Parent, R.; Zuhorn, I.S.; Salvati, A. Effect of Endothelial Cell Heterogeneity on Nanoparticle Uptake. Int. J. Pharm. 2020, 587, 119699. [Google Scholar] [CrossRef] [PubMed]
- Ozbakir, B.; Crielaard, B.J.; Metselaar, J.M.; Storm, G.; Lammers, T. Liposomal Corticosteroids for the Treatment of Inflammatory Disorders and Cancer. J. Control. Release 2014, 190, 624–636. [Google Scholar] [CrossRef] [PubMed]
- Rousseau, V.; Denizot, B.; Le Jeune, J.J.; Jallet, P. Early Detection of Liposome Brain Localization in Rat Experimental Allergic Encephalomyelitis. Exp. Brain Res. 1999, 125, 255–264. [Google Scholar] [CrossRef]
- Lu, L.; Qi, S.; Chen, Y.; Luo, H.; Huang, S.; Yu, X.; Luo, Q.; Zhang, Z. Targeted Immunomodulation of Inflammatory Monocytes across the Blood-Brain Barrier by Curcumin-Loaded Nanoparticles Delays the Progression of Experimental Autoimmune Encephalomyelitis. Biomaterials 2020, 245, 119987. [Google Scholar] [CrossRef]
- Doshi, A.; Chataway, J. Multiple Sclerosis, a Treatable Disease. Clin. Med. 2017, 17, 530–536. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, J.; Metselaar, J.M.; Wauben, M.H.M.; Toyka, K.V.; Storm, G.; Gold, R. Drug Targeting by Long-Circulating Liposomal Glucocorticosteroids Increases Therapeutic Efficacy in a Model of Multiple Sclerosis. Brain 2003, 126, 1895–1904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schweingruber, N.; Haine, A.; Tiede, K.; Karabinskaya, A.; van den Brandt, J.; Wüst, S.; Metselaar, J.M.; Gold, R.; Tuckermann, J.P.; Reichardt, H.M.; et al. Liposomal Encapsulation of Glucocorticoids Alters Their Mode of Action in the Treatment of Experimental Autoimmune Encephalomyelitis. J. Immunol. 2011, 187, 4310–4318. [Google Scholar] [CrossRef] [PubMed]
- Linker, R.A.; Weller, C.; Lühder, F.; Mohr, A.; Schmidt, J.; Knauth, M.; Metselaar, J.M.; Gold, R. Liposomal Glucocorticosteroids in Treatment of Chronic Autoimmune Demyelination: Long-Term Protective Effects and Enhanced Efficacy of Methylprednisolone Formulations. Exp. Neurol. 2008, 211, 397–406. [Google Scholar] [CrossRef]
- Gaillard, P.J.; Appeldoorn, C.C.M.; Rip, J.; Dorland, R.; van der Pol, S.M.A.; Kooij, G.; de Vries, H.E.; Reijerkerk, A. Enhanced Brain Delivery of Liposomal Methylprednisolone Improved Therapeutic Efficacy in a Model of Neuroinflammation. J. Control. Release 2012, 164, 364–369. [Google Scholar] [CrossRef]
- Ojha, S.; Kumar, B. Preparation and Statistical Modeling of Solid Lipid Nanoparticles of Dimethyl Fumarate for Better Management of Multiple Sclerosis. Adv. Pharm. Bull. 2018, 8, 225–233. [Google Scholar] [CrossRef] [Green Version]
- Kumar, P.; Sharma, G.; Gupta, V.; Kaur, R.; Thakur, K.; Malik, R.; Kumar, A.; Kaushal, N.; Raza, K. Preclinical Explorative Assessment of Dimethyl Fumarate-Based Biocompatible Nanolipoidal Carriers for the Management of Multiple Sclerosis. ACS Chem. Neurosci. 2018, 9, 1152–1158. [Google Scholar] [CrossRef] [PubMed]
- Pujol-Autonell, I.; Mansilla, M.J.; Rodriguez-Fernandez, S.; Cano-Sarabia, M.; Navarro-Barriuso, J.; Ampudia, R.M.; Rius, A.; Garcia-Jimeno, S.; Perna-Barrull, D.; Caceres, E.M.; et al. Liposome-Based Immunotherapy against Autoimmune Diseases: Therapeutic Effect on Multiple Sclerosis. Nanomedicine 2017, 12, 1231–1242. [Google Scholar] [CrossRef]
- Gholamzad, M.; Ebtekar, M.; Shafiee Ardestani, M. Intravenous Injection of Myelin Oligodendrocyte Glycoprotein-Coated PLGA Microparticles Have Tolerogenic Effects in Experimental Autoimmune Encephalomyelitis. Iran. J. Allergy Asthma Immunol. 2017, 16, 271–281. [Google Scholar] [PubMed]
- Pei, W.; Wan, X.; Shahzad, K.A.; Zhang, L.; Song, S.; Jin, X.; Wang, L.; Zhao, C.; Shen, C. Direct Modulation of Myelin-Autoreactive CD4+ and CD8+ T Cells in EAE Mice by a Tolerogenic Nanoparticle Co-Carrying Myelin Peptide-Loaded Major Histocompatibility Complexes, CD47 and Multiple Regulatory Molecules. Int. J. Nanomed. 2018, 13, 3731–3750. [Google Scholar] [CrossRef] [Green Version]
- Cappellano, G.; Woldetsadik, A.D.; Orilieri, E.; Shivakumar, Y.; Rizzi, M.; Carniato, F.; Gigliotti, C.L.; Boggio, E.; Clemente, N.; Comi, C.; et al. Subcutaneous Inverse Vaccination with PLGA Particles Loaded with a MOG Peptide and IL-10 Decreases the Severity of Experimental Autoimmune Encephalomyelitis. Vaccine 2014, 32, 5681–5689. [Google Scholar] [CrossRef]
- Hunter, Z.; McCarthy, D.P.; Yap, W.T.; Harp, C.T.; Getts, D.R.; Shea, L.D.; Miller, S.D. A Biodegradable Nanoparticle Platform for the Induction of Antigen-Specific Immune Tolerance for Treatment of Autoimmune Disease. ACS Nano 2014, 8, 2148–2160. [Google Scholar] [CrossRef]
- Kuo, R.; Saito, E.; Miller, S.D.; Shea, L.D. Peptide-Conjugated Nanoparticles Reduce Positive Co-Stimulatory Expression and T Cell Activity to Induce Tolerance. Mol. Ther. 2017, 25, 1676–1685. [Google Scholar] [CrossRef] [Green Version]
- Gammon, J.M.; Tostanoski, L.H.; Adapa, A.R.; Chiu, Y.C.; Jewell, C.M. Controlled Delivery of a Metabolic Modulator Promotes Regulatory T Cells and Restrains Autoimmunity. J. Control. Release 2015, 210, 169–178. [Google Scholar] [CrossRef] [Green Version]
- Osorio-Querejeta, I.; Carregal-Romero, S.; Ayerdi-Izquierdo, A.; Mäger, I.; Nash, L.A.; Wood, M.; Egimendia, A.; Betanzos, M.; Alberro, A.; Iparraguirre, L.; et al. MiR-219a-5p Enriched Extracellular Vesicles Induce OPC Differentiation and EAE Improvement More Efficiently than Liposomes and Polymeric Nanoparticles. Pharmaceutics 2020, 12, 186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belogurov, A.A.; Stepanov, A.V.; Smirnov, I.V.; Melamed, D.; Bacon, A.; Mamedov, A.E.; Boitsov, V.M.; Sashchenko, L.P.; Ponomarenko, N.A.; Sharanova, S.N.; et al. Liposome-Encapsulated Peptides Protect against Experimental Allergic Encephalitis. FASEB J. 2013, 27, 222–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belogurov, A.A.; Zargarova, T.A.; Turobov, V.I.; Novikova, N.I.; Favorova, O.O.; Ponomarenko, N.A.; Gabibov, A.G. Suppression of Ongoing Experimental Allergic Encephalomyelitis in Da Rats by Novel Peptide Drug, Structural Part of Human Myelin Basic Protein 4662. Autoimmunity 2009, 42, 362–364. [Google Scholar] [CrossRef]
- Ivanova, V.V.; Khaiboullina, S.F.; Gomzikova, M.O.; Martynova, E.V.; Ferreira, A.M.; Garanina, E.E.; Sakhapov, D.I.; Lomakin, Y.A.; Khaibullin, T.I.; Granatov, E.V.; et al. Divergent Immunomodulation Capacity of Individual Myelin Peptides-Components of Liposomal Therapeutic against Multiple Sclerosis. Front. Immunol. 2017, 1335, 8. [Google Scholar] [CrossRef] [Green Version]
- Lomakin, Y.; Belogurov, A.; Glagoleva, I.; Stepanov, A.; Zakharov, K.; Okunola, J.; Smirnov, I.; Genkin, D.; Gabibov, A. Administration of Myelin Basic Protein Peptides Encapsulated in Mannosylated Liposomes Normalizes Level of Serum TNF- α and IL-2 and Chemoattractants CCL2 and CCL4 in Multiple Sclerosis Patients. Mediators Inflamm. 2016, 2016, 2847232. [Google Scholar] [CrossRef] [Green Version]
- Rittchen, S.; Boyd, A.; Burns, A.; Park, J.; Fahmy, T.M.; Metcalfe, S.; Williams, A. Myelin Repair Invivo Is Increased by Targeting Oligodendrocyte Precursor Cells with Nanoparticles Encapsulating Leukaemia Inhibitory Factor (LIF). Biomaterials 2015, 56, 78–85. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Du, C.; Guo, N.; Teng, Y.; Meng, X.; Sun, H.; Li, S.; Yu, P.; Galons, H. Composition Design and Medical Application of Liposomes. Eur. J. Med. Chem. 2019, 164, 640–653. [Google Scholar] [CrossRef] [PubMed]
- Akbarzadeh, A.; Rezaei-Sadabady, R.; Davaran, S.; Joo, S.W.; Zarghami, N.; Hanifehpour, Y.; Samiei, M.; Kouhi, M.; Nejati-Koshki, K. Liposome: Classification, Preparation, and Applications. Nanoscale Res. Lett. 2013, 8, 102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milla, P.; Dosio, F.; Cattel, L. PEGylation of Proteins and Liposomes: A Powerful and Flexible Strategy to Improve the Drug Delivery. Curr. Drug Metab. 2011, 13, 105–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montes-Cobos, E.; Ring, S.; Fischer, H.J.; Heck, J.; Strauß, J.; Schwaninger, M.; Reichardt, S.D.; Feldmann, C.; Lühder, F.; Reichardt, H.M. Targeted Delivery of Glucocorticoids to Macrophages in a Mouse Model of Multiple Sclerosis Using Inorganic-Organic Hybrid Nanoparticles. J. Control. Release 2017, 245, 157–169. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulou, A.; D’Souza, M.; Kappos, L.; Yaldizli, O. Dimethyl Fumarate for Multiple Sclerosis. Expert Opin. Investig. Drugs 2010, 19, 1603–1612. [Google Scholar] [CrossRef] [PubMed]
- Bomprezzi, R. Dimethyl Fumarate in the Treatment of Relapsing-Remitting Multiple Sclerosis: An Overview. Ther. Adv. Neurol. Disord. 2015, 8, 20–30. [Google Scholar] [CrossRef] [Green Version]
- Belogurov, A.; Zakharov, K.; Lomakin, Y.; Surkov, K.; Avtushenko, S.; Kruglyakov, P.; Smirnov, I.; Makshakov, G.; Lockshin, C.; Gregoriadis, G.; et al. CD206-Targeted Liposomal Myelin Basic Protein Peptides in Patients with Multiple Sclerosis Resistant to First-Line Disease-Modifying Therapies: A First-in-Human, Proof-of-Concept Dose-Escalation Study. Neurotherapeutics 2016, 13, 895–904. [Google Scholar] [CrossRef] [Green Version]
- Park, K.; Skidmore, S.; Hadar, J.; Garner, J.; Park, H.; Otte, A.; Soh, B.K.; Yoon, G.; Yu, D.; Yun, Y.; et al. Injectable, Long-Acting PLGA Formulations: Analyzing PLGA and Understanding Microparticle Formation. J. Control. Release 2019, 304, 125–134. [Google Scholar] [CrossRef]
- Hua, Y.; Su, Y.; Zhang, H.; Liu, N.; Wang, Z.; Gao, X.; Gao, J.; Zheng, A. Poly(Lactic-Co-Glycolic Acid) Microsphere Production Based on Quality by Design: A Review. Drug Deliv. 2021, 28, 1342. [Google Scholar] [CrossRef] [PubMed]
- Pearson, R.M.; Podojil, J.R.; Shea, L.D.; King, N.J.C.; Miller, S.D.; Getts, D.R. Overcoming Challenges in Treating Autoimmuntity: Development of Tolerogenic Immune-Modifying Nanoparticles. Nanomed. Nanotechnol. Biol. Med. 2019, 18, 282–291. [Google Scholar] [CrossRef]
- Makadia, H.K.; Siegel, S.J. Poly Lactic-Co-Glycolic Acid (PLGA) as Biodegradable Controlled Drug Delivery Carrier. Polymers 2011, 3, 1377–1397. [Google Scholar] [CrossRef] [PubMed]
- Chiu, H.I.; Samad, N.A.; Fang, L.; Lim, V. Cytotoxicity of Targeted PLGA Nanoparticles: A Systematic Review. RSC Adv. 2021, 11, 9433–9449. [Google Scholar] [CrossRef]
- Hrkach, J.; Von Hoff, D.; Ali, M.M.; Andrianova, E.; Auer, J.; Campbell, T.; De Witt, D.; Figa, M.; Figueiredo, M.; Horhota, A.; et al. Preclinical Development and Clinical Translation of a PSMA-Targeted Docetaxel Nanoparticle with a Differentiated Pharmacological Profile. Sci. Transl. Med. 2012, 4, 128ra39. [Google Scholar] [CrossRef]
- Dargahi, N.; Katsara, M.; Tselios, T.; Androutsou, M.E.; De Courten, M.; Matsoukas, J.; Apostolopoulos, V. Multiple Sclerosis: Immunopathology and Treatment Update. Brain Sci. 2017, 7, 78. [Google Scholar] [CrossRef] [Green Version]
- Cavaletti, G.; Cassetti, A.; Canta, A.; Galbiati, S.; Gilardini, A.; Oggioni, N.; Rodriguez-Menendez, V.; Fasano, A.; Liuzzi, G.M.; Fattler, U.; et al. Cationic Liposomes Target Sites of Acute Neuroinflammation in Experimental Autoimmune Encephalomyelitis. Mol. Pharm. 2009, 6, 1363–1370. [Google Scholar] [CrossRef]
- Deverman, B.E.; Patterson, P.H. Exogenous Leukemia Inhibitory Factor Stimulates Oligodendrocyte Progenitor Cell Proliferation and Enhances Hippocampal Remyelination. J. Neurosci. 2012, 32, 2100–2109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butzkueven, H.; Zhang, J.G.; Soilu-Hanninen, M.; Hochrein, H.; Chionh, F.; Shipham, K.A.; Emery, B.; Turnley, A.M.; Petratos, S.; Ernst, M.; et al. LIF Receptor Signaling Limits Immune-Mediated Demyelination by Enhancing Oligodendrocyte Survival. Nat. Med. 2002, 8, 613–619. [Google Scholar] [CrossRef] [PubMed]
- Slaets, H.; Hendriks, J.J.A.; van den Haute, C.; Coun, F.; Baekelandt, V.; Stinissen, P.; Hellings, N. CNS-Targeted Lif Expression Improves Therapeutic Efficacy and Limits Autoimmune-Mediated Demyelination in a Model of Multiple Sclerosis. Mol. Ther. 2010, 18, 684–691. [Google Scholar] [CrossRef]
- Wang, P.; Xue, Y.; Shang, X.; Liu, Y. Diphtheria Toxin Mutant CRM197-Mediated Transcytosis across Blood-Brain Barrier in Vitro. Cell. Mol. Neurobiol. 2010, 30, 717–725. [Google Scholar] [CrossRef] [PubMed]
- Lu, Q.; Cai, X.; Zhang, X.; Li, S.; Song, Y.; Du, D.; Dutta, P.; Lin, Y. Synthetic Polymer Nanoparticles Functionalized with Different Ligands for Receptor-Mediated Transcytosis across the Blood-Brain Barrier. ACS Appl. Bio Mater. 2018, 1, 1687–1694. [Google Scholar] [CrossRef] [PubMed]
- Pardridge, W.M.; Kang, Y.S.; Buciak, J.L.; Yang, J. Human Insulin Receptor Monoclonal Antibody Undergoes High Affinity Binding to Human Brain Capillaries in Vitro and Rapid Transcytosis Through the Blood–Brain Barrier in Vivo in the Primate. Pharm. Res. 1995, 12, 807–816. [Google Scholar] [CrossRef] [PubMed]
- Stojanov, K.; Georgieva, J.V.; Brinkhuis, R.P.; van Hest, J.C.; Rutjes, F.P.; Dierckx, R.A.J.O.; De Vries, E.F.J.; Zuhorn, I.S. In Vivo Biodistribution of Prion- and GM1-Targeted Polymersomes Following Intravenous Administration in Mice. Mol. Pharm. 2012, 9, 1620–1627. [Google Scholar] [CrossRef]
- de Jong, E.; Williams, D.S.; Abdelmohsen, L.K.E.A.; van Hest, J.C.M.; Zuhorn, I.S. A Filter-Free Blood-Brain Barrier Model to Quantitatively Study Transendothelial Delivery of Nanoparticles by Fluorescence Spectroscopy. J. Control. 2018, 289, 14–22. [Google Scholar] [CrossRef]
- Dos Santos Rodrigues, B.; Lakkadwala, S.; Kanekiyo, T.; Singh, J. Development and Screening of Brain-Targeted Lipid-Based Nanoparticles with Enhanced Cell Penetration and Gene Delivery Properties. Int. J. Nanomed. 2019, 14, 6497–6517. [Google Scholar] [CrossRef] [Green Version]
- Georgieva, J.V.; Hoekstra, D.; Zuhorn, I.S. Smuggling Drugs into the Brain: An Overview of Ligands Targeting Transcytosis for Drug Delivery across the Blood–Brain Barrier. Pharmaceutics 2014, 6, 557–583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, C.H.; Tsai, C.Y.; Tomanek, B.; Chen, W.Y.; Cheng, F.Y. Evaluation of Blood-Brain Barrier-Stealth Nanocomposites for in Situ Glioblastoma Theranostics Applications. Nanoscale 2016, 8, 7866–7870. [Google Scholar] [CrossRef] [PubMed]
- Haroon, M.M.; Saba, K.; Harshavardhan Boddedda, V.; Kumar, J.M.; Patel, A.B.; Gopal, V. Delivery of BACE1 SiRNA Mediated by TARBP-BTP Fusion Protein Reduces b-Amyloid Deposits in a Transgenic Mouse Model of Alzheimer’s Disease. J. Biosci. 2019, 44, 1. [Google Scholar] [CrossRef]
- Haroon, M.M.; Dar, G.H.; Jeyalakshmi, D.; Venkatraman, U.; Saba, K.; Rangaraj, N.; Patel, A.B.; Gopal, V. A Designed Recombinant Fusion Protein for Targeted Delivery of SiRNA to the Mouse Brain. J. Control. Release 2016, 228, 120–131. [Google Scholar] [CrossRef]
- Zhang, H.; van Os, W.L.; Tian, X.; Zu, G.; Ribovski, L.; Bron, R.; Bussmann, J.; Kros, A.; Liu, Y.; Zuhorn, I.S. Development of Curcumin-Loaded Zein Nanoparticles for Transport across the Blood–Brain Barrier and Inhibition of Glioblastoma Cell Growth. Biomater. Sci. 2021, 26, 7092–7103. [Google Scholar] [CrossRef]
- Simionescu, M.; Gafencu, A.; Antohe, F. Transcytosis of Plasma Macromolecules in Endothelial Cells: A Cell Biological Survey. Microsc. Res. Tech. 2002, 57, 269–288. [Google Scholar] [CrossRef]
- Kreuter, J. Drug Delivery to the Central Nervous System by Polymeric Nanoparticles: What Do We Know? Adv. Drug Deliv. Rev. 2014, 71, 2–14. [Google Scholar] [CrossRef]
- Gong, Y.; Chowdhury, P.; Nagesh, P.K.B.; Rahman, M.A.; Zhi, K.; Yallapu, M.M.; Kumar, S. Novel Elvitegravir Nanoformulation for Drug Delivery across the Blood-Brain Barrier to Achieve HIV-1 Suppression in the CNS Macrophages. Sci. Rep. 2020, 10, 3835. [Google Scholar] [CrossRef] [Green Version]
- Gong, Y.; Zhi, K.; Nagesh, P.K.B.; Sinha, N.; Chowdhury, P.; Chen, H.; Gorantla, S.; Yallapu, M.M.; Kumar, S. An Elvitegravir Nanoformulation Crosses the Blood–Brain Barrier and Suppresses HIV-1 Replication in Microglia. Viruses 2020, 12, 564. [Google Scholar] [CrossRef] [PubMed]
- Pardridge, W.M.; Buciak, J.L.; Kang, Y.S.; Boado, R.J. Protamine-Mediated Transport of Albumin into Brain and Other Organs of the Rat. Binding and Endocytosis of Protamine-Albumin Complex by Microvascular Endothelium. J. Clin. Investig. 1993, 92, 2224–2229. [Google Scholar] [CrossRef] [PubMed]
- Hervé, F.; Ghinea, N.; Scherrmann, J.M. CNS Delivery via Adsorptive Transcytosis. AAPS J. 2008, 10, 455–472. [Google Scholar] [CrossRef] [Green Version]
- Kozielski, K.L.; Tzeng, S.Y.; Hurtado De Mendoza, B.A.; Green, J.J. Bioreducible Cationic Polymer-Based Nanoparticles for Efficient and Environmentally Triggered Cytoplasmic SiRNA Delivery to Primary Human Brain Cancer Cells. ACS Nano 2014, 8, 3232–3241. [Google Scholar] [CrossRef]
- Karlsson, J.; Rui, Y.; Kozielski, K.L.; Placone, A.L.; Choi, O.; Tzeng, S.Y.; Kim, J.; Keyes, J.J.; Bogorad, M.I.; Gabrielson, K.; et al. Engineered Nanoparticles for Systemic SiRNA Delivery to Malignant Brain Tumours. Nanoscale 2019, 11, 20045–20057. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Liu, L. Modern Methods for Delivery of Drugs across the Blood-Brain Barrier. Adv. Drug Deliv. Rev. 2012, 64, 640–665. [Google Scholar] [CrossRef] [PubMed]
- Shi, K.; Long, Y.; Xu, C.; Wang, Y.; Qiu, Y.; Yu, Q.; Liu, Y.; Zhang, Q.; Gao, H.; Zhang, Z.; et al. Liposomes Combined an Integrin αvβ3-Specific Vector with PH-Responsible Cell-Penetrating Property for Highly Effective Antiglioma Therapy through the Blood-Brain Barrier. ACS Appl. Mater. Interfaces 2015, 7, 21442–21454. [Google Scholar] [CrossRef]
- Shi, K.; Li, J.; Cao, Z.; Yang, P.; Qiu, Y.; Yang, B.; Wang, Y.; Long, Y.; Liu, Y.; Zhang, Q.; et al. A PH-Responsive Cell-Penetrating Peptide-Modified Liposomes with Active Recognizing of Integrin αvβ3 for the Treatment of Melanoma. J. Control. Release 2015, 217, 138–150. [Google Scholar] [CrossRef] [PubMed]
- Gartziandia, O.; Egusquiaguirre, S.P.; Bianco, J.; Pedraz, J.L.; Igartua, M.; Hernandez, R.M.; Préat, V.; Beloqui, A. Nanoparticle Transport across in Vitro Olfactory Cell Monolayers. Int. J. Pharm. 2016, 499, 81–89. [Google Scholar] [CrossRef]
- Liu, Y.; Ran, R.; Chen, J.; Kuang, Q.; Tang, J.; Mei, L.; Zhang, Q.; Gao, H.; Zhang, Z.; He, Q. Paclitaxel Loaded Liposomes Decorated with a Multifunctional Tandem Peptide for Glioma Targeting. Biomaterials 2014, 35, 4835–4847. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Ji, M.; Wong, M.K.; Il Joo, K.; Wang, P. Enhanced Therapeutic Efficacy of IRGD-Conjugated Crosslinked Multilayer Liposomes for Drug Delivery. Biomed Res. Int. 2013, 2013, 378380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Q.; Tang, J.; Fu, L.; Ran, R.; Liu, Y.; Yuan, M.; He, Q. A PH-Responsive α-Helical Cell Penetrating Peptide-Mediated Liposomal Delivery System. Biomaterials 2013, 34, 7980–7993. [Google Scholar] [CrossRef]
- Burgess, A.; Shah, K.; Hough, O.; Hynynen, K. Focused Ultrasound-Mediated Drug Delivery through the Blood-Brain Barrier. Expert Rev. Neurother. 2015, 15, 477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hynynen, K.; McDannold, N.; Vykhodtseva, N.; Jolesz, F.A. Noninvasive MR Imaging-Guided Focal Opening of the Blood-Brain Barrier in Rabbits. Radiology 2001, 220, 640–646. [Google Scholar] [CrossRef] [PubMed]
- Mesiwala, A.H.; Farrell, L.; Wenzel, H.J.; Silbergeld, D.L.; Crum, L.A.; Winn, H.R.; Mourad, H.R. High-Intensity Focused Ultrasound Selectively Disrupts the Blood-Brain Barrier in Vivo. Ultrasound Med. Biol. 2002, 28, 389–400. [Google Scholar] [CrossRef]
- Leinenga, G.; Götz, J. Scanning Ultrasound Removes Amyloid-β and Restores Memory in an Alzheimer’s Disease Mouse Model. Sci. Transl. Med. 2015, 7, 278ra33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mooney, S.J.; Shah, K.; Yeung, S.; Burgess, A.; Aubert, I.; Hynynen, K. Focused Ultrasound-Induced Neurogenesis Requires an Increase in Blood-Brain Barrier Permeability. PLoS ONE 2016, 11, e0159892. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.; Kong, C.; Lee, J.; Choi, B.Y.; Sim, J.; Koh, C.S.; Park, M.; Na, Y.C.; Suh, S.W.; Chang, W.S.; et al. Focused Ultrasound-Induced Blood-Brain Barrier Opening Improves Adult Hippocampal Neurogenesis and Cognitive Function in a Cholinergic Degeneration Dementia Rat Model. Alzheimers Res. Ther. 2019, 11, 110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, J.J.; Wang, S.; Brown, T.R.; Small, S.A.; Duff, K.E.K.; Konofagou, E.E. Noninvasive and Transient Blood-Brain Barrier Opening in the Hippocampus of Alzheimer’s Double Transgenic Mice Using Focused Ultrasound. Ultrason. Imaging 2008, 30, 189–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheikov, N.; McDannold, N.; Sharma, S.; Hynynen, K. Effect of Focused Ultrasound Applied with an Ultrasound Contrast Agent on the Tight Junctional Integrity of the Brain Microvascular Endothelium. Ultrasound Med. Biol. 2008, 34, 1093–1104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pandit, R.; Koh, W.K.; Sullivan, R.K.P.; Palliyaguru, T.; Parton, R.G.; Götz, J. Role for Caveolin-Mediated Transcytosis in Facilitating Transport of Large Cargoes into the Brain via Ultrasound. J. Control. Release 2020, 327, 667–675. [Google Scholar] [CrossRef] [PubMed]
- Meijering, B.D.M.; Juffermans, L.J.M.; van Wamel, A.; Henning, R.H.; Zuhorn, I.S.; Emmer, M.; Versteilen, A.M.G.; Paulus, W.J.; van Gilst, W.H.; Kooiman, K.; et al. Ultrasound and Microbubble-Targeted Delivery of Macromolecules Is Regulated by Induction of Endocytosis and Pore Formation. Circ. Res. 2009, 104, 679–687. [Google Scholar] [CrossRef] [PubMed]
- Crowe, T.P.; Greenlee, M.H.W.; Kanthasamy, A.G.; Hsu, W.H. Mechanism of Intranasal Drug Delivery Directly to the Brain. Life Sci. 2018, 195, 44–52. [Google Scholar] [CrossRef]
- Erdő, F.; Bors, L.A.; Farkas, D.; Bajza, Á.; Gizurarson, S. Evaluation of Intranasal Delivery Route of Drug Administration for Brain Targeting. Brain Res. Bull. 2018, 143, 155–170. [Google Scholar] [CrossRef]
- Chu, L.; Wanga, A.; Ni, L.; Yan, X.; Song, Y.; Zhao, M.; Sun, K.; Mu, H.; Liu, S.; Wu, Z.; et al. Nose-to-Brain Delivery of Temozolomide-Loaded Plga Nanoparticles Functionalized with Anti-Epha3 for Glioblastoma Targeting. Drug Deliv. 2018, 25, 1634–1641. [Google Scholar] [CrossRef] [Green Version]
- Nigam, K.; Kaur, A.; Tyagi, A.; Nematullah, M.; Khan, F.; Gabrani, R.; Dang, S. Nose-to-Brain Delivery of Lamotrigine-Loaded PLGA Nanoparticles. Drug Deliv. Transl. Res. 2019, 9, 879–890. [Google Scholar] [CrossRef]
- Musumeci, T.; Serapide, M.F.; Pellitteri, R.; Dalpiaz, A.; Ferraro, L.; Dal Magro, R.; Bonaccorso, A.; Carbone, C.; Veiga, F.; Sancini, G.; et al. Oxcarbazepine Free or Loaded PLGA Nanoparticles as Effective Intranasal Approach to Control Epileptic Seizures in Rodents. Eur. J. Pharm. Biopharm. 2018, 133, 309–320. [Google Scholar] [CrossRef]
- Zheng, X.; Shao, X.; Zhang, C.; Tan, Y.; Liu, Q.; Wan, X.; Zhang, Q.; Xu, S.; Jiang, X. Intranasal H102 Peptide-Loaded Liposomes for Brain Delivery to Treat Alzheimer’s Disease. Pharm. Res. 2015, 32, 3837–3849. [Google Scholar] [CrossRef]
- Narayan, R.; Singh, M.; Ranjan, O.P.; Nayak, Y.; Garg, S.; Shavi, G.V.; Nayak, U.Y. Development of Risperidone Liposomes for Brain Targeting through Intranasal Route. Life Sci. 2016, 163, 38–45. [Google Scholar] [CrossRef] [PubMed]
- Wei, H.; Liu, T.; Jiang, N.; Zhou, K.; Yang, K.; Ning, W.; Yu, Y. A Novel Delivery System of Cyclovirobuxine D for Brain Targeting: Angiopep-Conjugated Polysorbate 80-Coated Liposomes via Intranasal Administration. J. Biomed. Nanotechnol. 2018, 14, 1252–1262. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, M.; Saraf, S.; Saraf, S.; Antimisiaris, S.G.; Chougule, M.B.; Shoyele, S.A.; Alexander, A. Nose-to-Brain Drug Delivery: An Update on Clinical Challenges and Progress towards Approval of Anti-Alzheimer Drugs. J. Control. Release 2018, 281, 139–177. [Google Scholar] [CrossRef]
- Dhuria, S.V.; Hanson, L.R.; Frey, W.H. Intranasal Delivery to the Central Nervous System: Mechanisms and Experimental Considerations. J. Pharm. Sci. 2010, 99, 1654–1673. [Google Scholar] [CrossRef] [PubMed]
- Owens, D.R.; Zinman, B.; Bolli, G. Alternative Routes of Insulin Delivery. Diabet. Med. 2003, 20, 886–898. [Google Scholar] [CrossRef] [PubMed]
- Chow, H.-H.S.; Chen, Z.; Matsuura, G.T. Direct Transport of Cocaine from the Nasal Cavity to the Brain Following Intranasal Cocaine Administration in Rats. J. Pharm. Sci. 1999, 88, 754–758. [Google Scholar] [CrossRef] [PubMed]
- Li, W.Q.; Sun, L.P.; Xia, Y.; Hao, S.; Cheng, G.; Wang, Z.; Wan, Y.; Zhu, C.; He, H.; Zheng, S.Y. Preoccupation of Empty Carriers Decreases Endo-/Lysosome Escape and Reduces the Protein Delivery Efficiency of Mesoporous Silica Nanoparticles. ACS Appl. Mater. Interfaces 2018, 10, 5340–5347. [Google Scholar] [CrossRef]
- Choudhury, H.; Gorain, B.; Pandey, M.; Khurana, R.K.; Kesharwani, P. Strategizing Biodegradable Polymeric Nanoparticles to Cross the Biological Barriers for Cancer Targeting. Int. J. Pharm. 2019, 565, 509–522. [Google Scholar] [CrossRef] [PubMed]
- Selby, L.I.; Cortez-Jugo, C.M.; Such, G.K.; Johnston, A.P.R. Nanoescapology: Progress toward Understanding the Endosomal Escape of Polymeric Nanoparticles. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2017, 9, e1452. [Google Scholar] [CrossRef] [PubMed]
- Degors, I.M.S.; Wang, C.; Rehman, Z.U.; Zuhorn, I.S. Carriers Break Barriers in Drug Delivery: Endocytosis and Endosomal Escape of Gene Delivery Vectors. Acc. Chem. Res. 2019, 52, 1750–1760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rehman, Z.U.; Zuhorn, I.S.; Hoekstra, D. How Cationic Lipids Transfer Nucleic Acids into Cells and across Cellular Membranes: Recent Advances. J. Control. Release 2013, 166, 46–56. [Google Scholar] [CrossRef]
- Rehman, Z.U.; Hoekstra, D.; Zuhorn, I.S. Mechanism of Polyplex- and Lipoplex-Mediated Delivery of Nucleic Acids: Real-Time Visualization of Transient Membrane Destabilization without Endosomal Lysis. ACS Nano 2013, 7, 3767–3777. [Google Scholar] [CrossRef] [PubMed]
- Song, L.Y.; Ahkong, Q.F.; Rong, Q.; Wang, Z.; Ansell, S.; Hope, M.J.; Mui, B. Characterization of the Inhibitory Effect of PEG-Lipid Conjugates on the Intracellular Delivery of Plasmid and Antisense DNA Mediated by Cationic Lipid Liposomes. Biochim. Biophys. Acta 2002, 1558, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Remaut, K.; Lucas, B.; Braeckmans, K.; Demeester, J.; De Smedt, S.C. Pegylation of Liposomes Favours the Endosomal Degradation of the Delivered Phosphodiester Oligonucleotides. J. Control. Release 2007, 117, 256–266. [Google Scholar] [CrossRef]
- Shi, F.; Wasungu, L.; Nomden, A.; Stuart, M.C.A.; Polushkin, E.; Engberts, J.B.F.N.; Hoekstra, D. Interference of Poly(Ethylene Glycol)-Lipid Analogues with Cationic-Lipid-Mediated Delivery of Oligonucleotides; Role of Lipid Exchangeability and Non-Lamellar Transitions. Biochem. J. 2002, 366, 333–341. [Google Scholar] [CrossRef] [PubMed]
- Kirpotin, D.; Hong, K.; Mullah, N.; Papahadjopoulos, D.; Zalipsky, S. Liposomes with Detachable Polymer Coating: Destabilization and Fusion of Dioleoylphosphatidylethanolamine Vesicles Triggered by Cleavage of Surface-Grafted Poly(Ethylene Glycol). FEBS Lett. 1996, 388, 115–118. [Google Scholar] [CrossRef] [Green Version]
- Kuai, R.; Yuan, W.; Qin, Y.; Chen, H.; Tang, J.; Yuan, M.; Zhang, Z.; He, Q. Efficient Delivery of Payload into Tumor Cells in a Controlled Manner by TAT and Thiolytic Cleavable PEG Co-Modified Liposomes. Mol. Pharm. 2010, 7, 1816–1826. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.S.; MacKay, J.A.; Szoka, F.C. Low-PH-Sensitive PEG-Stabilized Plasmid-Lipid Nanoparticles: Preparation and Characterization. Bioconjug. Chem. 2003, 14, 420–429. [Google Scholar] [CrossRef]
- Joshi, B.S.; de Beer, M.A.; Giepmans, B.N.G.; Zuhorn, I.S. Endocytosis Extracellular Vesicles and Releaseof Their Cargo from Endosomes. ACS Nano 2020, 14, 4444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rayatpour, A.; Javan, M. Targeting the Brain Lesions Using Peptides: A Review Focused on the Possibility of Targeted Drug Delivery to Multiple Sclerosis Lesions. Pharmacol. Res. 2021, 167, 105441. [Google Scholar] [CrossRef] [PubMed]
- Kumra, H.; Reinhardt, D.P. Fibronectin-Targeted Drug Delivery in Cancer. Adv. Drug Deliv. Rev. 2016, 97, 101–110. [Google Scholar] [CrossRef] [PubMed]
- Han, Z.; Lu, Z.R. Targeting Fibronectin for Cancer Imaging and Therapy. J. Mater. Chem. B 2017, 5, 639–654. [Google Scholar] [CrossRef] [Green Version]
- Yang, C.H.; Wang, Y.; Sims, M.; Cai, C.; Pfeffer, L.M. MicroRNA-1 Suppresses Glioblastoma in Preclinical Models by Targeting Fibronectin. Cancer Lett. 2019, 465, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Saw, P.E.; Park, J.; Jon, S.; Farokhzad, O.C. A Drug-Delivery Strategy for Overcoming Drug Resistance in Breast Cancer through Targeting of Oncofetal Fibronectin. Nanomed. Nanotechnol. Biol. Med. 2017, 13, 713–722. [Google Scholar] [CrossRef] [PubMed]
- Han, Z.; Zhou, Z.; Shi, X.; Wang, J.; Wu, X.; Sun, D.; Chen, Y.; Zhu, H.; Magi-Galluzzi, C.; Lu, Z.-R. EDB Fibronectin Specific Peptide for Prostate Cancer Targeting. Bioconjug. Chem. 2015, 26, 830–838. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Kim, D.; Jung, H.H.; Lee, I.-H.; Kim, J.I.L.; Suh, J.-Y.; Jon, S. Bio-Inspired Design and Potential Biomedical Applications of a Novel Class of High-Affinity Peptides. Angew. Chem. Int. Ed. 2012, 51, 1890–1894. [Google Scholar] [CrossRef] [PubMed]
- Saw, P.E.; Kim, S.; Lee, I.; Park, J.; Yu, M.; Lee, J.; Kim, J.-I.; Jon, S. Aptide-Conjugated Liposome Targeting Tumor-Associated Fibronectin for Glioma Therapy. J. Mater. Chem. B 2013, 1, 4723–4726. [Google Scholar] [CrossRef]
- Saw, P.E.; Park, J.; Lee, E.; Ahn, S.; Lee, J.; Kim, H.; Kim, J.; Choi, M.; Farokhzad, O.C.; Jon, S. Effect of PEG Pairing on the Efficiency of Cancer-Targeting Liposomes. Theranostics 2015, 5, 746–754. [Google Scholar] [CrossRef] [Green Version]
- Gu, G.; Hu, Q.; Feng, X.; Gao, X.; Menglin, J.; Kang, T.; Jiang, D.; Song, Q.; Chen, H.; Chen, J. PEG-PLA Nanoparticles Modified with APTEDB Peptide for Enhanced Anti-Angiogenic and Anti-Glioma Therapy. Biomaterials 2014, 35, 8215–8226. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Lee, Y.; Lee, I.H.; Kim, S.; Kim, D.; Saw, P.E.; Lee, J.; Choi, M.; Kim, Y.C.; Jon, S. Synthesis and Therapeutic Evaluation of an Aptide–Docetaxel Conjugate Targeting Tumor-Associated Fibronectin. J. Control. Release 2014, 178, 118–124. [Google Scholar] [CrossRef] [PubMed]
- Mann, A.P.; Scodeller, P.; Hussain, S.; Joo, J.; Kwon, E.; Braun, G.B.; Mölder, T.; She, Z.G.; Kotamraju, V.R.; Ranscht, B.; et al. A Peptide for Targeted, Systemic Delivery of Imaging and Therapeutic Compounds into Acute Brain Injuries. Nat. Commun. 2016, 7, 11980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Labrak, Y.; Heurtault, B.; Frisch, B.; Saulnier, P.; Lepeltier, E.; Miron, V.E.; Muccioli, G.G.; des Rieux, A. Impact of Anti-PDGFRα Antibody Surface Functionalization on LNC Uptake by Oligodendrocyte Progenitor Cells. Int. J. Pharm. 2022, 618, 121623. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Wu, H.; Wang, S.; Koito, H.; Li, J.; Ye, F.; Hoang, J.; Escobar, S.S.; Gow, A.; Arnett, H.A.; et al. The Oligodendrocyte-Specific G Protein-Coupled Receptor GPR17 Is a Cell-Intrinsic Timer of Myelination. Nat. Neurosci. 2009, 12, 1398–1406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terashima, T.; Ogawa, N.; Nakae, Y.; Sato, T.; Katagi, M.; Okano, J.; Maegawa, H.; Kojima, H. Gene Therapy for Neuropathic Pain through siRNA-IRF5 Gene Delivery with Homing Peptides to Microglia. Mol. Ther. Nucleic Acids 2018, 11, 203–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, F.; Lin, Y.A.; Kannan, S.; Kannan, R.M. Targeting Specific Cells in the Brain with Nanomedicines for CNS Therapies. J. Control. Release 2016, 240, 212–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valori, C.F.; Possenti, A.; Brambilla, L.; Rossi, D. Challenges and Opportunities of Targeting Astrocytes to Halt Neurodegenerative Disorders. Cells 2021, 10, 2019. [Google Scholar] [CrossRef]
- Ramazani, F.; Chen, W.; van Nostrum, C.F.; Storm, G.; Kiessling, F.; Lammers, T.; Hennink, W.E.; Kok, R.J. Strategies for Encapsulation of Small Hydrophilic and Amphiphilic Drugs in PLGA Microspheres: State-of-the-Art and Challenges. Int. J. Pharm. 2016, 499, 358–367. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Li, X.; Zhao, C. Strategies to Obtain Encapsulation and Controlled Release of Small Hydrophilic Molecules. Front. Bioeng. Biotechnol. 2020, 8, 437. [Google Scholar] [CrossRef] [PubMed]
- van Horssen, J.; Brink, B.P.; de Vries, H.E.; van der Valk, P.; Bø, L. The Blood-Brain Barrier in Cortical Multiple Sclerosis Lesions. J. Neuropathol. Exp. Neurol. 2007, 66, 321–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishihara, H.; Perriot, S.; Gastfriend, B.D.; Steinfort, M.; Cibien, C.; Soldati, S.; Matsuo, K.; Guimbal, S.; Mathias, A.; Palecek, S.P.; et al. Intrinsic Blood-Brain Barrier Dysfunction Contributes to Multiple Sclerosis Pathogenesis. Brain 2022, 27, awac019. [Google Scholar] [CrossRef] [PubMed]
- Mallik, S.; Samson, R.S.; Wheeler-Kingshott, C.A.M.; Miller, D.H. Imaging Outcomes for Trials of Remyelination in Multiple Sclerosis. J. Neurol. Neurosurg. Psychiatry 2014, 85, 1396–1404. [Google Scholar] [CrossRef] [Green Version]
- Ben-Shalom, I.; Karni, A.; Kolb, H. The Role of Molecular Imaging as a Marker of Remyelination and Repair in Multiple Sclerosis. Int. J. Mol. Sci. 2021, 23, 474. [Google Scholar] [CrossRef] [PubMed]
- Ranzenberger, L.R.; Snyder, T. Diffusion Tensor Imaging. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Kuhle, J.; Kropshofer, H.; Haering, D.A.; Kundu, U.; Meinert, R.; Barro, C.; Dahlke, F.; Tomic, D.; Leppert, D.; Kappos, L. Blood Neurofilament Light Chain as a Biomarker of MS Disease Activity and Treatment Response. Neurology 2019, 92, e1007–e1015. [Google Scholar] [CrossRef] [PubMed]
- De Paula Faria, D. Myelin Positron Emission Tomography (PET) Imaging in Multiple Sclerosis. Neural Regen. Res. 2020, 15, 1842. [Google Scholar] [CrossRef]
- Klistorner, A.; Graham, S.L. Role of Multifocal Visually Evoked Potential as a Biomarker of Demyelination, Spontaneous Remyelination, and Myelin Repair in Multiple Sclerosis. Front. Neurosci. 2021, 15, 1410. [Google Scholar] [CrossRef] [PubMed]
- Muthupillai, R.; Lomas, D.J.; Rossman, P.J.; Greenleaf, J.F.; Manduca, A.; Ehman, R.L. Magnetic Resonance Elastography by Direct Visualization of Propagating Acoustic Strain Waves. Science 1995, 269, 1854–1857. [Google Scholar] [CrossRef]
- Hirsch, S.; Braun, J.; Sack, I. Magnetic Resonance Elastography-Physical Background and Medical Applications. Magn. Reson. Elastography 2017, 14, 263–281. [Google Scholar] [CrossRef]
- Wuerfel, J.; Paul, F.; Beierbach, B.; Hamhaber, U.; Klatt, D.; Papazoglou, S.; Zipp, F.; Martus, P.; Braun, J.; Sack, I. MR-Elastography Reveals Degradation of Tissue Integrity in Multiple Sclerosis. Neuroimage 2010, 49, 2520–2525. [Google Scholar] [CrossRef] [PubMed]
- Streitberger, K.J.; Sack, I.; Krefting, D.; Pfüller, C.; Braun, J.; Paul, F.; Wuerfel, J. Brain Viscoelasticity Alteration in Chronic-Progressive Multiple Sclerosis. PLoS ONE 2012, 7, e29888. [Google Scholar] [CrossRef] [Green Version]
- Schregel, K.; Née Tysiak, E.W.; Garteiser, P.; Gemeinhardt, I.; Prozorovski, T.; Aktas, O.; Merz, H.; Petersen, D.; Wuerfel, J.; Sinkus, R. Demyelination Reduces Brain Prenchymal Stiffness Quantified in Vivo by Magnetic Resonance Elastography. Proc. Natl. Acad. Sci. USA 2012, 109, 6650–6655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riek, K.; Millward, J.M.; Hamann, I.; Mueller, S.; Pfueller, C.F.; Paul, F.; Braun, J.; Infante-Duarte, C.; Sack, I. Magnetic Resonance Elastography Reveals Altered Brain Viscoelasticity in Experimental Autoimmune Encephalomyelitis. NeuroImage. Clin. 2012, 1, 81–90. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Millward, J.M.; Hanke-Vela, L.; Malla, B.; Pilch, K.; Gil-Infante, A.; Waiczies, S.; Mueller, S.; Boehm-Sturm, P.; Guo, J.; et al. MR Elastography-Based Assessment of Matrix Remodeling at Lesion Sites Associated with Clinical Severity in a Model of Multiple Sclerosis. Front. Neurol. 2019, 10, 1382. [Google Scholar] [CrossRef] [PubMed]
- Herthum, H.; Hetzer, S.; Scheel, M.; Shahryari, M.; Braun, J.; Paul, F.; Sack, I. In Vivo Stiffness of Multiple Sclerosis Lesions Is Similar to That of Normal-Appearing White Matter. Acta Biomater. 2022, 138, 410–421. [Google Scholar] [CrossRef] [PubMed]

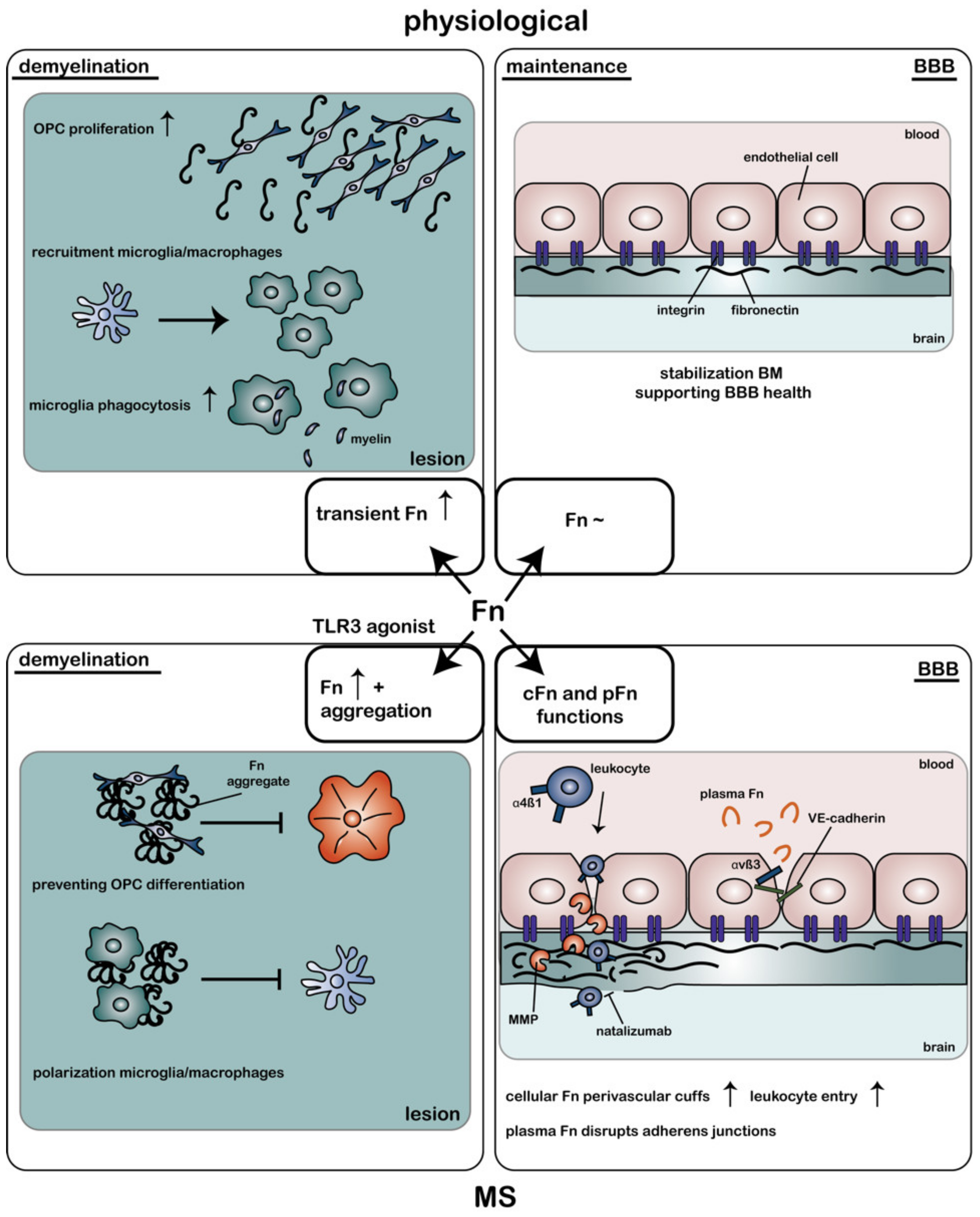
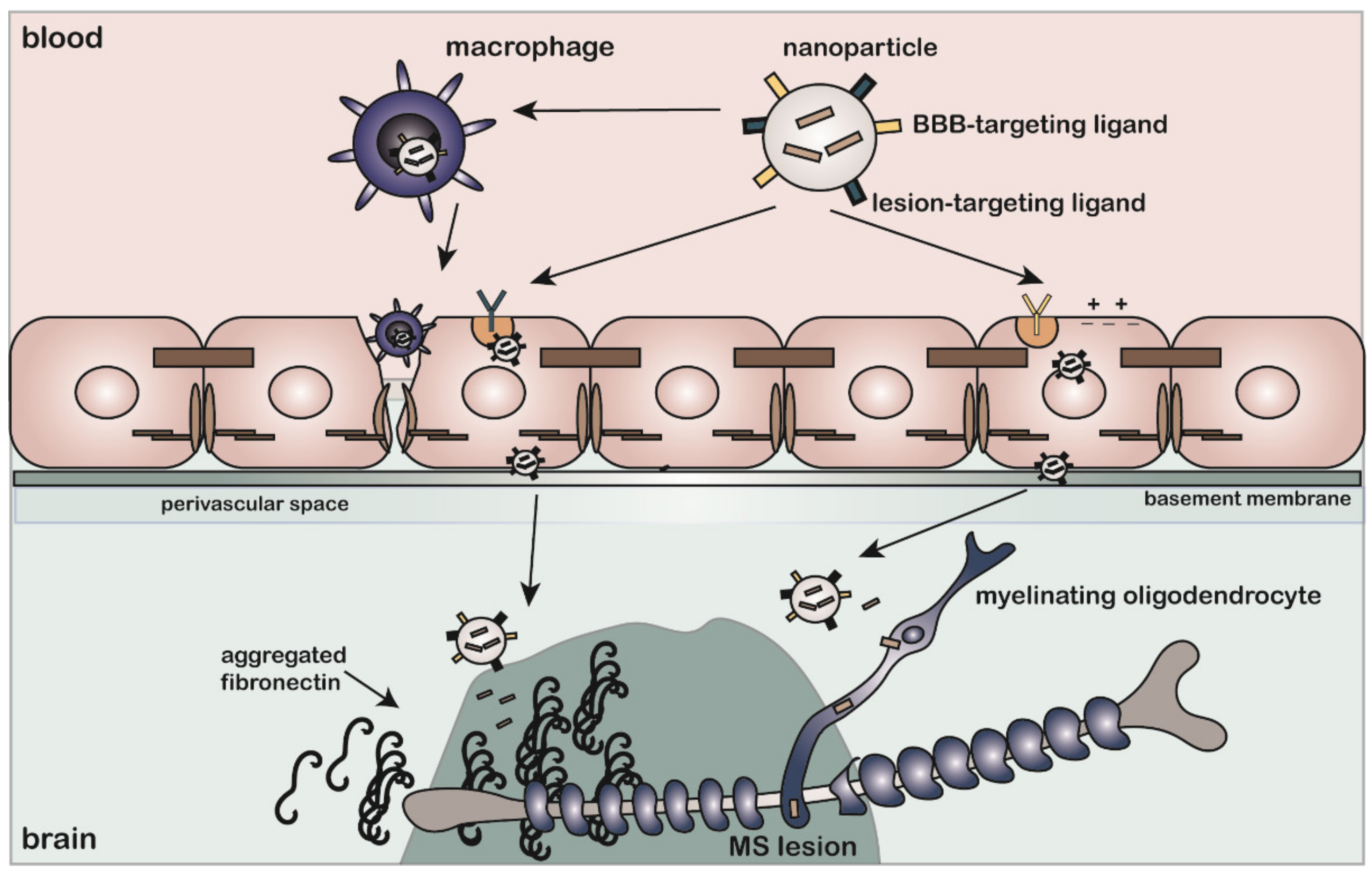
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
van Schaik, P.E.M.; Zuhorn, I.S.; Baron, W. Targeting Fibronectin to Overcome Remyelination Failure in Multiple Sclerosis: The Need for Brain- and Lesion-Targeted Drug Delivery. Int. J. Mol. Sci. 2022, 23, 8418. https://doi.org/10.3390/ijms23158418
van Schaik PEM, Zuhorn IS, Baron W. Targeting Fibronectin to Overcome Remyelination Failure in Multiple Sclerosis: The Need for Brain- and Lesion-Targeted Drug Delivery. International Journal of Molecular Sciences. 2022; 23(15):8418. https://doi.org/10.3390/ijms23158418
Chicago/Turabian Stylevan Schaik, Pauline E. M., Inge S. Zuhorn, and Wia Baron. 2022. "Targeting Fibronectin to Overcome Remyelination Failure in Multiple Sclerosis: The Need for Brain- and Lesion-Targeted Drug Delivery" International Journal of Molecular Sciences 23, no. 15: 8418. https://doi.org/10.3390/ijms23158418
APA Stylevan Schaik, P. E. M., Zuhorn, I. S., & Baron, W. (2022). Targeting Fibronectin to Overcome Remyelination Failure in Multiple Sclerosis: The Need for Brain- and Lesion-Targeted Drug Delivery. International Journal of Molecular Sciences, 23(15), 8418. https://doi.org/10.3390/ijms23158418