
ILB®, a Low Molecular Weight Dextran Sulphate, Restores Glutamate Homeostasis, Amino Acid Metabolism and Neurocognitive Functions in a Rat Model of Severe Traumatic Brain Injury

 ,
,  ,
,  , ,
, ,  ,
,  ,
,  and
and
Abstract
:1. Introduction
2. Results
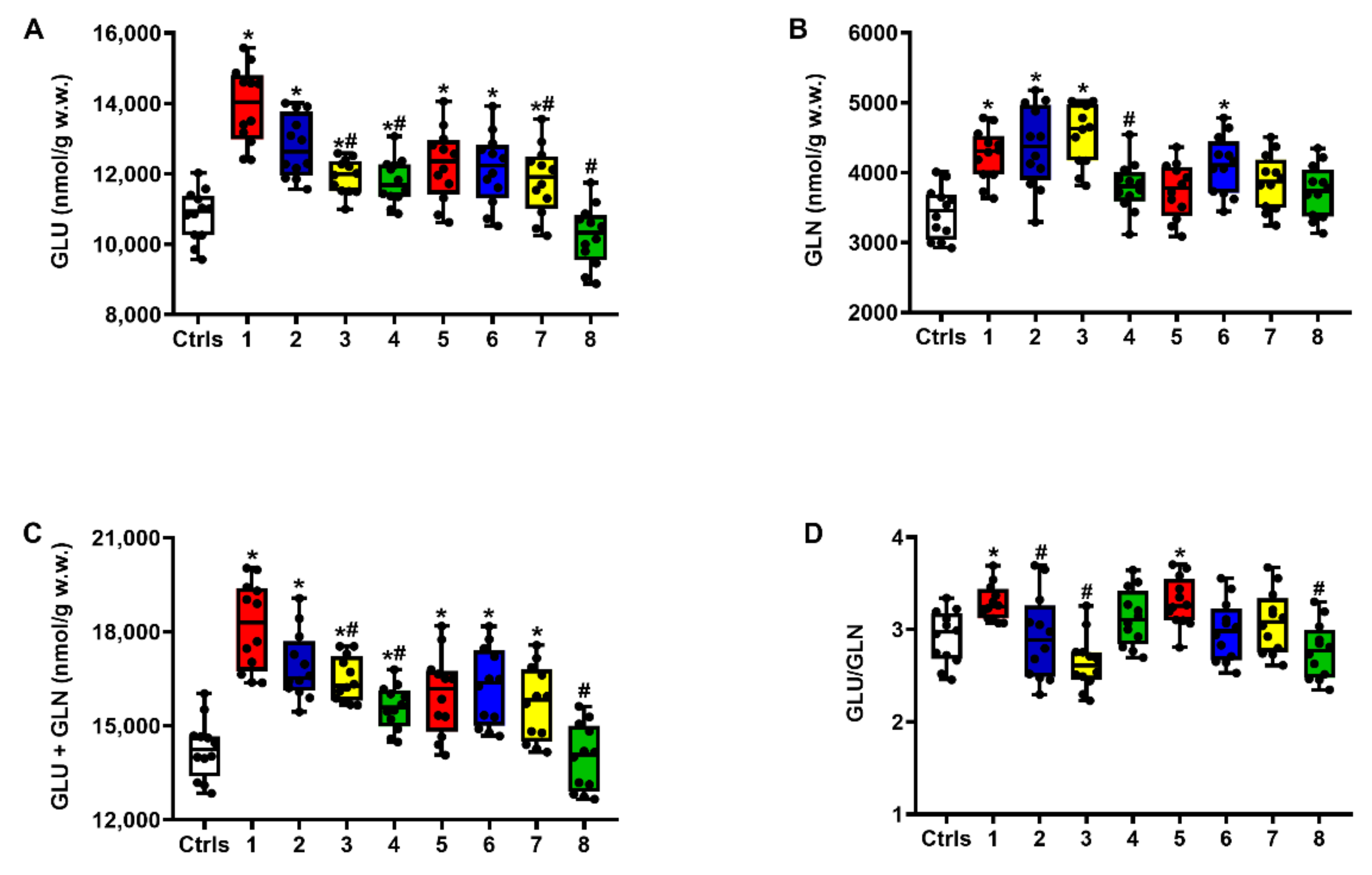
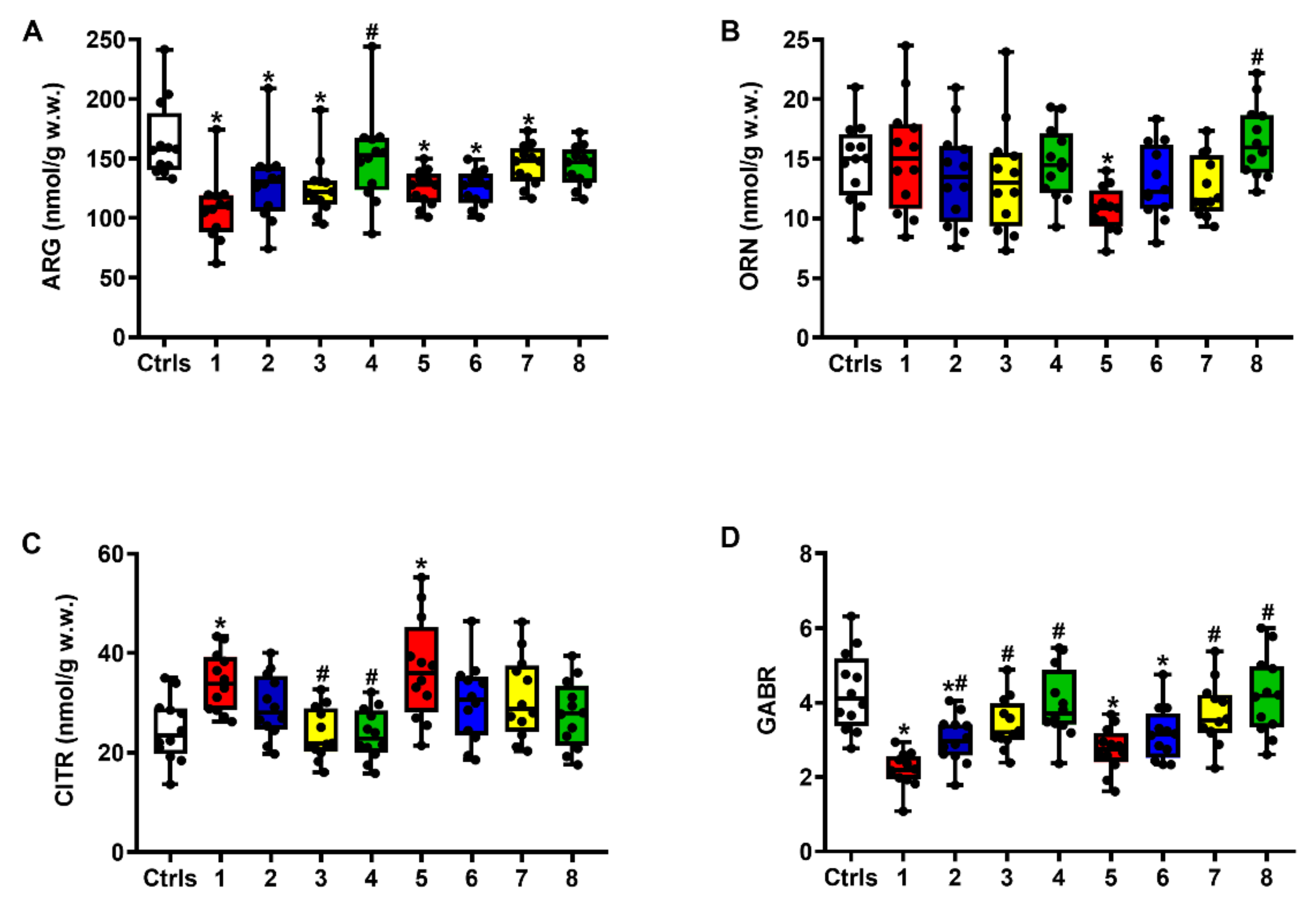
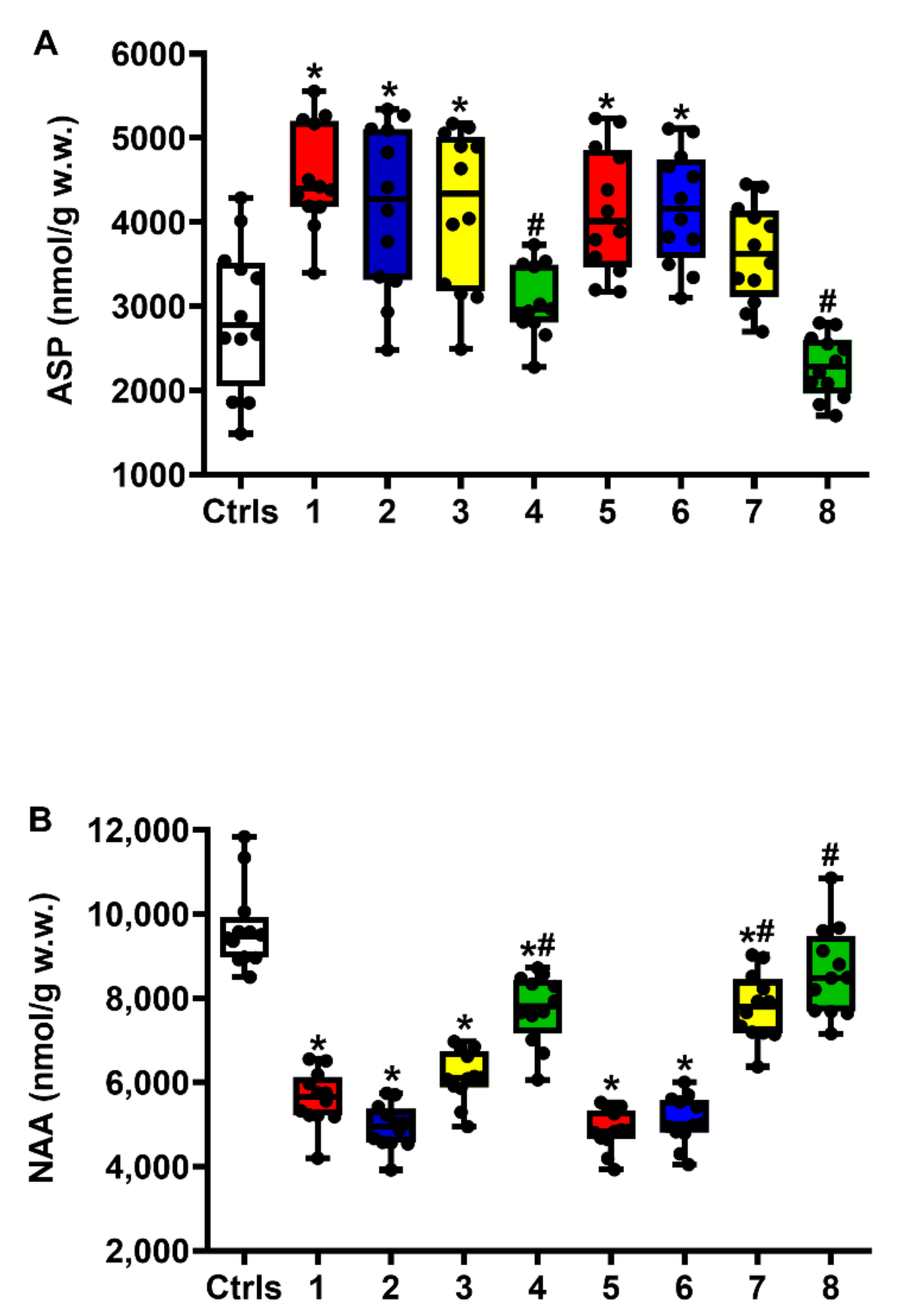
2.1. Effect of ILB® on Glutamate Excitotoxicity of the Post-sTBI Brain
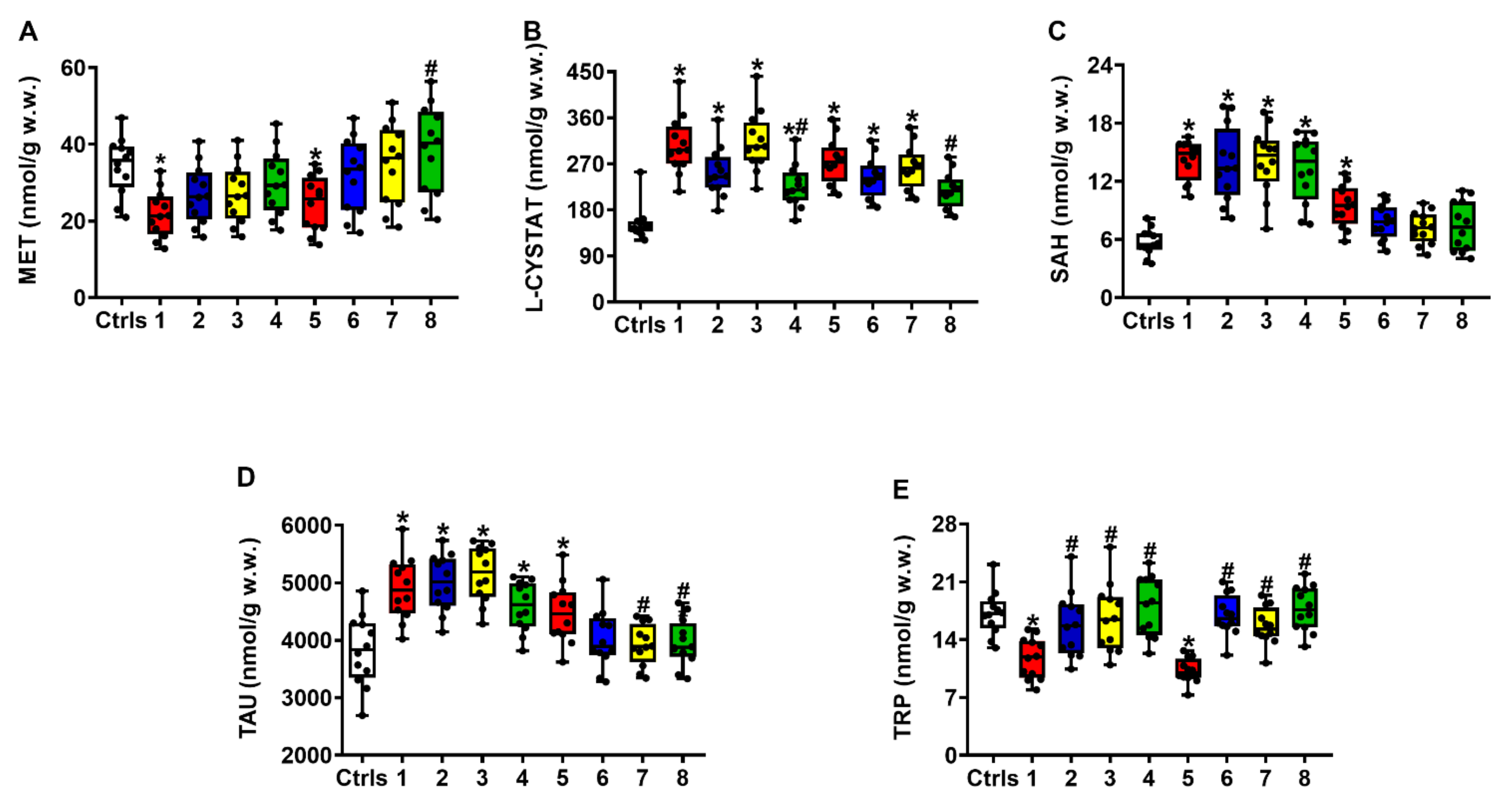
2.2. Effect of ILB® on Nitrosative Stress and Metabolism of Met, Tau and Trp
2.3. Effect of ILB® on NAA Metabolism
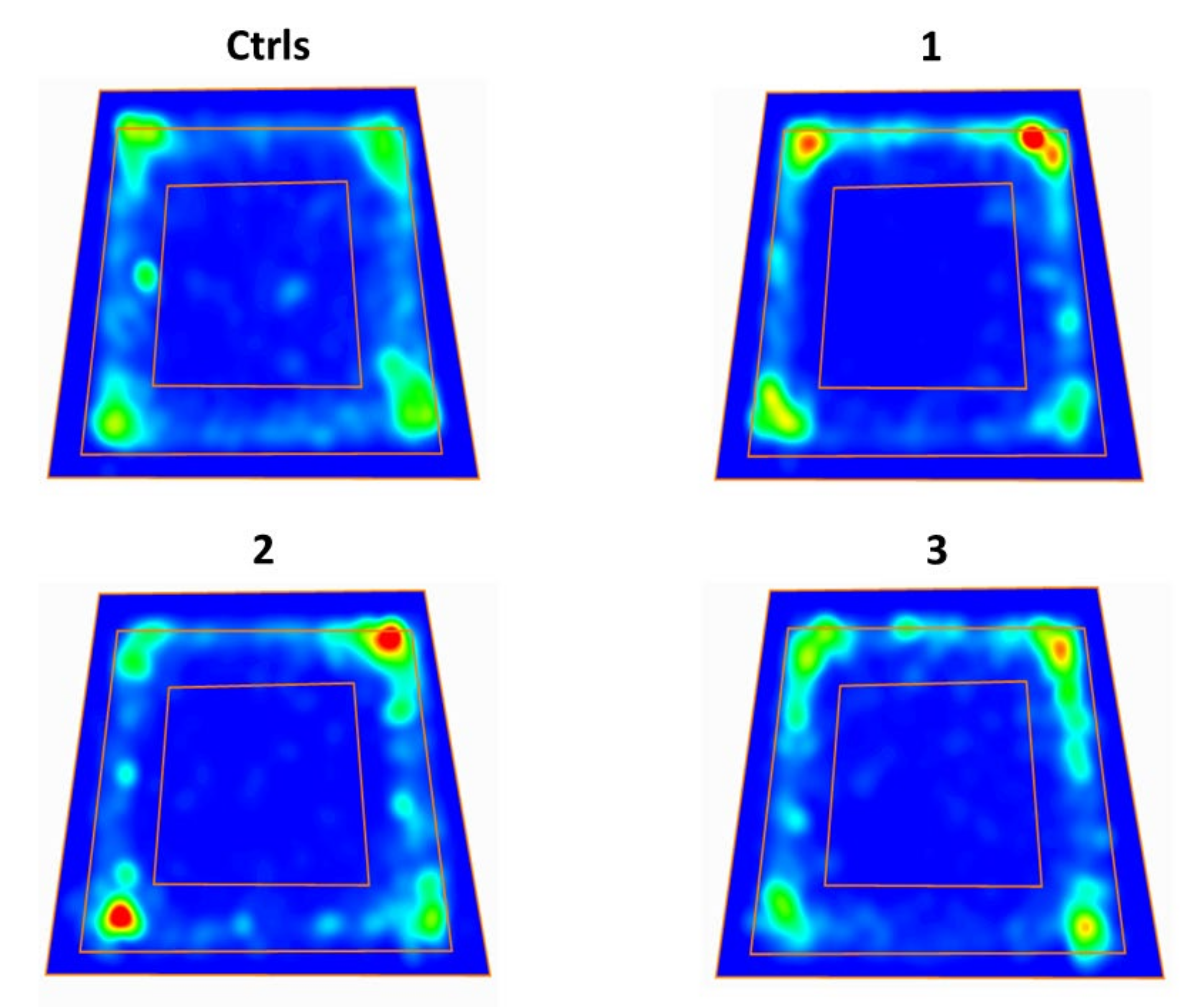
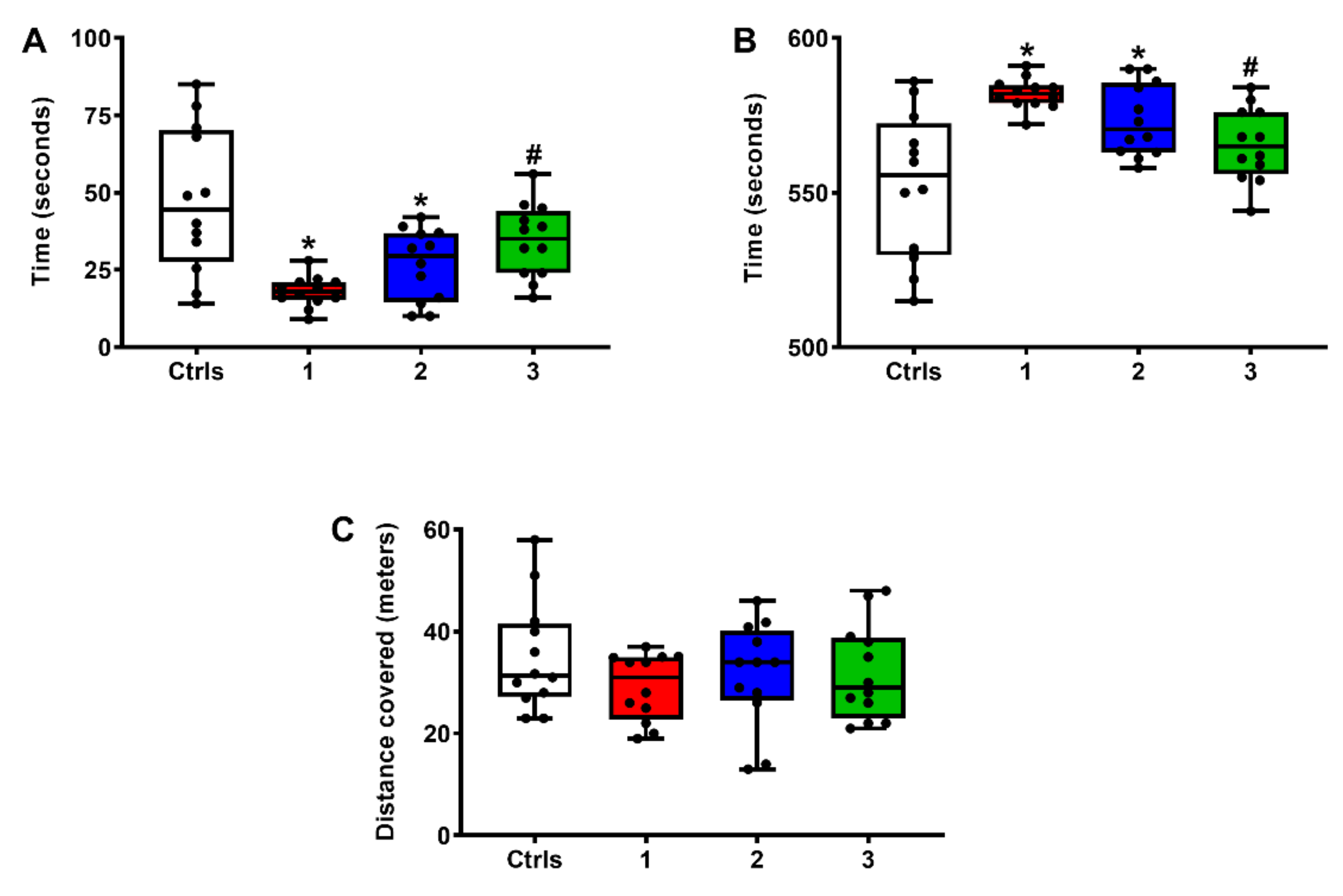
2.4. Effect of ILB® on Neurocognitive Functions of sTBI Rats: The OFT Test for Anxiety
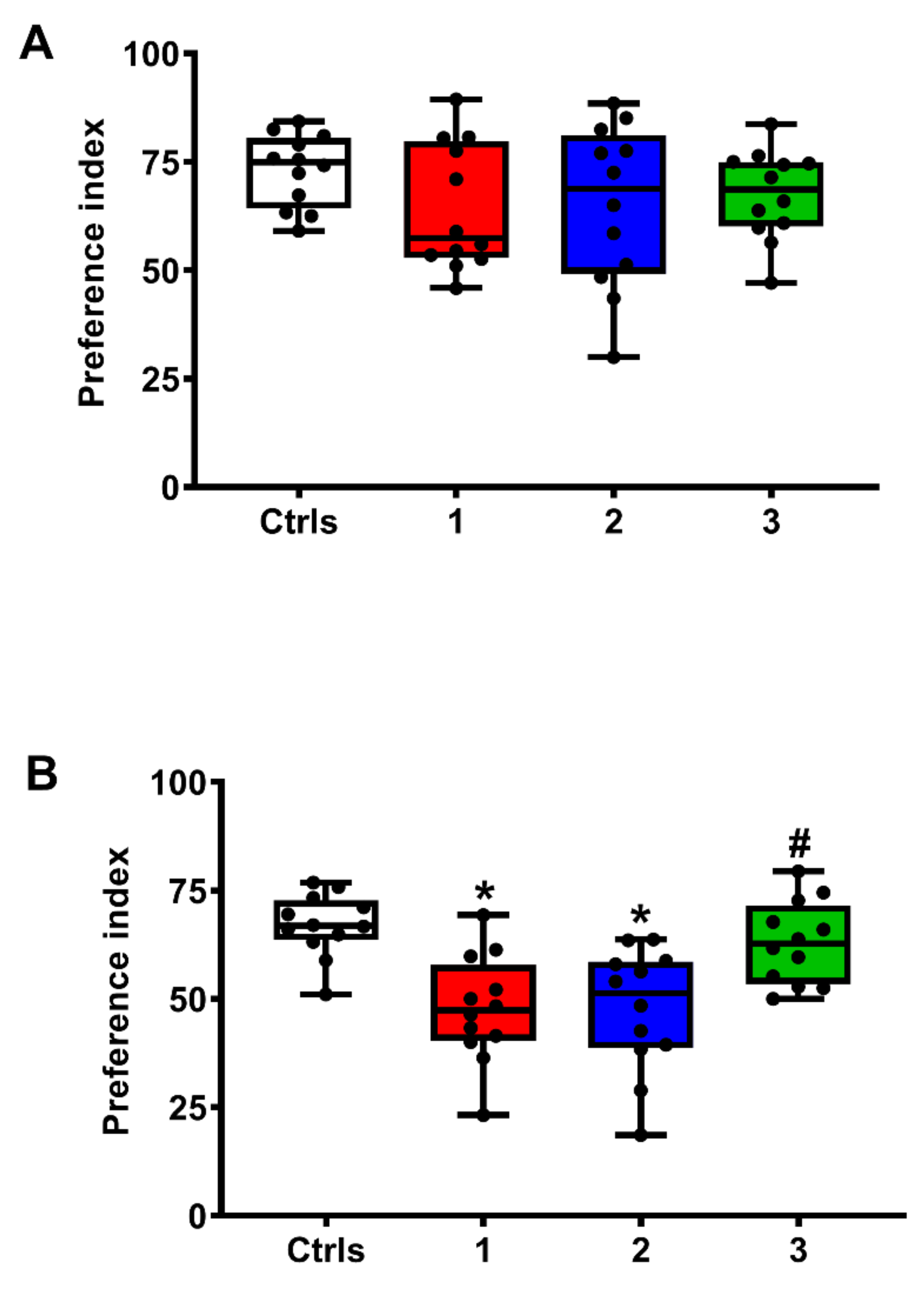
2.5. Effect of ILB® on Neurocognitive Functions of sTBI Rats: The NOR-STM and NOR-LTM Tests
2.6. Effect of ILB® on Selected Brain Metabolites Measured in the Neurocognitive Study Rats
3. Discussion
4. Materials and Methods
4.1. Animals, TBI Induction and Drug Administration Protocol
4.2. Tissue Preparation and HPLC Analysis of Free Amino Acids, Energy Metabolites and Antioxidants
4.3. Assessment of Neurocognitive Functions
4.4. Statistics
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Center for Disease Control and Prevention. Traumatic Brain Injury; CDC Injury Center; Centers for Disease Control and Prevention: Atlanta, GA, USA, 2017. Available online: https://www.cdc.gov/traumaticbraininjury/index.html (accessed on 14 April 2022).
- Dienel, G.A. Brain Glucose Metabolism: Integration of Energetics with Function. Physiol Rev. 2019, 99, 949–1045. [Google Scholar] [CrossRef] [PubMed]
- Di Pietro, V.; Yakoub, K.M.; Caruso, G.; Lazzarino, G.; Signoretti, S.; Barbey, A.K.; Tavazzi, B.; Lazzarino, G.; Belli, A.; Amorini, A.M. Antioxidant Therapies in Traumatic Brain Injury. Antioxidants 2020, 9, 260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, P.; Xiao, B.; Wang, L.P.; Li, Y.S.; Jin, H.; Jin, Q.H. Nitric oxide impairs spatial learning and memory in a rat model of Alzheimer’s disease via disturbance of glutamate response in the hippocampal dentate gyrus during spatial learning. Behav. Brain Res. 2022, 422, 113750. [Google Scholar] [CrossRef] [PubMed]
- Schousboe, A. Metabolic signaling in the brain and the role of astrocytes in control of glutamate and GABA neurotransmission. Neurosci. Lett. 2019, 689, 11–13. [Google Scholar] [CrossRef] [PubMed]
- Verma, M.; Lizama, B.N.; Chu, C.T. Excitotoxicity, calcium and mitochondria: A triad in synaptic neurodegeneration. Transl. Neurodegener. 2022, 11, 3. [Google Scholar] [CrossRef]
- Liao, R.; Wood, T.R.; Nance, E. Nanotherapeutic modulation of excitotoxicity and oxidative stress in acute brain injury. Nanobiomedicine 2020, 7, 1–18. [Google Scholar] [CrossRef]
- Meunier, C.N.; Dallérac, G.; Le Roux, N.; Sacchi, S.; Levasseur, G.; Amar, M.; Pollegioni, L.; Mothet, J.P.; Fossier, P. D-serine and glycine differentially control neurotransmission during visual cortex critical period. PLoS ONE 2016, 11, e0151233. [Google Scholar] [CrossRef] [Green Version]
- Sekine, A.; Okamoto, M.; Kanatani, Y.; Sano, M.; Shibata, K.; Fukuwatari, T. Amino acids inhibit kynurenic acid formation via suppression of kynurenine uptake or kynurenic acid synthesis in rat brain in vitro. Springerplus 2015, 4, 48. [Google Scholar] [CrossRef] [Green Version]
- Shnitko, T.A.; Taylor, S.C.; Stringfield, S.J.; Zandy, S.L.; Cofresí, R.U.; Doherty, J.M.; Lynch, W.B.; Boettiger, C.A.; Gonzales, R.A.; Robinson, D.L. Acute phenylalanine/tyrosine depletion of phasic dopamine in the rat brain. Psychopharmacology 2016, 233, 2045–2054. [Google Scholar] [CrossRef] [Green Version]
- De Simone, R.; Vissicchio, F.; Mingarelli, C.; De Nuccio, C.; Visentin, S.; Ajmone-Cat, M.A.; Minghetti, L. Branched-chain amino acids influence the immune properties of microglial cells and their responsiveness to pro-inflammatory signals. Biochim. Biophys. Acta 2013, 1832, 650–659. [Google Scholar] [CrossRef] [Green Version]
- Singhal, N.K.; Li, S.; Arning, E.; Alkhayer, K.; Clements, R.; Sarcyk, Z.; Dassanayake, R.S.; Brasch, N.E.; Freeman, E.J.; Bottiglieri, T.; et al. Changes in methionine metabolism and histone h3 trimethylation are linked to mitochondrial defects in multiple sclerosis. J. Neurosci 2015, 35, 15170–15186. [Google Scholar] [CrossRef] [Green Version]
- Rae, C.D.; Williams, S.R. Glutathione in the human brain: Review of its roles and measurement by magnetic resonance spectroscopy. Anal. Biochem 2017, 529, 127–143. [Google Scholar] [CrossRef]
- Cao, Y.; Gao, Y.; Xu, S.; Bao, J.; Lin, Y.; Luo, X.; Wang, Y.; Luo, Q.; Jiang, J.; Neale, J.H.; et al. Glutamate carboxypeptidase II gene knockout attenuates oxidative stress and cortical apoptosis after traumatic brain injury. BMC Neurosci. 2016, 17, 15. [Google Scholar] [CrossRef] [Green Version]
- Baslow, M.H. Evidence that the tri-cellular metabolism of N-acetylaspartate functions as the brain’s “operating system”: How NAA metabolism supports meaningful intercellular frequency-encoded communications. Amino Acids 2010, 39, 1139–1145. [Google Scholar] [CrossRef]
- Amorini, A.M.; Lazzarino, G.; Di Pietro, V.; Signoretti, S.; Lazzarino, G.; Belli, A.; Tavazzi, B. Metabolic, enzymatic and gene involvement in cerebral glucose dysmetabolism after traumatic brain injury. Biochim. Biophys. Acta 2016, 1862, 679–687. [Google Scholar] [CrossRef]
- Tavazzi, B.; Signoretti, S.; Lazzarino, G.; Amorini, A.M.; Delfini, R.; Cimatti, M.; Marmarou, A.; Vagnozzi, R. Cerebral oxidative stress and depression of energy metabolism correlate with severity of diffuse brain injury in rats. Neurosurgery 2005, 56, 582–589. [Google Scholar] [CrossRef]
- Di Pietro, V.; Lazzarino, G.; Amorini, A.M.; Signoretti, S.; Hill, L.J.; Porto, E.; Tavazzi, B.; Lazzarino, G.; Belli, A. Fusion or fission: The destiny of mitochondria in traumatic brain injury of different severities. Sci. Rep. 2017, 7, 9189. [Google Scholar] [CrossRef]
- Vagnozzi, R.; Tavazzi, B.; Signoretti, S.; Amorini, A.M.; Belli, A.; Cimatti, M.; Delfini, R.; Di Pietro, V.; Finocchiaro, A.; Lazzarino, G. Temporal window of metabolic brain vulnerability to concussions: Mitochondrial-related impairment--part I. Neurosurgery 2007, 61, 379–388. [Google Scholar] [CrossRef] [Green Version]
- Tavazzi, B.; Vagnozzi, R.; Signoretti, S.; Amorini, A.M.; Belli, A.; Cimatti, M.; Delfini, R.; Di Pietro, V.; Finocchiaro, A.; Lazzarino, G. Temporal window of metabolic brain vulnerability to concussions: Oxidative and nitrosativestresses--part II. Neurosurgery 2007, 61, 390–395. [Google Scholar] [CrossRef] [Green Version]
- Di Pietro, V.; Lazzarino, G.; Amorini, A.M.; Tavazzi, B.; D’Urso, S.; Longo, S.; Vagnozzi, R.; Signoretti, S.; Clementi, E.; Giardina, B.; et al. Neuroglobin expression and oxidant/antioxidant balance after graded traumatic brain injury in the rat. Free Radic Biol. Med. 2014, 69, 258–264. [Google Scholar] [CrossRef]
- Di Pietro, V.; Amorini, A.M.; Tavazzi, B.; Vagnozzi, R.; Logan, A.; Lazzarino, G.; Signoretti, S.; Lazzarino, G.; Belli, A. The molecular mechanisms affecting N-acetylaspartate homeostasis following experimental graded traumatic brain injury. Mol. Med. 2014, 20, 147–157. [Google Scholar] [CrossRef] [PubMed]
- Amorini, A.M.; Lazzarino, G.; Di Pietro, V.; Signoretti, S.; Lazzarino, G.; Belli, A.; Tavazzi, B. Severity of experimental traumatic brain injury modulates changes in concentrations of cerebral free amino acids. J. Cell. Mol. Med. 2017, 21, 530–542. [Google Scholar] [CrossRef] [PubMed]
- Lazzarino, G.; Amorini, A.M.; Barnes, N.M.; Bruce, L.; Mordente, A.; Lazzarino, G.; Di Pietro, V.; Tavazzi, B.; Belli, A.; Logan, A. Low Molecular Weight Dextran Sulfate (ILB®) Administration Restores Brain Energy Metabolism Following Severe Traumatic Brain Injury in the Rat. Antioxidants 2020, 9, 850. [Google Scholar] [CrossRef] [PubMed]
- Ali-Sisto, T.; Tolmunen, T.; Viinamäki, H.; Mäntyselkä, P.; Valkonen-Korhonen, M.; Koivumaa-Honkanen, H.; Honkalampi, K.; Ruusunen, A.; Nandania, J.; Velagapudi, V.; et al. Global arginine bioavailability ratio is decreased in patients with major depressive disorder. J. Affect. Disord. 2018, 229, 145–151. [Google Scholar] [CrossRef] [Green Version]
- Bersani, F.S.; Wolkowitz, O.M.; Lindqvist, D.; Yehuda, R.; Flory, J.; Bierer, L.M.; Makotine, I.; Abu-Amara, D.; Coy, M.; Reus, V.I.; et al. Global arginine bioavailability, a marker of nitric oxide synthetic capacity, is decreased in PTSD and correlated with symptom severity and markers of inflammation. Brain Behav. Immun. 2016, 52, 153–160. [Google Scholar] [CrossRef] [Green Version]
- Andersen, J.V.; Markussen, K.H.; Jakobsen, E.; Schousboe, A.; Waagepetersen, H.S.; Rosenberg, P.A.; Aldana, B.I. Glutamate metabolism and recycling at the excitatory synapse in health and neurodegeneration. Neuropharmacology 2021, 196, 108719. [Google Scholar] [CrossRef]
- Luo, P.; Li, X.; Wu, X.; Dai, S.; Yang, Y.; Xu, H.; Jing, D.; Rao, W.; Xu, H.; Gao, X.; et al. Preso regulates NMDA receptor-mediated excitotoxicity via modulating nitric oxide and calcium responses after traumatic brain injury. Cell Death Dis. 2019, 10, 496. [Google Scholar] [CrossRef]
- Tehse, J.; Taghibiglou, C. The overlooked aspect of excitotoxicity: Glutamate-independent excitotoxicity in traumatic brain injuries. Eur. J. Neurosci. 2019, 49, 1157–1170. [Google Scholar] [CrossRef]
- Singh, K.; Trivedi, R.; Verma, A.; D’souza, M.M.; Koundal, S.; Rana, P.; Baishya, B.; Khushu, S. Altered metabolites of the rat hippocampus after mild and moderate traumatic brain injury—A combined in vivo and in vitro 1H-MRS study. NMR Biomed. 2017, 30, e3764. [Google Scholar] [CrossRef]
- Kumar Sahel, D.; Kaira, M.; Raj, K.; Sharma, S.; Singh, S. Mitochondrial dysfunctioning and neuroinflammation: Recent highlights on the possible mechanisms involved in Traumatic Brain Injury. Neurosci. Lett. 2019, 710, 134347. [Google Scholar] [CrossRef]
- Yonutas, H.M.; Hubbard, W.B.; Pandya, J.D.; Vekaria, H.J.; Geldenhuys, W.J.; Sullivan, P.G. Bioenergetic restoration and neuroprotection after therapeutic targeting of mitoNEET: New mechanism of pioglitazone following traumatic brain injury. Exp. Neurol. 2020, 327, 113243. [Google Scholar] [CrossRef]
- Sowers, J.L.; Sowers, M.L.; Shavkunov, A.S.; Hawkins, B.E.; Wu, P.; DeWitt, D.S.; Prough, D.S.; Zhang, K. Traumatic brain injury induces region-specific glutamate metabolism changes as measured by multiple mass spectrometry methods. iScience 2021, 24, 103108. [Google Scholar] [CrossRef]
- Lazzarino, G.; Mangione, R.; Belli, A.; Di Pietro, V.; Nagy, Z.; Barnes, N.M.; Bruce, L.; Ropero, B.M.; Persson, L.I.; Manca, B.; et al. ILB® Attenuates Clinical Symptoms and Serum Biomarkers of Oxidative/Nitrosative Stress and Mitochondrial Dysfunction in Patients with Amyotrophic Lateral Sclerosis. J. Pers. Med. 2021, 11, 794. [Google Scholar] [CrossRef]
- Fustin, J.M.; Ye, S.; Rakers, C.; Kaneko, K.; Fukumoto, K.; Yamano, M.; Versteven, M.; Grünewald, E.; Cargill, S.J.; Tamai, T.K.; et al. Methylation deficiency disrupts biological rhythms from bacteria to humans. Commun. Biol. 2020, 3, 211. [Google Scholar] [CrossRef]
- Balasubramanian, N.; Jadhav, G.; Sakharkar, A.J. Repeated mild traumatic brain injuries perturb the mitochondrial biogenesis via DNA methylation in the hippocampus of rat. Mitochondrion 2021, 61, 11–24. [Google Scholar] [CrossRef]
- Arun, P.; Rittase, W.B.; Wilder, D.M.; Wang, Y.; Gist, I.D.; Long, J.B. Defective methionine metabolism in the brain after repeated blast exposures might contribute to increased oxidative stress. Neurochem. Int. 2018, 112, 234–238. [Google Scholar] [CrossRef]
- Dash, P.K.; Hergenroeder, G.W.; Jeter, C.B.; Choi, H.A.; Kobori, N.; Moore, A.N. Traumatic Brain Injury Alters Methionine Metabolism: Implications for Pathophysiology. Front. Syst. Neurosci. 2016, 10, 36. [Google Scholar] [CrossRef] [Green Version]
- Pushchina, E.V.; Zharikova, E.I.; Varaksin, A.A. Mechanical Brain Injury Increases Cells’ Production of Cystathionine β-Synthase and Glutamine Synthetase, but Reduces Pax2 Expression in the Telencephalon of Juvenile Chum Salmon, Oncorhynchus keta. Int. J. Mol. Sci. 2021, 22, 1279. [Google Scholar] [CrossRef]
- Ignowski, E.; Winter, A.N.; Duval, N.; Fleming, H.; Wallace, T.; Manning, E.; Koza, L.; Huber, K.; Serkova, N.J.; Linseman, D.A. The cysteine-rich whey protein supplement, Immunocal®, preserves brain glutathione and improves cognitive, motor, and histopathological indices of traumatic brain injury in a mouse model of controlled cortical impact. Free Radic Biol. Med. 2018, 124, 328–341. [Google Scholar] [CrossRef]
- Signoretti, S.; Marmarou, A.; Tavazzi, B.; Lazzarino, G.; Beaumont, A.; Vagnozzi, R. N-Acetylaspartate reduction as a measure of injury severity and mitochondrial dysfunction following diffuse traumatic brain injury. J. Neurotrauma 2001, 18, 977–991. [Google Scholar] [CrossRef]
- Sinson, G.; Bagley, L.J.; Cecil, K.M.; Torchia, M.; McGowan, J.C.; Lenkinski, R.E.; McIntosh, T.K.; Grossman, R.I. Magnetization transfer imaging and proton MR spectroscopy in the evaluation of axonal injury: Correlation with clinical outcome after traumatic brain injury. AJNR Am. J. Neuroradiol. 2001, 22, 143–151. [Google Scholar] [PubMed]
- Signoretti, S.; Marmarou, A.; Aygok, G.A.; Fatouros, P.P.; Portella, G.; Bullock, R.M. Assessment of mitochondrial impairment in traumatic brain injury using high-resolution proton magnetic resonance spectroscopy. J. Neurosurg. 2008, 108, 42–52. [Google Scholar] [CrossRef] [PubMed]
- Babikian, T.; Alger, J.R.; Ellis-Blied, M.U.; Giza, C.C.; Dennis, E.; Olsen, A.; Mink, R.; Babbitt, C.; Johnson, J.; Thompson, P.M.; et al. Whole Brain Magnetic Resonance Spectroscopic Determinants of Functional Outcomes in Pediatric Moderate/Severe Traumatic Brain Injury. J. Neurotrauma 2018, 35, 1637–1645. [Google Scholar] [CrossRef] [PubMed]
- Yang, A.; Xiao, X.H.; Liu, Z.H.; Wan, Z.L.; Wang, Z.Y. A Multimodal Magnetic Resonance Imaging Study of Recovery of Consciousness in Severe Traumatic Brain Injury: Preliminary Results. J. Neurotrauma 2018, 35, 308–313. [Google Scholar] [CrossRef]
- Lin, J.C.; Mueller, C.; Campbell, K.A.; Thannickal, H.H.; Daredia, A.F.; Sheriff, S.; Maudsley, A.A.; Brunner, R.C.; Younger, J.W. Investigating whole-brain metabolite abnormalities in the chronic stages of moderate or severe traumatic brain injury. PM R 2022, 14, 472–485. [Google Scholar] [CrossRef]
- Babikian, T.; Freier, M.C.; Ashwal, S.; Riggs, M.L.; Burley, T.; Holshouser, B.A. MR spectroscopy: Predicting long-term neuropsychological outcome following pediatric TBI. J. Magn. Reason. Imaging 2006, 24, 801–811. [Google Scholar] [CrossRef]
- Babikian, T.; Marion, S.D.; Copeland, S.; Alger, J.R.; O’Neill, J.; Cazalis, F.; Mink, R.; Giza, C.C.; Vu, J.A.; Hilleary, S.M.; et al. Metabolic levels in the corpus callosum and their structural and behavioral correlates after moderate to severe pediatric TBI. J. Neurotrauma 2010, 27, 473–481. [Google Scholar] [CrossRef] [Green Version]
- George, E.O.; Roys, S.; Sours, C.; Rosenberg, J.; Zhuo, J.; Shanmuganathan, K.; Gullapalli, R.P. Longitudinal and prognostic evaluation of mild traumatic brain injury: A 1H-magnetic resonance spectroscopy study. J. Neurotrauma 2014, 31, 1018–1028. [Google Scholar] [CrossRef]
- Sikoglu, E.M.; Liso Navarro, A.A.; Czerniak, S.M.; McCafferty, J.; Eisenstock, J.; Stevenson, J.H.; King, J.A.; Moore, C.M. Effects of Recent Concussion on Brain Bioenergetics: A Phosphorus-31 Magnetic Resonance Spectroscopy Study. Cogn. Behav. Neurol. 2015, 28, 181–187. [Google Scholar] [CrossRef]
- Rozas, N.S.; Redell, J.B.; Hill, J.L.; McKenna, J., 3rd; Moore, A.N.; Gambello, M.J.; Dash, P.K. Genetic activation of mTORC1 signaling worsens neurocognitive outcome after traumatic brain injury. J. Neurotrauma 2015, 32, 149–158. [Google Scholar] [CrossRef] [Green Version]
- Xing, G.; Ren, M.; Verma, A. Divergent Induction of Branched-Chain Aminotransferases and Phosphorylation of Branched Chain Keto-Acid Dehydrogenase Is a Potential Mechanism Coupling Branched-Chain Keto-Acid-Mediated-Astrocyte Activation to Branched-Chain Amino Acid Depletion-Mediated Cognitive Deficit after Traumatic Brain Injury. J. Neurotrauma 2018, 35, 2482–2494. [Google Scholar]
- Josephine, A.; Veena, C.K.; Amudha, G.; Preetha, S.P.; Varalakshmi, P. Evaluating the effect of sulphated polysaccharides on cyclosporine a induced oxidative renal injury. Mol. Cell Biochem. 2006, 287, 101–108. [Google Scholar] [CrossRef]
- Chen, L.; Huang, G. Antioxidant activities of sulfated pumpkin polysaccharides. Int. J. Biol. Macromol. 2019, 126, 743–746. [Google Scholar] [CrossRef]
- Presa, F.B.; Marques, M.L.M.; Viana, R.L.S.; Nobre, L.T.D.B.; Costa, L.S.; Rocha, H.A.O. The Protective Role of Sulfated Polysaccharides from Green Seaweed Udotea flabellum in Cells Exposed to Oxidative Damage. Mar. Drugs 2018, 16, 135. [Google Scholar] [CrossRef] [Green Version]
- Ma, X.T.; Sun, X.Y.; Yu, K.; Gui, B.S.; Gui, Q.; Ouyang, J.M. Effect of Content of Sulfate Groups in Seaweed Polysaccharides on Antioxidant Activity and Repair Effect of Subcellular Organelles in Injured HK-2 Cells. Oxid. Med. Cell Longev. 2017, 2017, 2542950. [Google Scholar] [CrossRef]
- Logan, A.; Nagy, Z.; Barnes, N.M.; Belli, A.; Di Pietro, V.; Tavazzi, B.; Lazzarino, G.; Lazzarino, G.; Bruce, L.; Persson, L.I. A phase II open label clinical study of the safety, tolerability and efficacy of ILB® for Amyotrophic Lateral Sclerosis. PLoS ONE 2022, 17, e0267183. [Google Scholar] [CrossRef]
- Patel, R.K.; Prasad, N.; Kuwar, R.; Haldar, D.; Abdul-Muneer, P.M. Transforming growth factor-beta 1 signaling regulates neuroinflammation and apoptosis in mild traumatic brain injury. Brain Behav. Immun. 2017, 64, 244–258. [Google Scholar] [CrossRef]
- Vasantharaja, R.; Stanley Abraham, L.; Gopinath, V.; Hariharan, D.; Smita, K.M. Attenuation of oxidative stress induced mitochondrial dysfunction and cytotoxicity in fibroblast cells by sulfated polysaccharide from Padina gymnospora. Int. J. Biol. Macromol. 2019, 124, 50–59. [Google Scholar] [CrossRef]
- Josephine, A.; Amudha, G.; Veena, C.K.; Preetha, S.P.; Rajeswari, A.; Varalakshmi, P. Beneficial effects of sulfated polysaccharides from Sargassum wightii against mitochondrial alterations induced by Cyclosporine A in rat kidney. Mol. Nutr. Food Res. 2007, 51, 1413–1422. [Google Scholar] [CrossRef]
- Wang, T.; Zhu, M.; He, Z.Z. Low-Molecular-Weight Fucoidan Attenuates Mitochondrial Dysfunction and Improves Neurological Outcome After Traumatic Brain Injury in Aged Mice: Involvement of Sirt3. Cell Mol. Neurobiol. 2016, 36, 1257–1268. [Google Scholar] [CrossRef]
- Marmarou, A.; Foda, M.A.; van den Brink, W.; Campbell, J.; Kita, H.; Demetriadou, K. A new model of diffuse brain injury in rats. Part I: Pathophysiology and biomechanics. J. Neurosurg. 1994, 80, 291–300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amorini, A.M.; Giorlandino, C.; Longo, S.; D’Urso, S.; Mesoraca, A.; Santoro, M.L.; Picardi, M.; Gullotta, S.; Cignini, P.; Lazzarino, D.; et al. Metabolic profile of amniotic fluid as a biochemical tool to screen for inborn errors of metabolism and fetal anomalies. Mol. Cell Biochem. 2012, 359, 205–216. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| nmol/g w.w. | Controls | sTBI | sTBI + ILB® 1 mg/kg b.w. | sTBI + ILB® 5 mg/kg b.w. |
|---|---|---|---|---|
| ATP | 2278 ± 356 | 1472 ± 226 a | 1689 ± 193 a | 1805 ± 237 a,b |
| GTP | 621 ± 76 | 401 ± 59 a | 452 ± 63 a | 511 ± 70 a,b |
| NAD+ | 542 ± 39 | 304 ± 44 a | 296 ± 38 a | 395 ± 48 a,b |
| Ascorbate | 3005 ± 412 | 2126 ± 378 a | 2314 ± 404 a | 2456 ± 512 a |
| GSH | 3656 ± 592 | 2014 ± 194 a | 2166 ± 388 a | 2704 ± 259 a,b |
| NAA | 9756 ± 753 | 5031 ± 642 a | 5389 ± 703 a | 6976 ± 775 a,b |
| Glu | 10358 ± 1202 | 12548 ± 1311 a | 12001 ± 1154 a | 11782 ± 1096 a,b |
| GABR | 3.94 ± 0.91 | 2.55 ± 0.55 a | 2.89 ± 0.78 a | 3.21 ± 0.85 b |
| Met | 42.50 ± 7.33 | 28.72 ± 6.02 a | 33.14 ± 5.70 | 36.41 ± 6.95 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lazzarino, G.; Di Pietro, V.; Rinaudo, M.; Nagy, Z.; Barnes, N.M.; Bruce, L.; Signoretti, S.; Mangione, R.; Saab, M.W.; Tavazzi, B.; et al. ILB®, a Low Molecular Weight Dextran Sulphate, Restores Glutamate Homeostasis, Amino Acid Metabolism and Neurocognitive Functions in a Rat Model of Severe Traumatic Brain Injury. Int. J. Mol. Sci. 2022, 23, 8460. https://doi.org/10.3390/ijms23158460
Lazzarino G, Di Pietro V, Rinaudo M, Nagy Z, Barnes NM, Bruce L, Signoretti S, Mangione R, Saab MW, Tavazzi B, et al. ILB®, a Low Molecular Weight Dextran Sulphate, Restores Glutamate Homeostasis, Amino Acid Metabolism and Neurocognitive Functions in a Rat Model of Severe Traumatic Brain Injury. International Journal of Molecular Sciences. 2022; 23(15):8460. https://doi.org/10.3390/ijms23158460
Chicago/Turabian StyleLazzarino, Giacomo, Valentina Di Pietro, Marco Rinaudo, Zsuzsanna Nagy, Nicholas M. Barnes, Lars Bruce, Stefano Signoretti, Renata Mangione, Miriam Wissam Saab, Barbara Tavazzi, and et al. 2022. "ILB®, a Low Molecular Weight Dextran Sulphate, Restores Glutamate Homeostasis, Amino Acid Metabolism and Neurocognitive Functions in a Rat Model of Severe Traumatic Brain Injury" International Journal of Molecular Sciences 23, no. 15: 8460. https://doi.org/10.3390/ijms23158460
APA StyleLazzarino, G., Di Pietro, V., Rinaudo, M., Nagy, Z., Barnes, N. M., Bruce, L., Signoretti, S., Mangione, R., Saab, M. W., Tavazzi, B., Belli, A., Lazzarino, G., Amorini, A. M., & Logan, A. (2022). ILB®, a Low Molecular Weight Dextran Sulphate, Restores Glutamate Homeostasis, Amino Acid Metabolism and Neurocognitive Functions in a Rat Model of Severe Traumatic Brain Injury. International Journal of Molecular Sciences, 23(15), 8460. https://doi.org/10.3390/ijms23158460