
The Genome of the Mimosoid Legume Prosopis cineraria, a Desert Tree


 ,
,  and
and
Abstract
:1. Introduction
2. Results
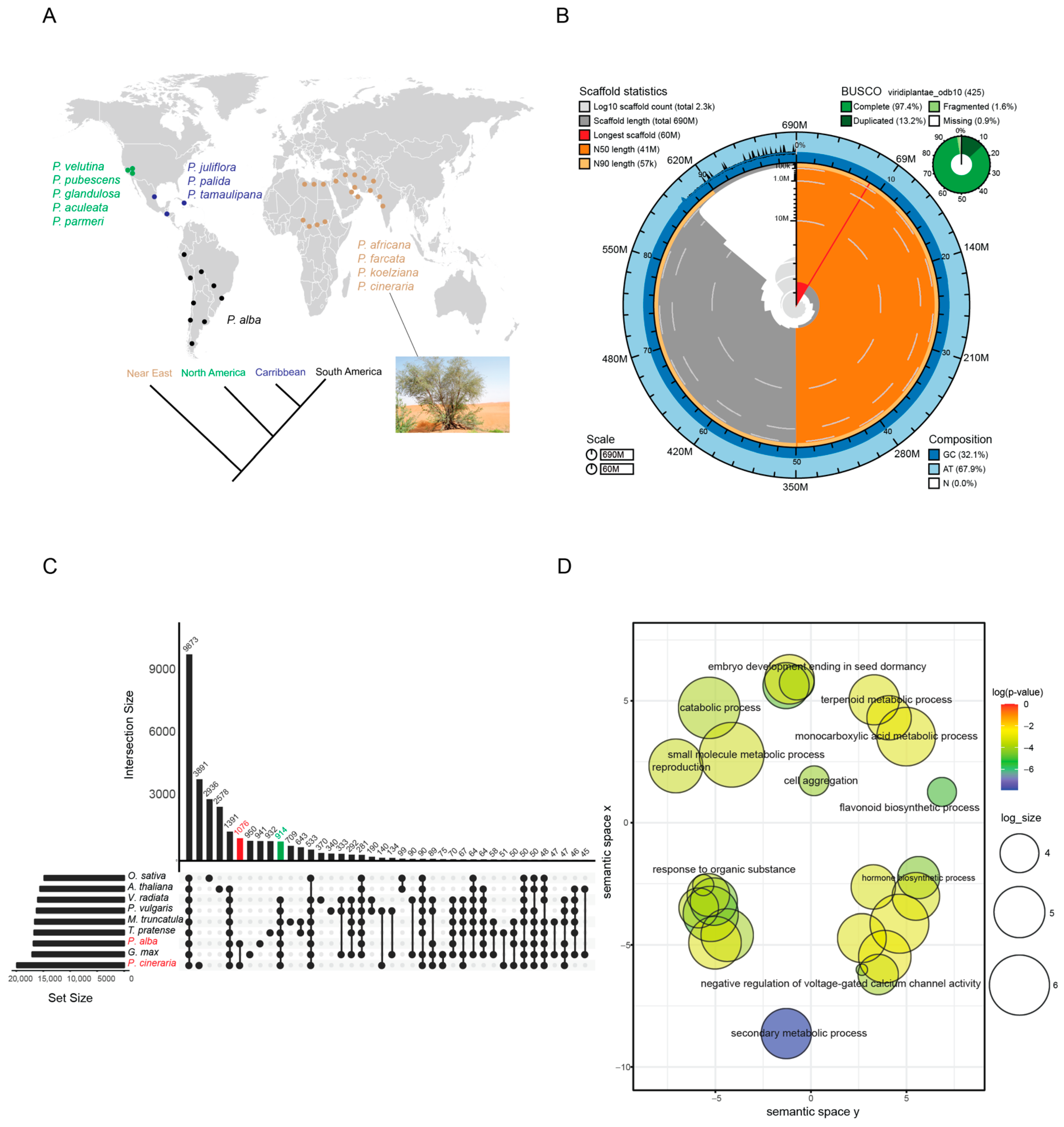
2.1. Reference Genome Sequencing and Assembly
2.2. Gene prediction and Annotation
2.3. Orthologous Group Analysis
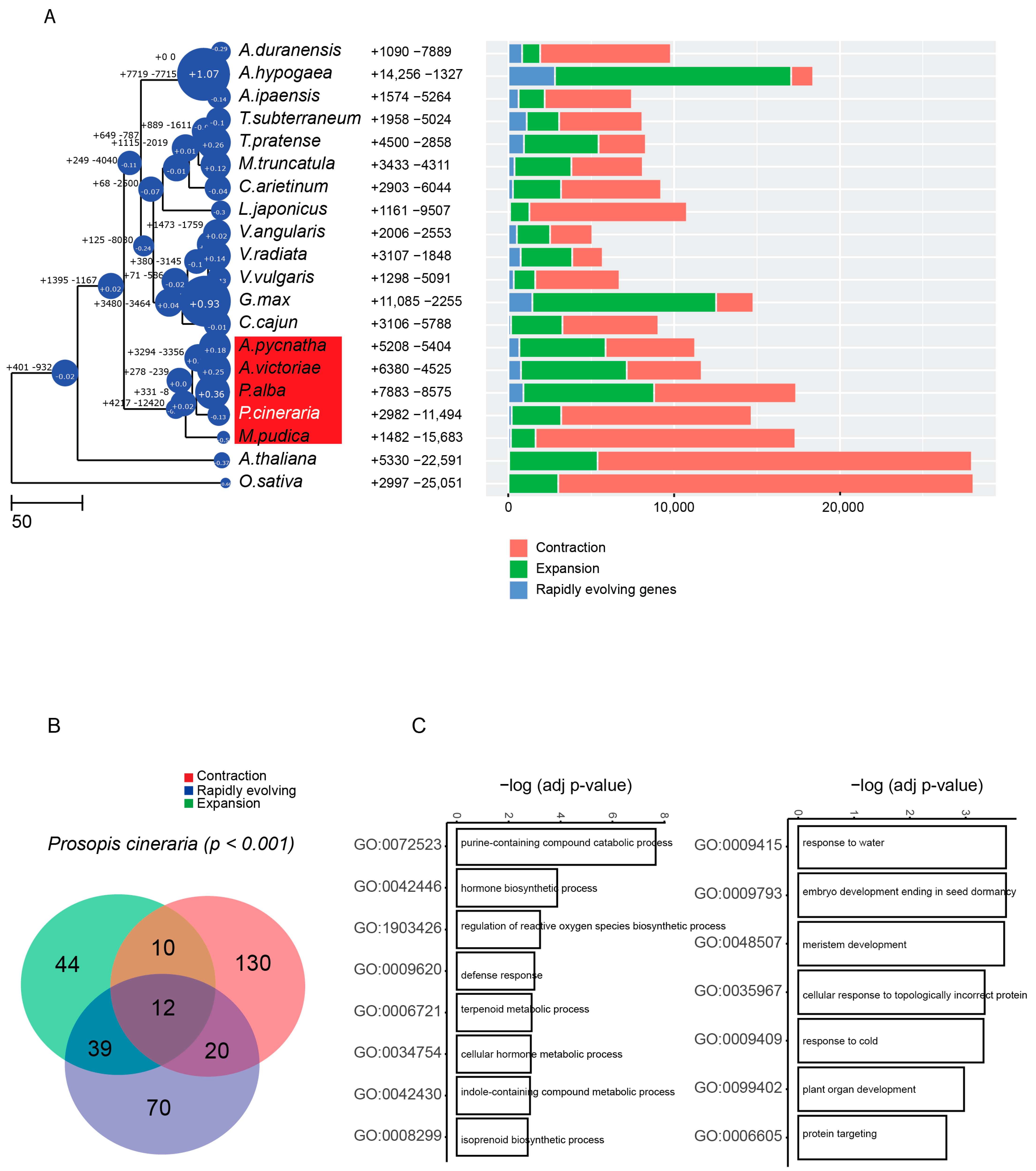
2.4. Genome Evolution in P. cineraria
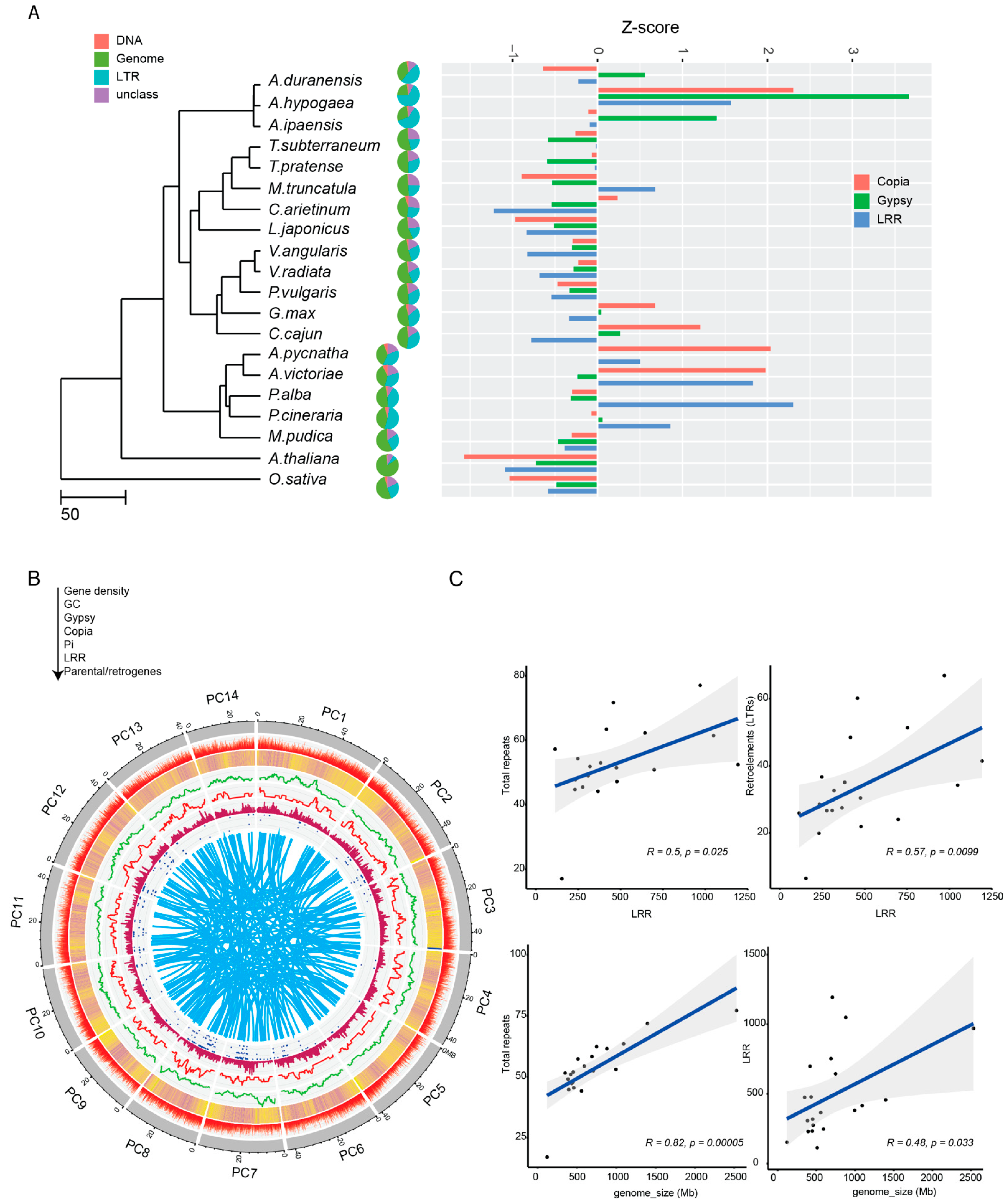
2.5. Comparative Analysis of Repeats, including Disease-Resistance Genes
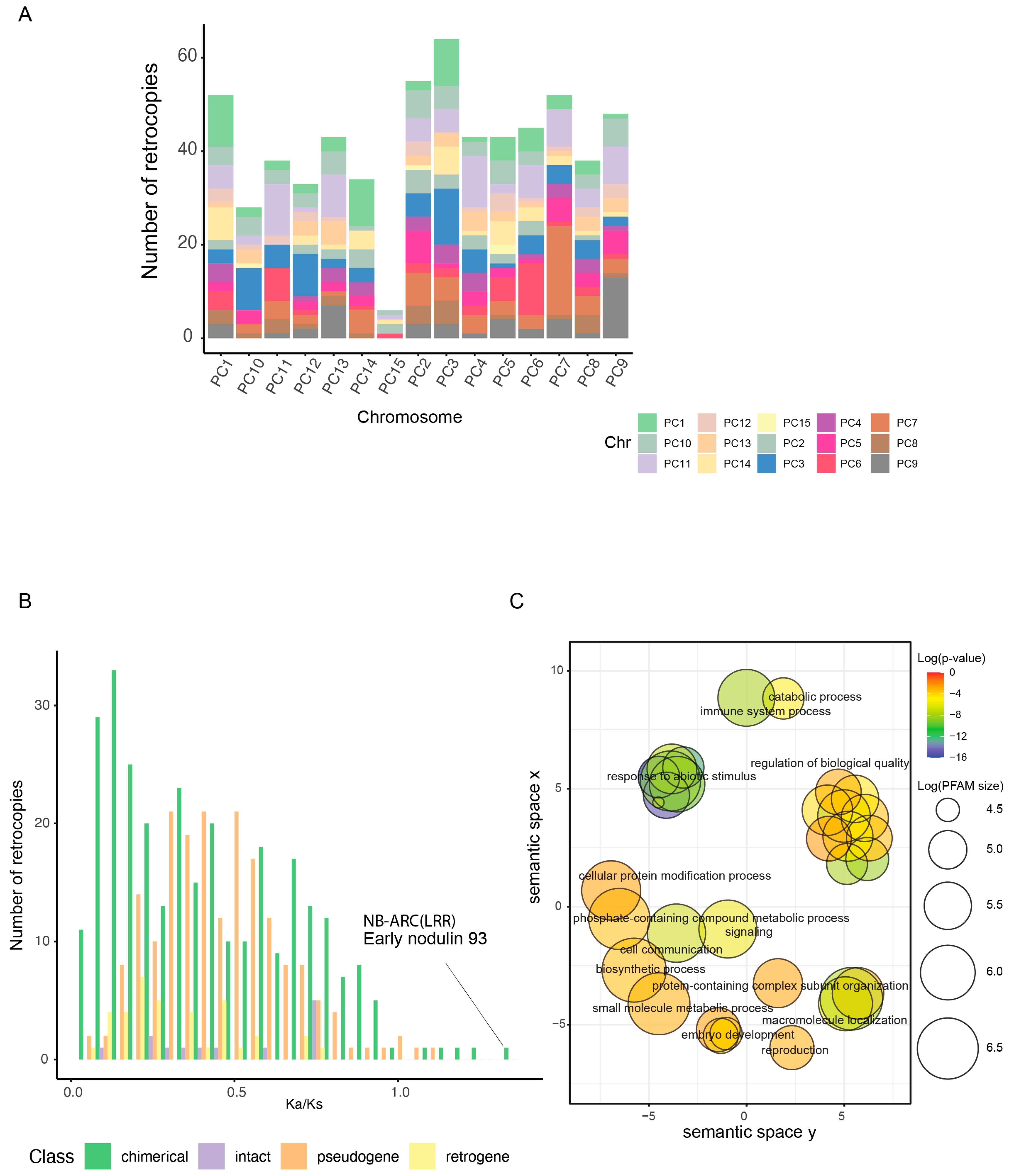
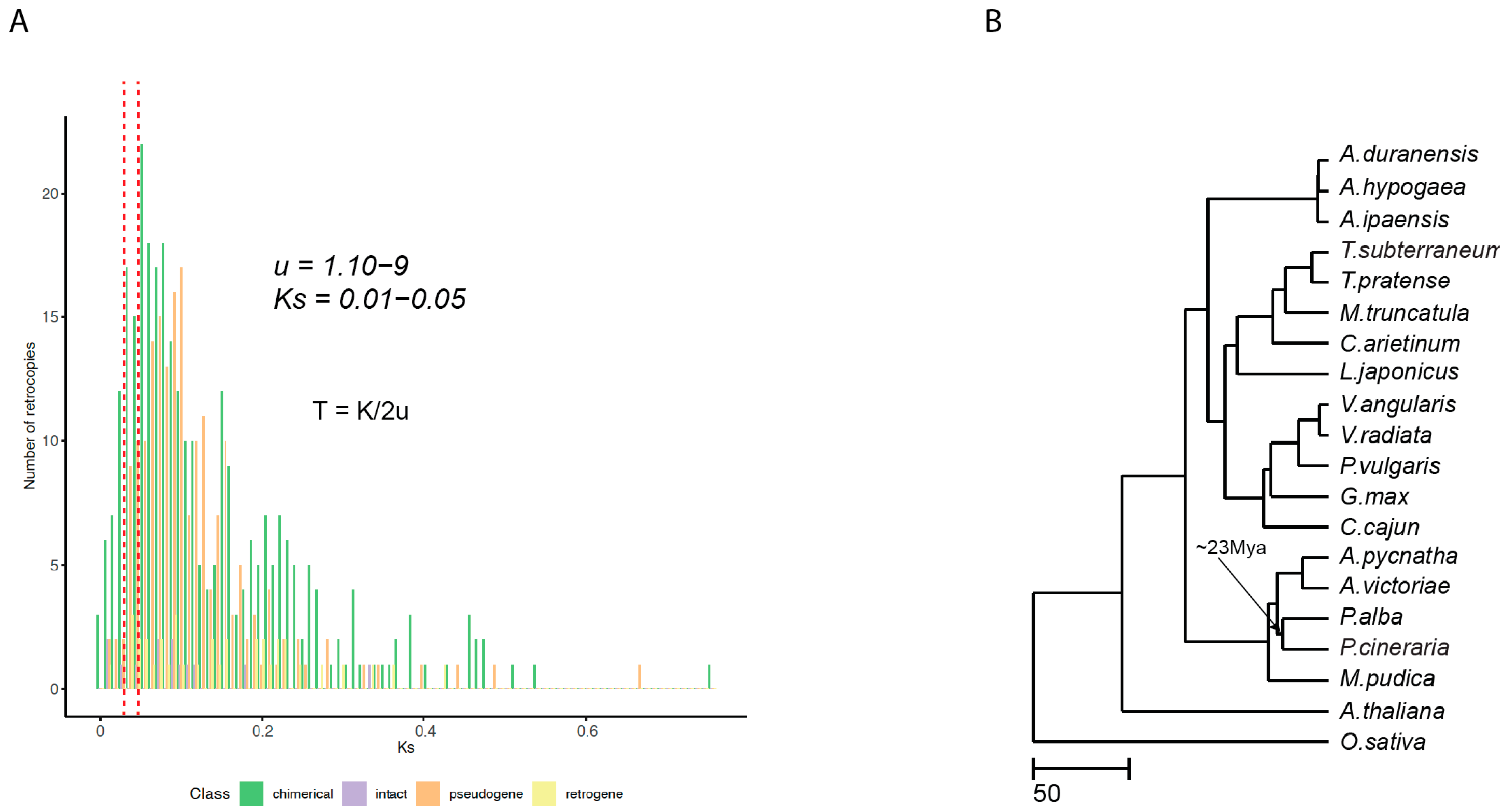
2.6. Retrogene Identification, Selection, and Activity in P. cineraria
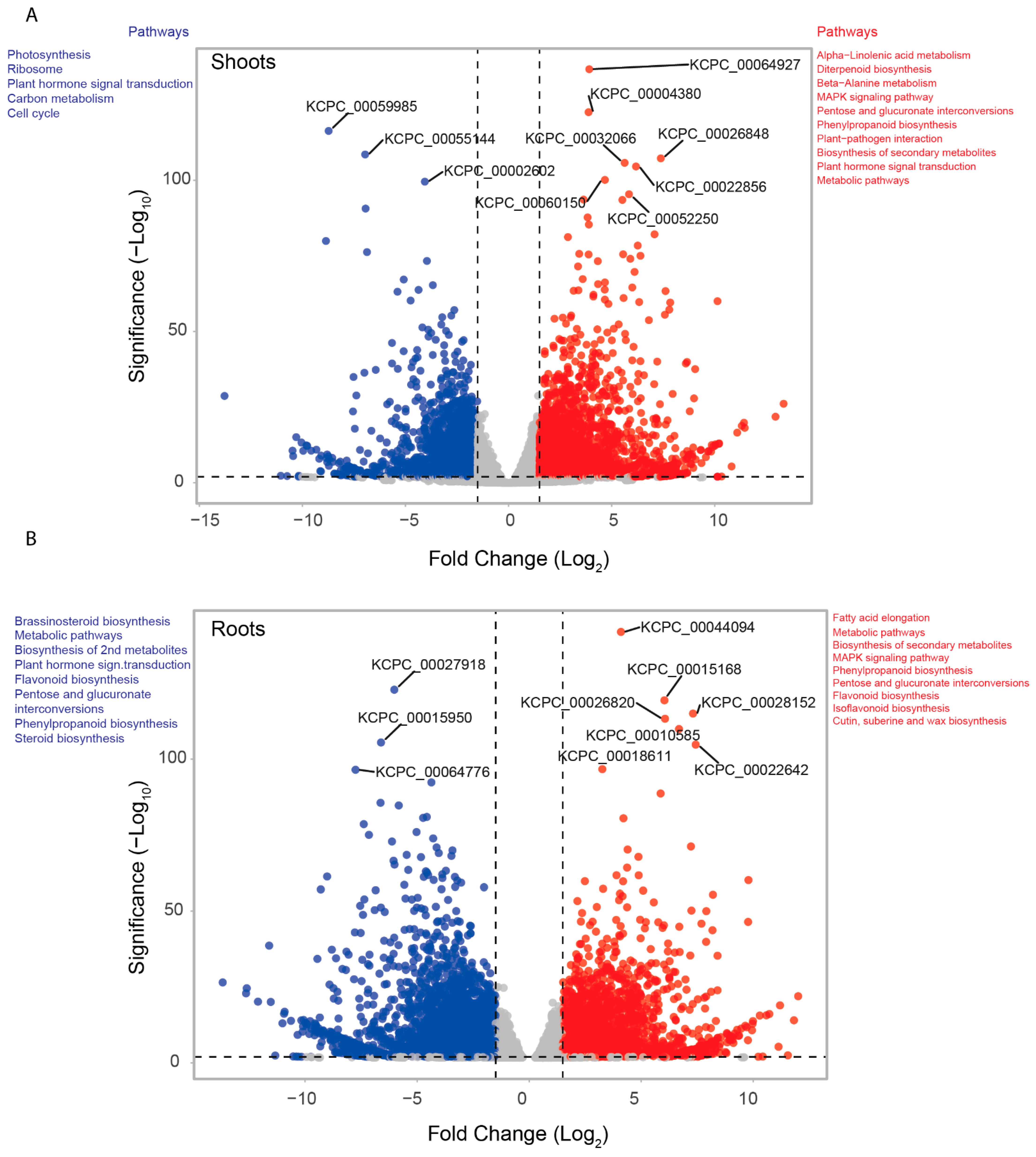
2.7. Differential Gene Expression under Salt Stress
3. Discussion
4. Materials and Methods
4.1. P. cineraria Sample Collection and Sequencing
4.2. Reference Genome Assembly and Genome Size Estimation
4.3. Gene Prediction and Genome Annotation
4.4. Orthogroup Analysis
4.5. Phylogenetic Analysis and Divergence Times
4.6. Genome Evolution of P. cineraria
4.7. Comparative Analysis of Repeats, including NBS-LRR Disease-Resistance Genes
4.8. Retrogene Identification and Expression in P. cineraria
4.9. Differential Gene Expression in P. cineraria
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Polhill, R. Classification of the Leguminosae. In Phytochemical Dictionary of the Leguminosae; Bisby, F.A., Buckingham, J., Harborne, J.B., Eds.; Chapman and Hall: New York, NY, USA, 1994; pp. 16–48. [Google Scholar]
- Cardoso, D.; De Queiroz, L.P.; Pennington, R.T.; De Lima, H.C.; Fonty, É.; Wojciechowski, M.F.; Lavin, M. Revisiting the phylogeny of papilionoid legumes: New insights from comprehensively sampled early-branching lineages. Am. J. Bot. 2012, 99, 1991–2013. [Google Scholar] [CrossRef] [PubMed]
- Lavin, M.; Herendeen, P.S.; Wojciechowski, M.F. Evolutionary rates analysis of Leguminosae implicates a rapid diversification of lineages during the tertiary. Syst. Biol. 2005, 54, 575–594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cannon, S.B.; Sterck, L.; Rombauts, S.; Sato, S.; Cheung, F.; Gouzy, J.; Wang, X.; Mudge, J.; Vasdewani, J.; Schiex, T. Legume genome evolution viewed through the Medicago truncatula and Lotus japonicus genomes. Proc. Natl. Acad. Sci. USA 2006, 103, 14959–14964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, S.; Nakamura, Y.; Kaneko, T.; Asamizu, E.; Kato, T.; Nakao, M.; Sasamoto, S.; Watanabe, A.; Ono, A.; Kawashima, K. Genome structure of the legume, Lotus japonicus. DNA Res. 2008, 15, 227–239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmutz, J.; Cannon, S.B.; Schlueter, J.; Ma, J.; Mitros, T.; Nelson, W.; Hyten, D.L.; Song, Q.; Thelen, J.J.; Cheng, J. Genome sequence of the palaeopolyploid soybean. Nature 2010, 463, 178–183. [Google Scholar] [CrossRef] [Green Version]
- Varshney, R.K.; Chen, W.; Li, Y.; Bharti, A.K.; Saxena, R.K.; Schlueter, J.A.; Donoghue, M.T.; Azam, S.; Fan, G.; Whaley, A.M. Draft genome sequence of pigeonpea (Cajanus cajan), an orphan legume crop of resource-poor farmers. Nat. Biotechnol. 2012, 30, 83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jain, M.; Misra, G.; Patel, R.K.; Priya, P.; Jhanwar, S.; Khan, A.W.; Shah, N.; Singh, V.K.; Garg, R.; Jeena, G. A draft genome sequence of the pulse crop chickpea (Cicer arietinum L.). Plant J. 2013, 74, 715–729. [Google Scholar] [CrossRef]
- Varshney, R.K.; Song, C.; Saxena, R.K.; Azam, S.; Yu, S.; Sharpe, A.G.; Cannon, S.; Baek, J.; Rosen, B.D.; Tar’an, B. Draft genome sequence of chickpea (Cicer arietinum) provides a resource for trait improvement. Nat. Biotechnol. 2013, 31, 240–246. [Google Scholar] [CrossRef] [Green Version]
- Kang, Y.J.; Kim, S.K.; Kim, M.Y.; Lestari, P.; Kim, K.H.; Ha, B.-K.; Jun, T.H.; Hwang, W.J.; Lee, T.; Lee, J. Genome sequence of mungbean and insights into evolution within Vigna species. Nat. Commun. 2014, 5, 5443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, N.D.; Bharti, A.K. Genome-enabled insights into legume biology. Annu. Rev. Plant Biol. 2012, 63, 283–305. [Google Scholar] [CrossRef]
- Blanc, G.; Wolfe, K.H. Widespread paleopolyploidy in model plant species inferred from age distributions of duplicate genes. Plant Cell 2004, 16, 1667–1678. [Google Scholar] [CrossRef] [Green Version]
- Bertioli, D.J.; Moretzsohn, M.C.; Madsen, L.H.; Sandal, N.; Leal-Bertioli, S.C.; Guimarães, P.M.; Hougaard, B.K.; Fredslund, J.; Schauser, L.; Nielsen, A.M. An analysis of synteny of Arachis with Lotus and Medicago sheds new light on the structure, stability and evolution of legume genomes. BMC Genom. 2009, 10, 45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soltis, D.E.; Albert, V.A.; Leebens-Mack, J.; Bell, C.D.; Paterson, A.H.; Zheng, C.; Sankoff, D.; de Pamphilis, C.W.; Wall, P.K.; Soltis, P.S. Polyploidy and angiosperm diversification. Am. J. Bot. 2009, 96, 336–348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sprent, J.I.; James, E.K. Legume evolution: Where do nodules and mycorrhizas fit in? Plant Physiol. 2007, 144, 575–581. [Google Scholar] [CrossRef] [Green Version]
- Afifi, H.S.A.; Al-rub, I.A. Prosopis cineraria as an unconventional legumes, nutrition and health benefits. In Legume Seed Nutraceutical Research; IntechOpen: Rijeka, Croatia, 2018; Available online: https://www.intechopen.com/chapters/62401 (accessed on 10 March 2021). [CrossRef] [Green Version]
- Panwar, D.; Pareek, K.; Bharti, C. Unripe Pods of Prosopis cineraria used as a vegetable (sangri) in Shekhawati region. Int. J. Sci. Eng. Res. 2014, 5, 892–895. [Google Scholar]
- Riveros, F. The genus Prosopis and its potential to improve livestock production in arid and semi arid regions. In Legume Trees and Other Fodder Trees as Protein Sources for Livestock; FAO: Roma, Italy, 1992; pp. 257–276. [Google Scholar]
- Rani, B.; Singh, U.; Sharma, R.; GUPTA, A.A.; Dhawan, N.G.; Sharma, A.K.; Sharma, S.; Maheshwari, R.K. Prosopis cineraria (L.) Druce: A desert tree to brace livelihood in Rajasthan. Asian J. Pharm. Res. Health Care 2013, 5, 58–64. [Google Scholar]
- Ramoliya, P.; Patel, H.; Joshi, J.; Pandey, A. Effect of salinization of soil on growth and nutrient accumulation in seedlings of Prosopis cineraria. J. Plant Nutr. 2006, 29, 283–303. [Google Scholar] [CrossRef]
- Mann, H.; Shankarnarayan, K. The role of Prosopis cineraria in an agro-pastoral system in western Rajasthan [augmenting fertility status and soil moisture, use as fodder]. In Proceedings of the International Symposium on Browse in Africa, Addis Ababa, Ethiopia, 8–12 April 1980. [Google Scholar]
- Quadri, S.; Iyer, R. Prosopis cineraria: Treasure of Arid Region. Editor. Board Memb. 2021, 2, 1–6. [Google Scholar]
- Lee, S.G.; Felker, P. Influence of water/heat stress on flowering and fruiting of mesquite (Prosopis glandulosa var. glandulosa). J. Arid Environ. 1992, 23, 309–319. [Google Scholar] [CrossRef]
- Kumar, A.; Rawat, D.; Rao, S. Studies on cytogenetical variation in Prosopis cineraia (Linn.) druce-a key stone tree species of Indian desert. Silvae Genet. 2007, 56, 184–189. [Google Scholar]
- Gurib-Fakim, A. Medicinal plants: Traditions of yesterday and drugs of tomorrow. Mol. Asp. Med. 2006, 27, 1–93. [Google Scholar] [CrossRef] [PubMed]
- Karunanithi, P.S.; Zerbe, P. Terpene synthases as metabolic gatekeepers in the evolution of plant terpenoid chemical diversity. Front. Plant Sci. 2019, 10, 1166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmelz, E.A.; Huffaker, A.; Sims, J.W.; Christensen, S.A.; Lu, X.; Okada, K.; Peters, R.J. Biosynthesis, elicitation and roles of monocot terpenoid phytoalexins. Plant J. 2014, 79, 659–678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaughan, M.M.; Christensen, S.; Schmelz, E.A.; Huffaker, A.; Mcauslane, H.J.; Alborn, H.T.; Romero, M.; Allen, L.H.; Teal, P.E. Accumulation of terpenoid phytoalexins in maize roots is associated with drought tolerance. Plant Cell Environ. 2015, 38, 2195–2207. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Tholl, D.; Bohlmann, J.; Pichersky, E. The family of terpene synthases in plants: A mid-size family of genes for specialized metabolism that is highly diversified throughout the kingdom. Plant J. 2011, 66, 212–229. [Google Scholar] [CrossRef]
- Tholl, D. Biosynthesis and biological functions of terpenoids in plants. Biotechnol. Isoprenoids 2015, 148, 63–106. [Google Scholar]
- Zerbe, P.; Bohlmann, J. Plant diterpene synthases: Exploring modularity and metabolic diversity for bioengineering. Trends Biotechnol. 2015, 33, 419–428. [Google Scholar] [CrossRef]
- Garg, A.; Mittal, S.K. Review on Prosopis cineraria: A potential herb of Thar desert. Drug Invent. Today 2013, 5, 60–65. [Google Scholar] [CrossRef]
- Guo, Y.-L.; Fitz, J.; Schneeberger, K.; Ossowski, S.; Cao, J.; Weigel, D. Genome-wide comparison of nucleotide-binding site-leucine-rich repeat-encoding genes in Arabidopsis. Plant Physiol. 2011, 157, 757–769. [Google Scholar] [CrossRef] [Green Version]
- Ratnaparkhe, M.B.; Wang, X.; Li, J.; Compton, R.O.; Rainville, L.K.; Lemke, C.; Kim, C.; Tang, H.; Paterson, A.H. Comparative analysis of peanut NBS-LRR gene clusters suggests evolutionary innovation among duplicated domains and erosion of gene microsynteny. New Phytol. 2011, 192, 164–178. [Google Scholar] [CrossRef]
- Seo, E.; Kim, S.; Yeom, S.-I.; Choi, D. Genome-wide comparative analyses reveal the dynamic evolution of nucleotide-binding leucine-rich repeat gene family among Solanaceae plants. Front. Plant Sci. 2016, 7, 1205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, S.; Zhang, X.; Yue, J.-X.; Tian, D.; Chen, J.-Q. Recent duplications dominate NBS-encoding gene expansion in two woody species. Mol. Genet. Genom. 2008, 280, 187–198. [Google Scholar] [CrossRef] [PubMed]
- Richter, T.E.; Pryor, T.J.; Bennetzen, J.L.; Hulbert, S.H. New rust resistance specificities associated with recombination in the Rp1 complex in maize. Genetics 1995, 141, 373–381. [Google Scholar] [CrossRef]
- Nagy, E.D.; Bennetzen, J.L. Pathogen corruption and site-directed recombination at a plant disease resistance gene cluster. Genome Res. 2008, 18, 1918–1923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Long, M.; Betrán, E.; Thornton, K.; Wang, W. The origin of new genes: Glimpses from the young and old. Nat. Rev. Genet. 2003, 4, 865–875. [Google Scholar] [CrossRef] [PubMed]
- Brumfield, R.T.; Beerli, P.; Nickerson, D.A.; Edwards, S.V. The utility of single nucleotide polymorphisms in inferences of population history. Trends Ecol. Evol. 2003, 18, 249–256. [Google Scholar] [CrossRef]
- Buschiazzo, E.; Ritland, C.; Bohlmann, J.; Ritland, K. Slow but not low: Genomic comparisons reveal slower evolutionary rate and higher dN/dS in conifers compared to angiosperms. BMC Evol. Biol. 2012, 12, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.; Park, J.; Yeom, S.-I.; Kim, Y.-M.; Seo, E.; Kim, K.-T.; Kim, M.-S.; Lee, J.M.; Cheong, K.; Shin, H.-S. New reference genome sequences of hot pepper reveal the massive evolution of plant disease-resistance genes by retroduplication. Genome Biol. 2017, 18, 210. [Google Scholar] [CrossRef] [Green Version]
- Hoen, D.R.; Park, K.C.; Elrouby, N.; Yu, Z.; Mohabir, N.; Cowan, R.K.; Bureau, T.E. Transposon-mediated expansion and diversification of a family of ULP-like genes. Mol. Biol. Evol. 2006, 23, 1254–1268. [Google Scholar] [CrossRef] [Green Version]
- Kong, H.; Landherr, L.L.; Frohlich, M.W.; Leebens-Mack, J.; Ma, H.; DePamphilis, C.W. Patterns of gene duplication in the plant SKP1 gene family in angiosperms: Evidence for multiple mechanisms of rapid gene birth. Plant J. 2007, 50, 873–885. [Google Scholar] [CrossRef]
- Hayashi, K.; Yoshida, H. Refunctionalization of the ancient rice blast disease resistance gene Pit by the recruitment of a retrotransposon as a promoter. Plant J. 2009, 57, 413–425. [Google Scholar] [CrossRef] [PubMed]
- Kuykendall, D.; Shao, J.; Trimmer, K. A nest of LTR retrotransposons adjacent the disease resistance-priming gene NPR1 in Beta vulgaris LUS Hybrid H20. Int. J. Plant Genom. 2009, 2009, 576742. [Google Scholar]
- Khan, J.A.; Wang, Q.; Sjolund, R.D.; Schulz, A.; Thompson, G.A. An early nodulin-like protein accumulates in the sieve element plasma membrane of Arabidopsis. Plant Physiol. 2007, 143, 1576–1589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- BI, Y.M.; Kant, S.; Clark, J.; Gidda, S.; Ming, F.; Xu, J.; Rochon, A.; Shelp, B.J.; Hao, L.; Zhao, R. Increased nitrogen-use efficiency in transgenic rice plants over-expressing a nitrogen-responsive early nodulin gene identified from rice expression profiling. Plant Cell Environ. 2009, 32, 1749–1760. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Zou, Y.; Hu, J.; Ding, Y. Genome-wide analysis of the rice PPR gene family and their expression profiles under different stress treatments. BMC Genom. 2018, 19, 1–14. [Google Scholar] [CrossRef]
- Takashima, S.; Abe, T.; Yoshida, S.; Kawahigashi, H.; Saito, T.; Tsuji, S.; Tsujimoto, M. Analysis of sialyltransferase-like proteins from Oryza sativa. J. Biochem. 2006, 139, 279–287. [Google Scholar] [CrossRef]
- Liu, H.; Ma, Y.; Chen, N.; Guo, S.; Liu, H.; Guo, X.; Chong, K.; Xu, Y. Overexpression of stress-inducible OsBURP16, the β subunit of polygalacturonase 1, decreases pectin content and cell adhesion and increases abiotic stress sensitivity in rice. Plant Cell Environ. 2014, 37, 1144–1158. [Google Scholar] [CrossRef] [Green Version]
- Kohorn, B.D.; Kohorn, S.L. The cell wall-associated kinases, WAKs, as pectin receptors. Front. Plant Sci. 2012, 3, 88. [Google Scholar] [CrossRef] [Green Version]
- Keeling, C.I.; Bohlmann, J. Diterpene resin acids in conifers. Phytochemistry 2006, 67, 2415–2423. [Google Scholar] [CrossRef]
- Keeling, C.I.; Bohlmann, J. Genes, enzymes and chemicals of terpenoid diversity in the constitutive and induced defence of conifers against insects and pathogens. New Phytol. 2006, 170, 657–675. [Google Scholar] [CrossRef]
- Chang, S.; Puryear, J.; Cairney, J. A simple and efficient method for isolating RNA from pine trees. Plant Mol. Biol. Report. 1993, 11, 113–116. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrews, S. FastQC: A Quality Control Tool for High throughput Sequence Data. 2014. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 15 May 2021).
- Marcais, G.; Kingsford, C. Jellyfish: A fast k-mer counter. Tutor. E Manuais 2012, 1, 1–8. [Google Scholar]
- Vurture, G.W.; Sedlazeck, F.J.; Nattestad, M.; Underwood, C.J.; Fang, H.; Gurtowski, J.; Schatz, M.C. GenomeScope: Fast reference-free genome profiling from short reads. Bioinformatics 2017, 33, 2202–2204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koren, S.; Walenz, B.P.; Berlin, K.; Miller, J.R.; Bergman, N.H.; Phillippy, A.M. Canu: Scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 2017, 27, 722–736. [Google Scholar] [CrossRef] [Green Version]
- Walker, B.J.; Abeel, T.; Shea, T.; Priest, M.; Abouelliel, A.; Sakthikumar, S.; Cuomo, C.A.; Zeng, Q.; Wortman, J.; Young, S.K. Pilon: An integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE 2014, 9, e112963. [Google Scholar] [CrossRef]
- Putnam, N.H.; O’Connell, B.L.; Stites, J.C.; Rice, B.J.; Blanchette, M.; Calef, R.; Troll, C.J.; Fields, A.; Hartley, P.D.; Sugnet, C.W. Chromosome-scale shotgun assembly using an in vitro method for long-range linkage. Genome Res. 2016, 26, 342–350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- Seppey, M.; Manni, M.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness. In Gene Prediction; Springer: Berlin/Heidelberg, Germany, 2019; pp. 227–245. [Google Scholar]
- Hoff, K.J.; Lomsadze, A.; Borodovsky, M.; Stanke, M. Whole-genome annotation with BRAKER. In Gene Prediction; Springer: Berlin/Heidelberg, Germany, 2019; pp. 65–95. [Google Scholar]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef]
- Gremme, G. GenomeThreader Gene Prediction Software. Ph.D. Thesis, Universität Hamburg, Hamburg, Germany, 2013. [Google Scholar]
- Bruna, T.; Lomsadze, A.; Borodovsky, M. GeneMark-EP+: Eukaryotic gene prediction with self-training in the space of genes and proteins. NAR Genom. Bioinform. 2020, 2, lqaa026. [Google Scholar] [CrossRef]
- Stanke, M.; Keller, O.; Gunduz, I.; Hayes, A.; Waack, S.; Morgenstern, B. AUGUSTUS: Ab initio prediction of alternative transcripts. Nucleic Acids Res. 2006, 34, W435–W439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campbell, M.S.; Holt, C.; Moore, B.; Yandell, M. Genome annotation and curation using MAKER and MAKER-P. Curr. Protoc. Bioinform. 2014, 48, 4–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korf, I. Gene finding in novel genomes. BMC Bioinform. 2004, 5, 59. [Google Scholar] [CrossRef] [Green Version]
- Haas, B.J.; Salzberg, S.L.; Zhu, W.; Pertea, M.; Allen, J.E.; Orvis, J.; White, O.; Buell, C.R.; Wortman, J.R. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol. 2008, 9, R7. [Google Scholar] [CrossRef] [Green Version]
- Lagesen, K.; Hallin, P.; Rødland, E.A.; Stærfeldt, H.-H.; Rognes, T.; Ussery, D.W. RNAmmer: Consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 2007, 35, 3100–3108. [Google Scholar] [CrossRef] [PubMed]
- Chan, P.P.; Lowe, T.M. tRNAscan-SE: Searching for tRNA genes in genomic sequences. In Gene Prediction; Springer: Berlin/Heidelberg, Germany, 2019; pp. 1–14. [Google Scholar]
- Geer, L.Y.; Marchler-Bauer, A.; Geer, R.C.; Han, L.; He, J.; He, S.; Liu, C.; Shi, W.; Bryant, S.H. The NCBI biosystems database. Nucleic Acids Res. 2010, 38, D492–D496. [Google Scholar] [CrossRef] [Green Version]
- Bairoch, A.; Apweiler, R.; Wu, C.H.; Barker, W.C.; Boeckmann, B.; Ferro, S.; Gasteiger, E.; Huang, H.; Lopez, R.; Magrane, M. The universal protein resource (UniProt). Nucleic Acids Res. 2005, 33, D154–D159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bateman, A.; Coin, L.; Durbin, R.; Finn, R.D.; Hollich, V.; Griffiths-Jones, S.; Khanna, A.; Marshall, M.; Moxon, S.; Sonnhammer, E.L. The Pfam protein families database. Nucleic Acids Res. 2004, 32, D138–D141. [Google Scholar] [CrossRef]
- Hunter, S.; Apweiler, R.; Attwood, T.K.; Bairoch, A.; Bateman, A.; Binns, D.; Bork, P.; Das, U.; Daugherty, L.; Duquenne, L. InterPro: The integrative protein signature database. Nucleic Acids Res. 2009, 37, D211–D215. [Google Scholar] [CrossRef] [Green Version]
- Consortium, G.O. The gene ontology resource: 20 years and still GOing strong. Nucleic Acids Res. 2019, 47, D330–D338. [Google Scholar]
- Mulder, N.; Apweiler, R. Interpro and interproscan. In Comparative Genomics; Springer: Berlin/Heidelberg, Germany, 2007; pp. 59–70. [Google Scholar]
- Challis, R.; Richards, E.; Rajan, J.; Cochrane, G.; Blaxter, M. BlobToolKit–interactive quality assessment of genome assemblies. G3 Genes Genomes Genet. 2020, 10, 1361–1374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moriya, Y.; Itoh, M.; Okuda, S.; Yoshizawa, A.C.; Kanehisa, M. KAAS: An automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 2007, 35, W182–W185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Zhang, H.; Jin, J.; Tang, L.; Zhao, Y.; Gu, X.; Gao, G.; Luo, J. PlantTFDB 2.0: Update and improvement of the comprehensive plant transcription factor database. Nucleic Acids Res. 2011, 39, D1114–D1117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emms, D.M.; Kelly, S. OrthoFinder: Phylogenetic orthology inference for comparative genomics. Genome Biol. 2019, 20, 238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conway, J.R.; Lex, A.; Gehlenborg, N. UpSetR: An R package for the visualization of intersecting sets and their properties. Bioinformatics 2017, 33, 2938–2940. [Google Scholar] [CrossRef] [Green Version]
- Supek, F.; Bošnjak, M.; Škunca, N.; Šmuc, T. REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS ONE 2011, 6, e21800. [Google Scholar] [CrossRef] [Green Version]
- Sanderson, M.J. r8s: Inferring absolute rates of molecular evolution and divergence times in the absence of a molecular clock. Bioinformatics 2003, 19, 301–302. [Google Scholar] [CrossRef] [Green Version]
- Mendes, F.K.; Vanderpool, D.; Fulton, B.; Hahn, M.W. CAFE 5 models variation in evolutionary rates among gene families. Bioinformatics 2021, 36, 5516–5518. [Google Scholar] [CrossRef]
- Walter, W.; Sánchez-Cabo, F.; Ricote, M. GOplot: An R package for visually combining expression data with functional analysis. Bioinformatics 2015, 31, 2912–2914. [Google Scholar] [CrossRef]
- Flynn, J.M.; Hubley, R.; Goubert, C.; Rosen, J.; Clark, A.G.; Feschotte, C.; Smit, A.F. RepeatModeler2 for automated genomic discovery of transposable element families. Proc. Natl. Acad. Sci. USA 2020, 117, 9451–9457. [Google Scholar] [CrossRef]
- Ellinghaus, D.; Kurtz, S.; Willhoeft, U. LTRharvest, an efficient and flexible software for de novo detection of LTR retrotransposons. BMC Bioinform. 2008, 9, 18. [Google Scholar] [CrossRef] [Green Version]
- Ou, S.; Jiang, N. LTR_retriever: A highly accurate and sensitive program for identification of long terminal repeat retrotransposons. Plant Physiol. 2018, 176, 1410–1422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, N. Using Repeat Masker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinform. 2004, 5, 4–10. [Google Scholar] [CrossRef] [PubMed]
- Toda, N.; Rustenholz, C.; Baud, A.; Le Paslier, M.-C.; Amselem, J.; Merdinoglu, D.; Faivre-Rampant, P. NLGenomeSweeper: A tool for genome-wide NBS-LRR resistance gene identification. Genes 2020, 11, 333. [Google Scholar] [CrossRef] [Green Version]
- Krzywinski, M.; Schein, J.; Birol, I.; Connors, J.; Gascoyne, R.; Horsman, D.; Jones, S.J.; Marra, M.A. Circos: An information aesthetic for comparative genomics. Genome Res. 2009, 19, 1639–1645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, Z.; Sun, J.; Li, Q.; Yao, T.; Zeng, H.; Wang, Y. RetroScan: An Easy-to-Use Pipeline for Retrocopy Annotation and Visualization. Front. Genet. 2021, 16, 719204. [Google Scholar] [CrossRef] [PubMed]
- Pertea, M.; Kim, D.; Pertea, G.M.; Leek, J.T.; Salzberg, S.L. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 2016, 11, 1650–1667. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.-C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, H.; Gough, J. DcGO: Database of domain-centric ontologies on functions, phenotypes, diseases and more. Nucleic Acids Res. 2013, 41, D536–D544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ge, S.X.; Jung, D.; Yao, R. ShinyGO: A graphical gene-set enrichment tool for animals and plants. Bioinformatics 2020, 36, 2628–2629. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Features | Values |
|---|---|
| Total scaffolds | 2265 |
| Total genome size | 691,392,202 bp |
| Pseudochromosome | 14 |
| Pseudochromosome coverage | ~86% |
| (A + T) percentage | 67.8% |
| (G + C) percentage | 32.1% |
| N percentage | 2.44% |
| Min sequence length | 4999 bp |
| Max sequence length | 59,799,197 bp |
| Average sequence length | 305,250.42 bp |
| N50 length | 41,482,946 bp |
| L50 number | 8 |
| Repeat % | 58% |
| Number of genes | 76,554 |
| Number of exons | 344,680 |
| Number of rRNA genes | 361 |
| Number of tRNA genes | 664 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sudalaimuthuasari, N.; Ali, R.; Kottackal, M.; Rafi, M.; Al Nuaimi, M.; Kundu, B.; Al-Maskari, R.S.; Wang, X.; Mishra, A.K.; Balan, J.; et al. The Genome of the Mimosoid Legume Prosopis cineraria, a Desert Tree. Int. J. Mol. Sci. 2022, 23, 8503. https://doi.org/10.3390/ijms23158503
Sudalaimuthuasari N, Ali R, Kottackal M, Rafi M, Al Nuaimi M, Kundu B, Al-Maskari RS, Wang X, Mishra AK, Balan J, et al. The Genome of the Mimosoid Legume Prosopis cineraria, a Desert Tree. International Journal of Molecular Sciences. 2022; 23(15):8503. https://doi.org/10.3390/ijms23158503
Chicago/Turabian StyleSudalaimuthuasari, Naganeeswaran, Rashid Ali, Martin Kottackal, Mohammed Rafi, Mariam Al Nuaimi, Biduth Kundu, Raja Saeed Al-Maskari, Xuewen Wang, Ajay Kumar Mishra, Jithin Balan, and et al. 2022. "The Genome of the Mimosoid Legume Prosopis cineraria, a Desert Tree" International Journal of Molecular Sciences 23, no. 15: 8503. https://doi.org/10.3390/ijms23158503
APA StyleSudalaimuthuasari, N., Ali, R., Kottackal, M., Rafi, M., Al Nuaimi, M., Kundu, B., Al-Maskari, R. S., Wang, X., Mishra, A. K., Balan, J., Chaluvadi, S. R., Al Ansari, F., Bennetzen, J. L., Purugganan, M. D., Hazzouri, K. M., & Amiri, K. M. A. (2022). The Genome of the Mimosoid Legume Prosopis cineraria, a Desert Tree. International Journal of Molecular Sciences, 23(15), 8503. https://doi.org/10.3390/ijms23158503