
Reduced VDAC1, Maintained Mitochondrial Dynamics and Enhanced Mitochondrial Biogenesis in a Transgenic Tau Mouse Model of Alzheimer’s Disease


{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. VDAC1+/− Heterozygous Knockout Mice Decrease Mitochondrial Fission and Increase Mitochondrial Fusion Protein Expression
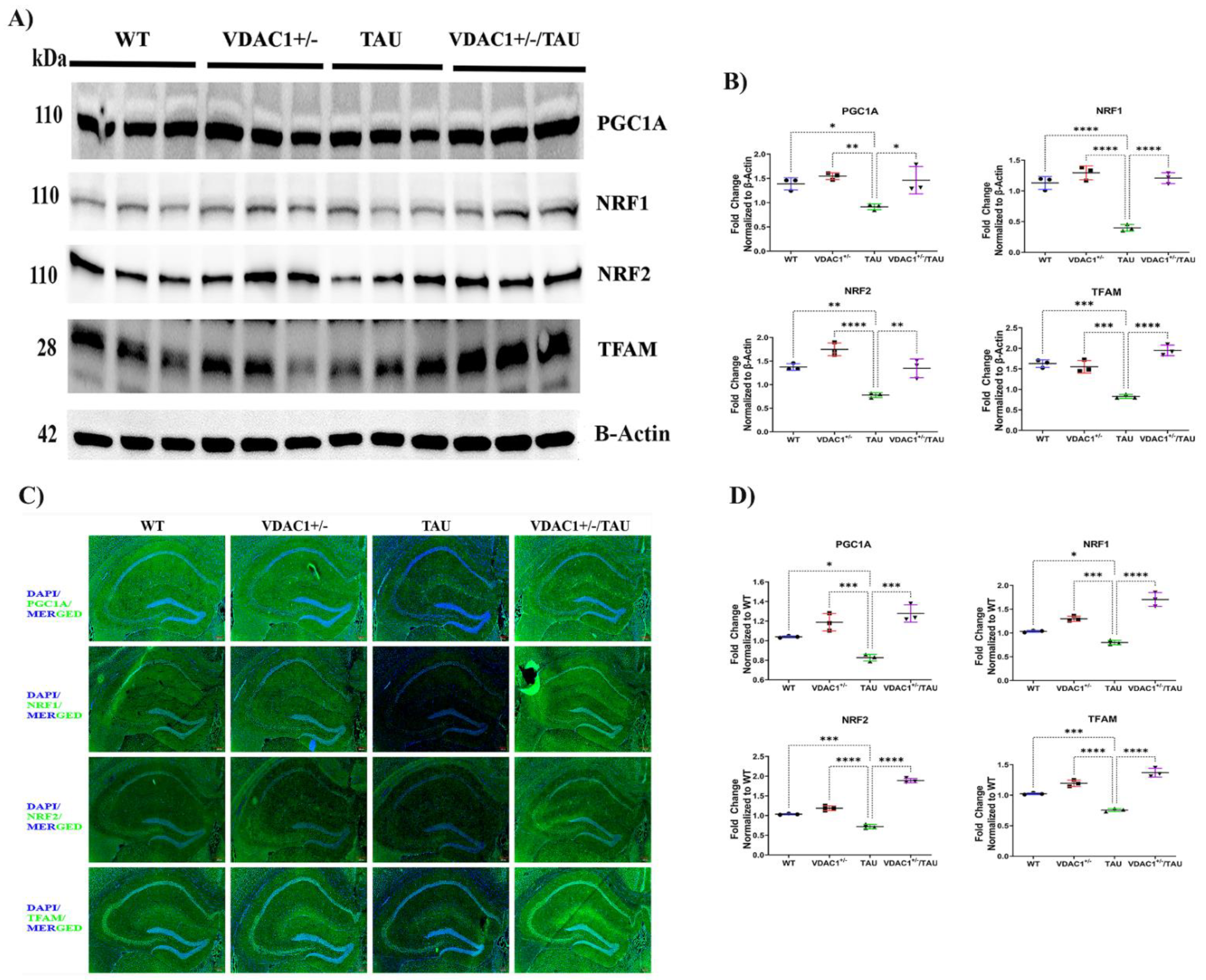
2.2. Reduced Expression of VDAC1 Induces Mitochondrial Biogenesis in VDAC1+/−/TAU Mice
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Immunoblotting
4.3. Immunofluorescence
4.4. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Selkoe, D.J. Alzheimer’s disease: Genes, proteins, and therapy. Physiol. Rev. 2001, 81, 741–766. [Google Scholar] [CrossRef] [PubMed]
- Reddy, P.H.; Beal, M.F. Amyloid beta, mitochondrial dysfunction and synaptic damage: Implications for cognitive decline in aging and Alzheimer’s disease. Trends Mol. Med. 2008, 14, 45–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Zhao, F.; Ma, X.; Perry, G.; Zhu, X. Mitochondria dysfunction in the pathogenesis of Alzheimer’s disease: Recent advances. Mol. Neurodegener. 2020, 15, 30. [Google Scholar] [CrossRef] [PubMed]
- Cornutiu, G. The Epidemiological Scale of Alzheimer’s Disease. J. Clin. Med. Res. 2015, 7, 657–666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiu, C.; Kivipelto, M.; von Strauss, E. Epidemiology of Alzheimer’s disease: Occurrence, determinants, and strategies toward intervention. Dialogues Clin. Neurosci. 2009, 11, 111–128. [Google Scholar] [CrossRef] [PubMed]
- Reddy, P.H. Mitochondrial medicine for aging and neurodegenerative diseases. Neuromolecular Med. 2008, 10, 291–315. [Google Scholar] [CrossRef] [Green Version]
- Ishihara, N.; Nomura, M.; Jofuku, A.; Kato, H.; Suzuki, S.O.; Masuda, K.; Otera, H.; Nakanishi, Y.; Nonaka, I.; Goto, Y.; et al. Mitochondrial fission factor Drp1 is essential for embryonic development and synapse formation in mice. Nat. Cell. Biol. 2009, 11, 958–966. [Google Scholar] [CrossRef]
- Swerdlow, R.H.; Kish, S.J. Mitochondria in Alzheimer’s disease. Int. Rev. Neurobiol. 2002, 53, 341–385. [Google Scholar] [CrossRef] [PubMed]
- Hirai, K.; Aliev, G.; Nunomura, A.; Fujioka, H.; Russell, R.L.; Atwood, C.S.; Johnson, A.B.; Kress, Y.; Vinters, H.V.; Tabaton, M.; et al. Mitochondrial abnormalities in Alzheimer’s disease. J. Neurosci. 2001, 21, 3017–3023. [Google Scholar] [CrossRef] [Green Version]
- Chan, D.C. Mitochondrial fusion and fission in mammals. Annu. Rev. Cell Dev. Biol. 2006, 22, 79–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Su, B.; Lee, H.G.; Li, X.; Perry, G.; Smith, M.A.; Zhu, X. Impaired balance of mitochondrial fission and fusion in Alzheimer’s disease. J. Neurosci. 2009, 29, 9090–9103. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.T.; Beal, M.F. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 2006, 443, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Manczak, M.; Calkins, M.J.; Reddy, P.H. Impaired mitochondrial dynamics and abnormal interaction of amyloid beta with mitochondrial protein Drp1 in neurons from patients with Alzheimer’s disease: Implications for neuronal damage. Hum. Mol. Genet. 2011, 20, 2495–2509. [Google Scholar] [CrossRef] [PubMed]
- Colombini, M. VDAC structure, selectivity, and dynamics. Biochim. Biophys. Acta 2012, 1818, 1457–1465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manczak, M.; Reddy, P.H. Abnormal interaction of VDAC1 with amyloid beta and phosphorylated tau causes mitochondrial dysfunction in Alzheimer’s disease. Hum. Mol. Genet. 2012, 21, 5131–5146. [Google Scholar] [CrossRef]
- Reddy, P.H. Is the mitochondrial outermembrane protein VDAC1 therapeutic target for Alzheimer’s disease? Biochim. Biophys. Acta 2013, 1832, 67–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manczak, M.; Reddy, P.H. RNA silencing of genes involved in Alzheimer’s disease enhances mitochondrial function and synaptic activity. Biochim. Biophys. Acta 2013, 1832, 2368–2378. [Google Scholar] [CrossRef] [Green Version]
- Manczak, M.; Sheiko, T.; Craigen, W.J.; Reddy, P.H. Reduced VDAC1 protects against Alzheimer’s disease, mitochondria, and synaptic deficiencies. J. Alzheimers Dis. 2013, 37, 679–690. [Google Scholar] [CrossRef] [Green Version]
- Vijayan, M.; Alvir, R.V.; Alvir, R.V.; Bunquin, L.E.; Pradeepkiran, J.A.; Reddy, P.H. A partial reduction of VDAC1 enhances mitophagy, autophagy, synaptic activities in a transgenic Tau mouse model. Aging Cell 2022, e13663. [Google Scholar] [CrossRef]
- Santos, R.X.; Correia, S.C.; Wang, X.; Perry, G.; Smith, M.A.; Moreira, P.I.; Zhu, X. A synergistic dysfunction of mitochondrial fission/fusion dynamics and mitophagy in Alzheimer’s disease. J. Alzheimers Dis. 2010, 20 (Suppl. 2), S401–S412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, B.; Wang, X.; Zheng, L.; Perry, G.; Smith, M.A.; Zhu, X. Abnormal mitochondrial dynamics and neurodegenerative diseases. Biochim. Biophys. Acta 2010, 1802, 135–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calkins, M.J.; Manczak, M.; Mao, P.; Shirendeb, U.; Reddy, P.H. Impaired mitochondrial biogenesis, defective axonal transport of mitochondria, abnormal mitochondrial dynamics and synaptic degeneration in a mouse model of Alzheimer’s disease. Hum. Mol. Genet. 2011, 20, 4515–4529. [Google Scholar] [CrossRef] [PubMed]
- Kerr, J.S.; Adriaanse, B.A.; Greig, N.H.; Mattson, M.P.; Cader, M.Z.; Bohr, V.A.; Fang, E.F. Mitophagy and Alzheimer’s Disease: Cellular and Molecular Mechanisms. Trends Neurosci. 2017, 40, 151–166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reddy, P.H.; Yin, X.; Manczak, M.; Kumar, S.; Pradeepkiran, J.A.; Vijayan, M.; Reddy, A.P. Mutant APP and amyloid beta-induced defective autophagy, mitophagy, mitochondrial structural and functional changes and synaptic damage in hippocampal neurons from Alzheimer’s disease. Hum. Mol. Genet. 2018, 27, 2502–2516. [Google Scholar] [CrossRef]
- Fang, E.F.; Hou, Y.; Palikaras, K.; Adriaanse, B.A.; Kerr, J.S.; Yang, B.; Lautrup, S.; Hasan-Olive, M.M.; Caponio, D.; Dan, X.; et al. Mitophagy inhibits amyloid-beta and tau pathology and reverses cognitive deficits in models of Alzheimer’s disease. Nat. Neurosci. 2019, 22, 401–412. [Google Scholar] [CrossRef]
- Wu, Z.; Zhu, Y.; Cao, X.; Sun, S.; Zhao, B. Mitochondrial toxic effects of Abeta through mitofusins in the early pathogenesis of Alzheimer’s disease. Mol Neurobiol 2014, 50, 986–996. [Google Scholar] [CrossRef] [PubMed]
- Cipolat, S.; Rudka, T.; Hartmann, D.; Costa, V.; Serneels, L.; Craessaerts, K.; Metzger, K.; Frezza, C.; Annaert, W.; D’Adamio, L.; et al. Mitochondrial rhomboid PARL regulates cytochrome c release during apoptosis via OPA1-dependent cristae remodeling. Cell 2006, 126, 163–175. [Google Scholar] [CrossRef]
- Weeber, E.J.; Levy, M.; Sampson, M.J.; Anflous, K.; Armstrong, D.L.; Brown, S.E.; Sweatt, J.D.; Craigen, W.J. The role of mitochondrial porins and the permeability transition pore in learning and synaptic plasticity. J. Biol. Chem. 2002, 277, 18891–18897. [Google Scholar] [CrossRef] [Green Version]
- Lewis, J.; McGowan, E.; Rockwood, J.; Melrose, H.; Nacharaju, P.; Van Slegtenhorst, M.; Gwinn-Hardy, K.; Paul Murphy, M.; Baker, M.; Yu, X.; et al. Neurofibrillary tangles, amyotrophy and progressive motor disturbance in mice expressing mutant (P301L) tau protein. Nat. Genet. 2000, 25, 402–405. [Google Scholar] [CrossRef]
- Vijayan, M.; Bose, C.; Reddy, P.H. Protective effects of a small molecule inhibitor, DDQ against amyloid beta in Alzheimer’s disease. Mitochondrion 2021, 59, 17–29. [Google Scholar] [CrossRef]
- Vijayan, M.; Bose, C.; Reddy, P.H. Anti-brain Aging Effects of Small Molecule Inhibitor DDQ. Mol. Neurobiol. 2021, 58, 3588–3600. [Google Scholar] [CrossRef] [PubMed]
- Vijayan, M.; George, M.; Bunquin, L.E.; Bose, C.; Reddy, P.H. Protective effects of a small-molecule inhibitor DDQ against tau-induced toxicities in a transgenic tau mouse model of Alzheimer’s disease. Hum. Mol. Genet. 2021, 31, 1022–1034. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vijayan, M.; Reddy, P.H. Reduced VDAC1, Maintained Mitochondrial Dynamics and Enhanced Mitochondrial Biogenesis in a Transgenic Tau Mouse Model of Alzheimer’s Disease. Int. J. Mol. Sci. 2022, 23, 8561. https://doi.org/10.3390/ijms23158561
Vijayan M, Reddy PH. Reduced VDAC1, Maintained Mitochondrial Dynamics and Enhanced Mitochondrial Biogenesis in a Transgenic Tau Mouse Model of Alzheimer’s Disease. International Journal of Molecular Sciences. 2022; 23(15):8561. https://doi.org/10.3390/ijms23158561
Chicago/Turabian StyleVijayan, Murali, and P. Hemachandra Reddy. 2022. "Reduced VDAC1, Maintained Mitochondrial Dynamics and Enhanced Mitochondrial Biogenesis in a Transgenic Tau Mouse Model of Alzheimer’s Disease" International Journal of Molecular Sciences 23, no. 15: 8561. https://doi.org/10.3390/ijms23158561