
Uncovering a Phenomenon of Active Hormone Transcriptional Regulation during Early Somatic Embryogenesis in Medicago sativa


Abstract
:1. Introduction
2. Results
2.1. Morphological Comparison of SE at Different Developmental Stages
2.2. Transcriptome Sequencing and Assembly
2.3. Gene Annotation and Functional Classification in M. sativa
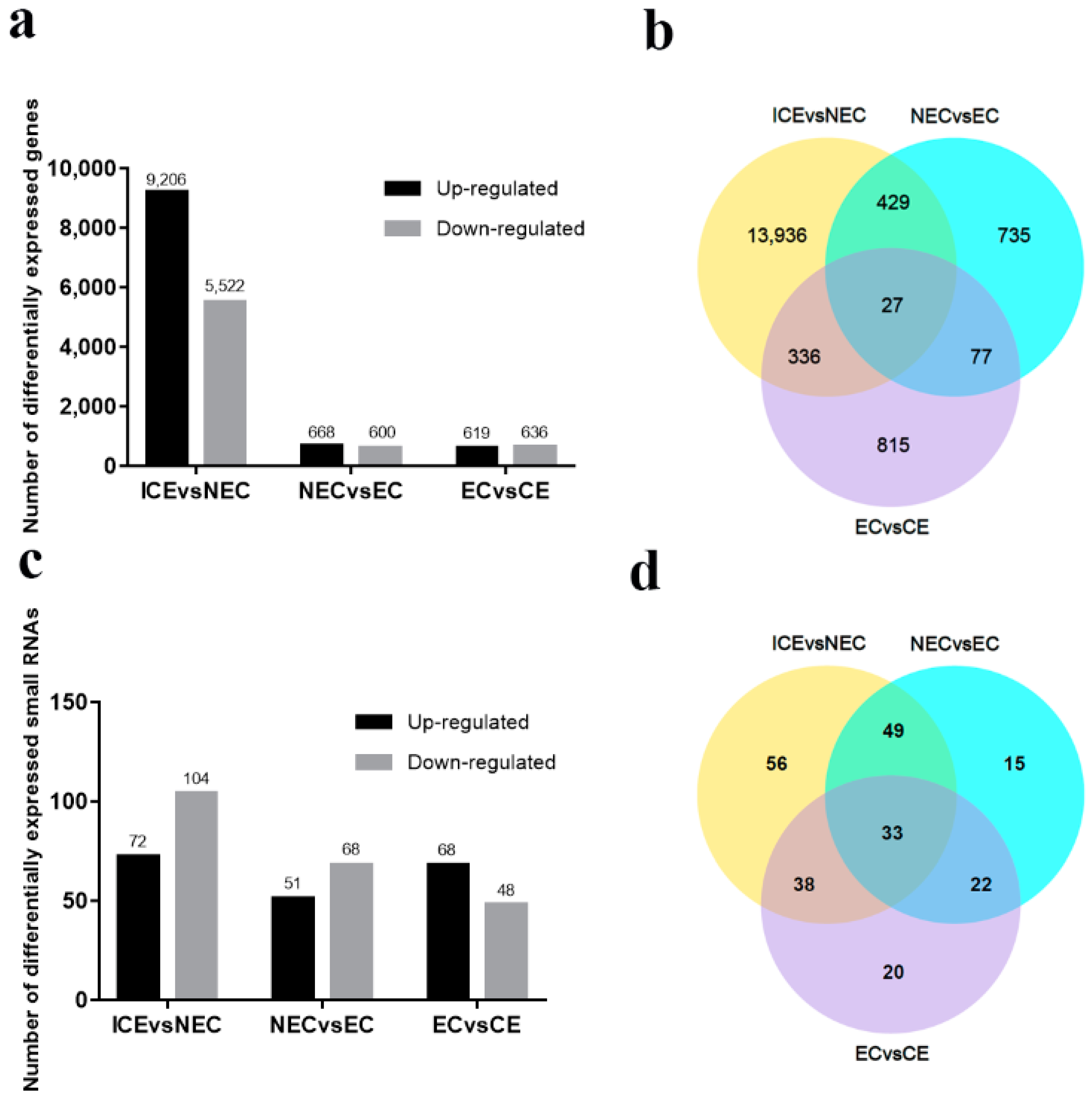
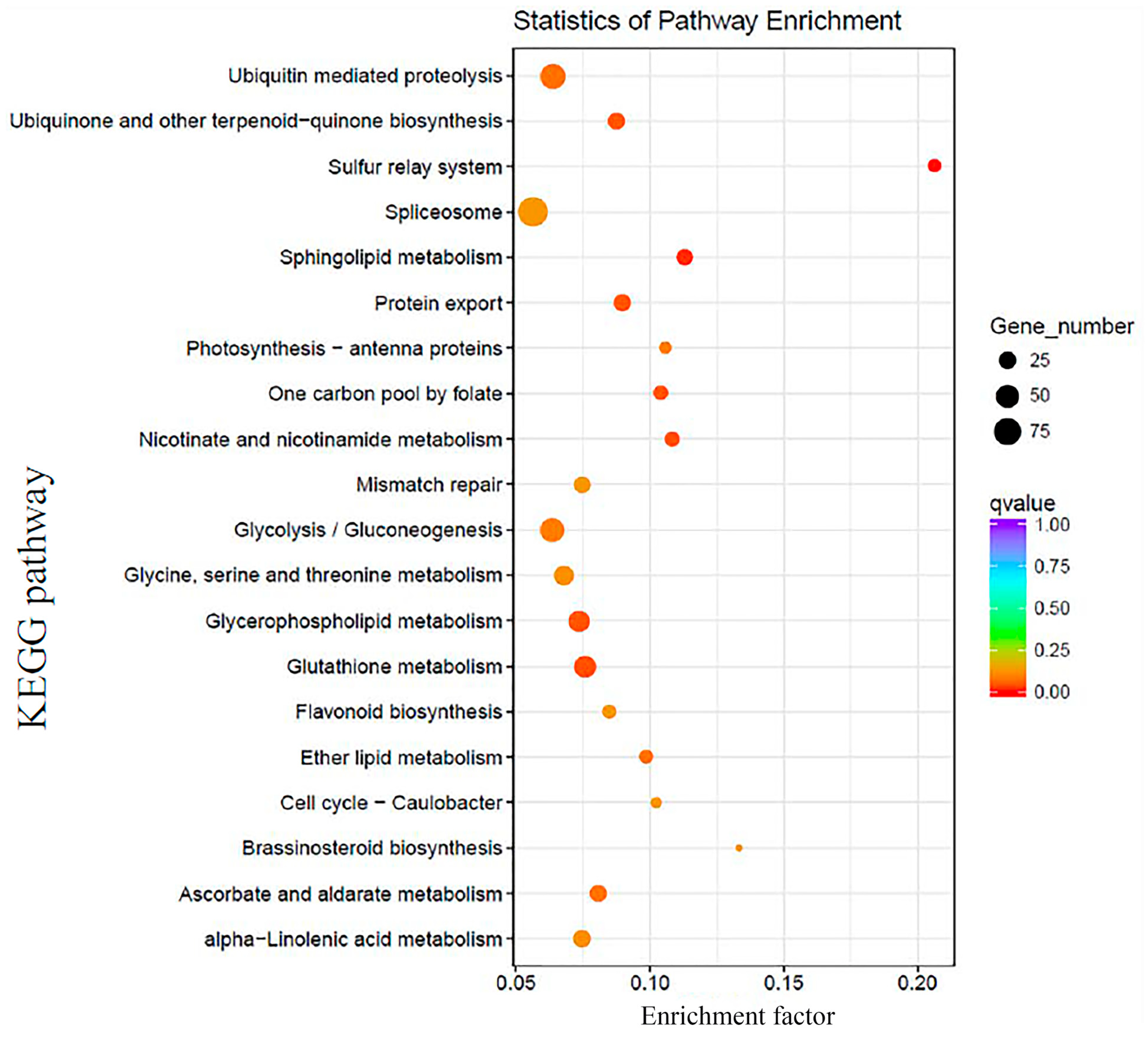
2.4. Identification and Analysis of DEGs in SE
2.5. Some DEGs Involved in Embryonic Induction, Embryonic and Maturation
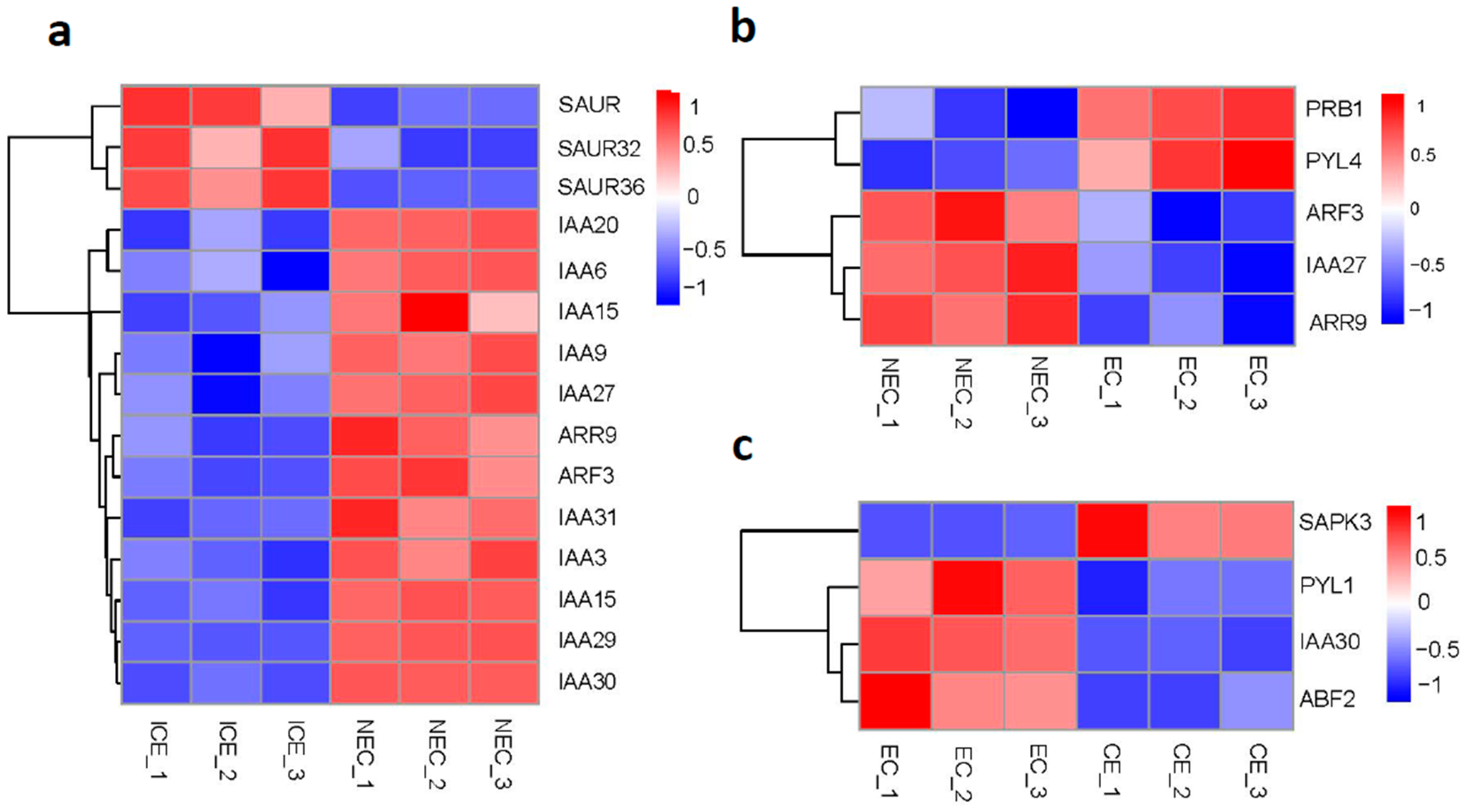
2.6. Expression of Plant Hormone Signal Transduction Genes in SE
2.7. Annotation of Small RNA-Seq Data
2.8. Identification and Expression Analysis of Known miRNAs in M. sativa
2.9. Identification of Potential Novel miRNAs in M. sativa
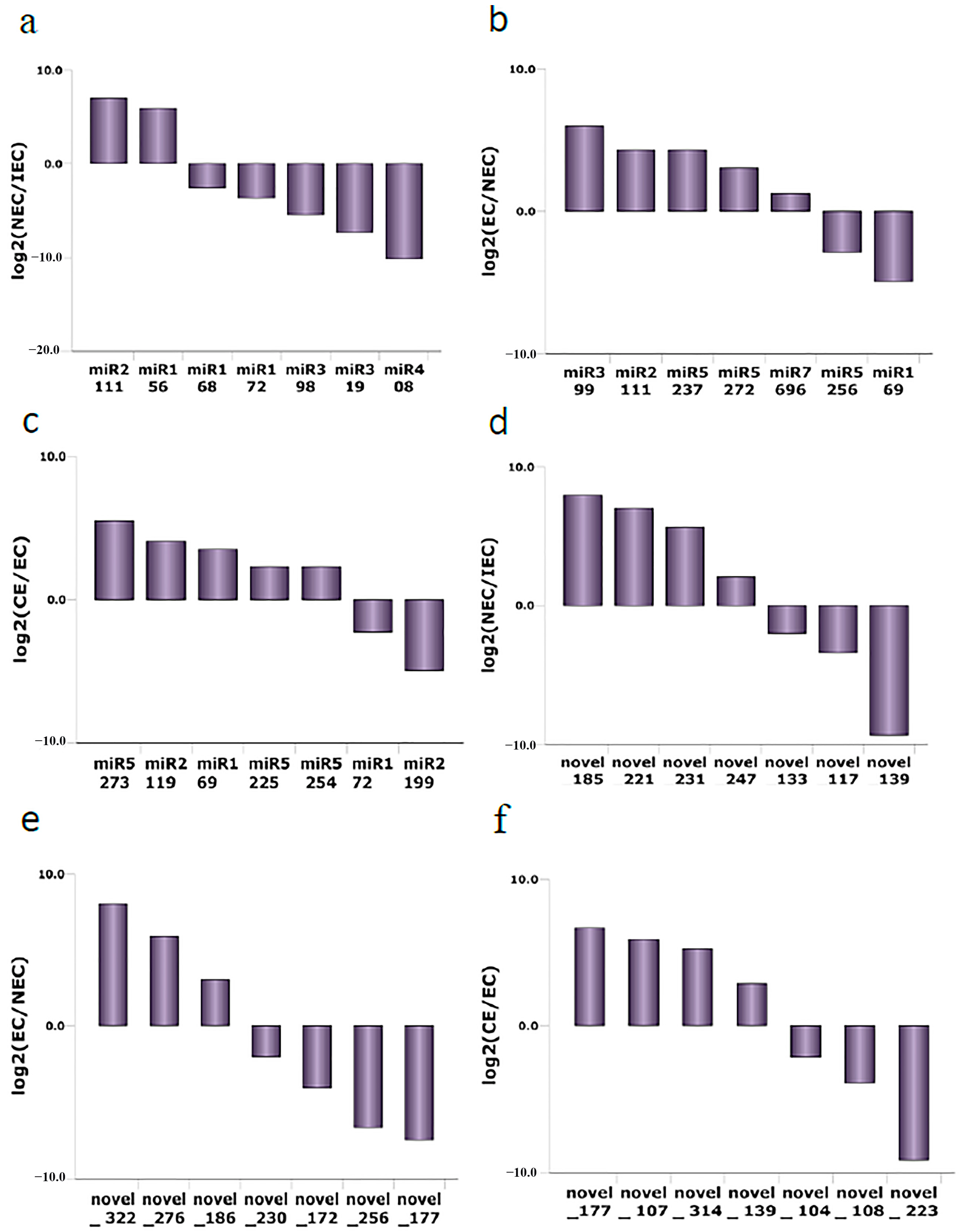
2.10. Expression of Known and Novel miRNAs during SE in M. sativa
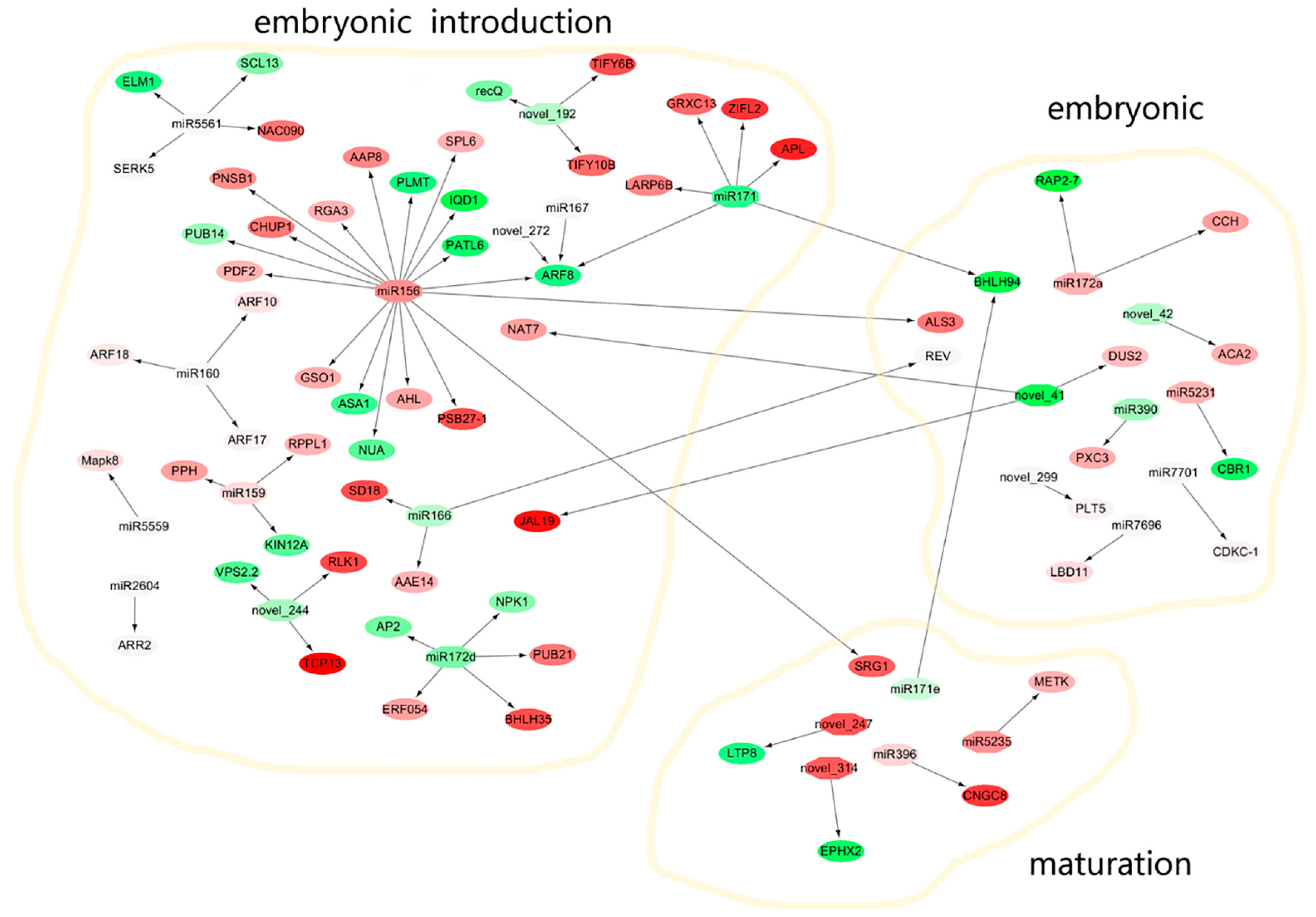
2.11. Analysis of Target Genes of miRNAs
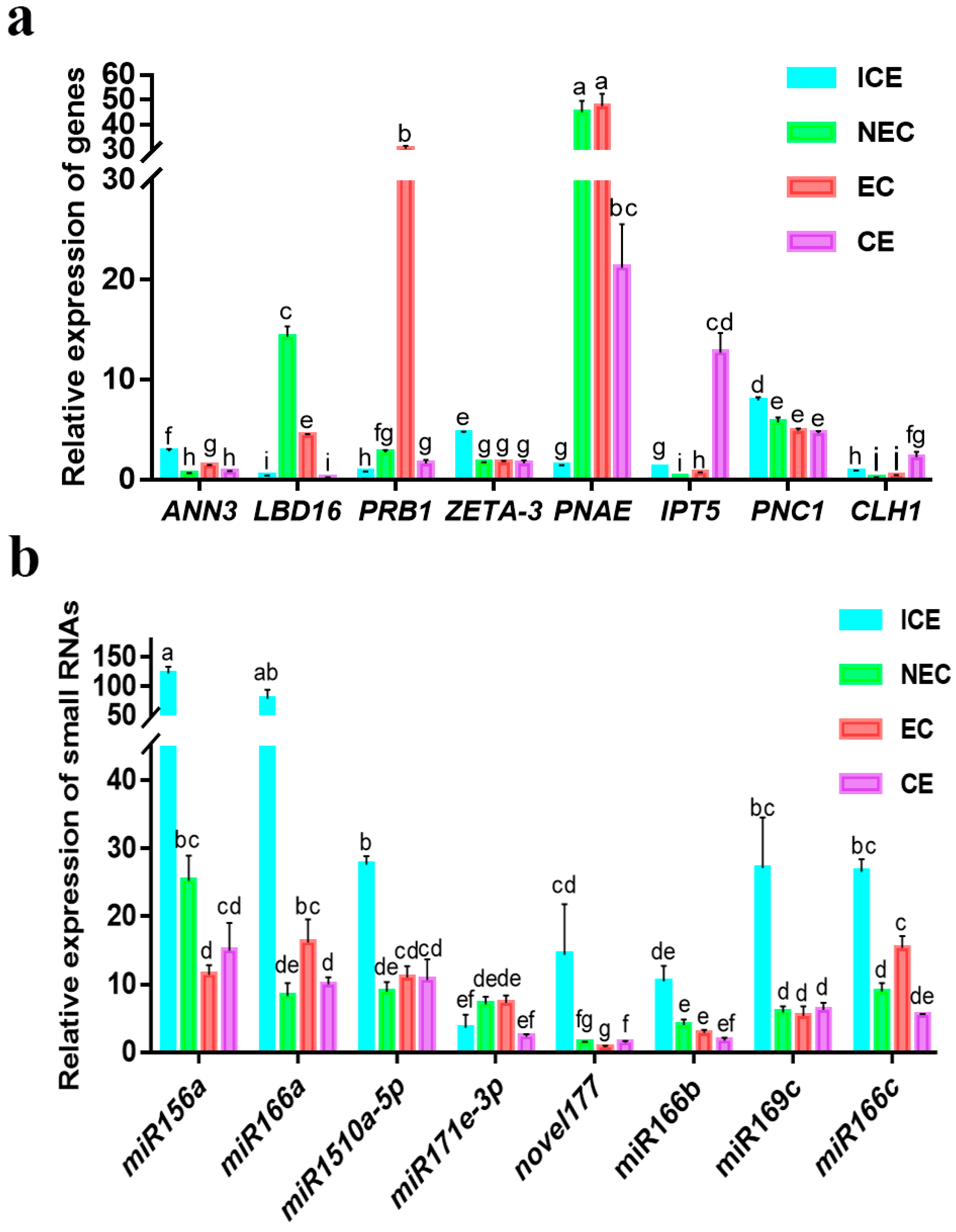
2.12. Validation of Various Expression Patterns of DEGs and miRNAs
3. Discussion
3.1. The Early Phase of SE Is an Active Process of Molecular Regulation
3.2. Identification of Hormonal Signal Transduction during SE in M. sativa
3.3. miRNA and Target Genes Form a Potential Molecular Regulatory Network in SE in M. sativa
4. Materials and Methods
4.1. Plant Materials
4.2. Morphological Analysis
4.3. RNA Extraction
4.4. Sample Preparation for Sequencing and Data Quality Control
4.5. Annotation of DEGs and miRNAs
4.6. Functional Annotation of DEGs and miRNAs
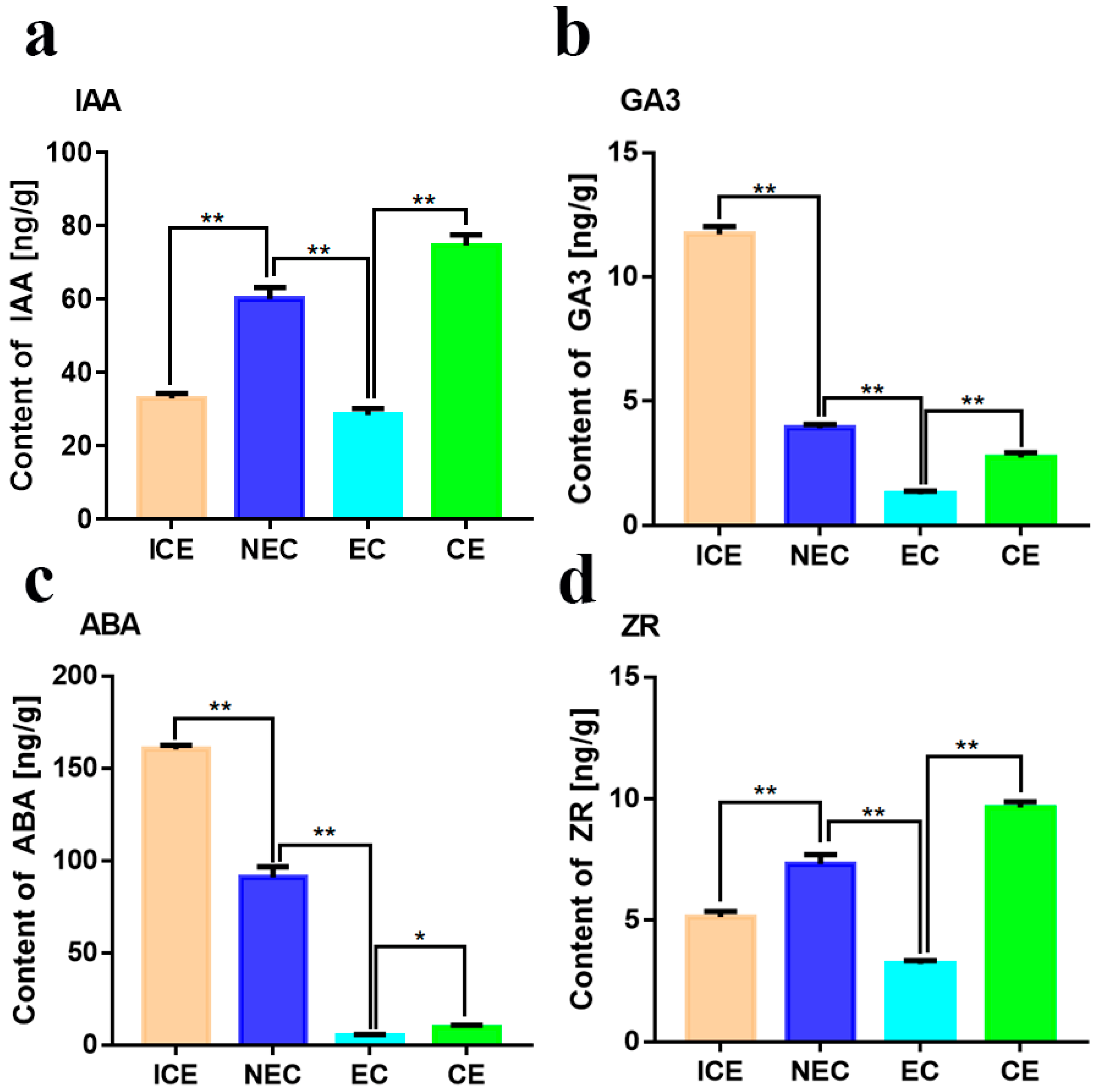
4.7. Indirect Competitive Enzyme-Linked Immunoassay (icELISA) for Detection of Indole-3-Acetic Acid (IAA), Gibberellin (GA3), Zeatin Riboside (ZR) and Abscisic Acid (ABA)
4.8. Quantitative RT-PCR
4.9. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zimmerman, J.L. Somatic embryogenesis: A model for early development in higher plants. Plant Cell 1993, 5, 1411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, X.M.; Kou, S.J.; Liu, Y.L.; Fang, Y.N.; Xu, Q.; Guo, W.W. Genomewide analysis of small RNAs in nonembryogenic and embryogenic tissues of citrus: MicroRNA- and siRNA-mediated transcript cleavage involved in somatic embryogenesis. Plant Biotechnol. J. 2015, 13, 383–394. [Google Scholar] [CrossRef]
- Altamura, M.M.; Della Rovere, F.; Fattorini, L.; D’Angeli, S.; Falasca, G. Recent Advances on Genetic and Physiological Bases of In Vitro Somatic Embryo Formation. Methods Mol. Biol. 2016, 1359, 47–85. [Google Scholar] [PubMed]
- Feher, A.; Pasternak, T.P.; Dudits, D. Transition of somatic plant cells to an embryogenic state. Plant Cell Tissue Organ Cult. 2003, 74, 201–228. [Google Scholar] [CrossRef]
- Zhao, X.Y.; Su, Y.H.; Cheng, Z.J.; Zhang, X.S. Cell fate switch during in vitro plant organogenesis. J. Integr Plant Biol. 2008, 50, 816–824. [Google Scholar] [CrossRef]
- Mantiri, F.R.; Kurdyukov, S.; Lohar, D.P.; Sharopova, N.; Saeed, N.A.; Wang, X.D.; Vandenbosch, K.A.; Rose, R.J. The transcription factor MtSERF1 of the ERF subfamily identified by transcriptional profiling is required for somatic embryogenesis induced by auxin plus cytokinin in Medicago truncatula. Plant Physiol. 2008, 146, 1622–1636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Z.; Andersen, S.U.; Ljung, K.; Dolezal, K.; Miotk, A.; Schultheiss, S.J.; Lohmann, J.U. Hormonal control of the shoot stem-cell niche. Nature 2010, 465, 1089–1092. [Google Scholar] [CrossRef] [PubMed]
- Arnholdt-Schmitt, B. Physiological aspects of genome variability in tissue culture. II. Growth phase-dependent quantitative variability of repetitive BstNI fragments of primary cultures of Daucus carota L. Theor. Appl. Genet. 1995, 91, 816–823. [Google Scholar] [CrossRef]
- Thibaud-Nissen, F.; Shealy, R.T.; Khanna, A.; Vodkin, L.O. Clustering of microarray data reveals transcript patterns associated with somatic embryogenesis in soybean. Plant Physiol. 2003, 132, 118–136. [Google Scholar] [CrossRef] [Green Version]
- Sharma, S.K.; Millam, S.; Hein, I.; Bryan, G.J. Cloning and molecular characterisation of a potato SERK gene transcriptionally induced during initiation of somatic embryogenesis. Planta 2008, 228, 319–330. [Google Scholar] [CrossRef] [PubMed]
- Quiroz-Figueroa, F.R.; Rojas-Herrera, R.; Galaz-Avalos, R.M.; Loyola-Vargas, V.M. Embryo production through somatic embryogenesis can be used to study cell differentiation in plants. Plant Cell Tissue Organ Cult. 2006, 86, 285–301. [Google Scholar] [CrossRef]
- Gliwicka, M.; Nowak, K.; Balazadeh, S.; Mueller-Roeber, B.; Gaj, M.D. Extensive modulation of the transcription factor transcriptome during somatic embryogenesis in Arabidopsis thaliana. PLoS ONE 2013, 8, e69261. [Google Scholar] [CrossRef] [Green Version]
- Boutilier, K.; Offringa, R.; Sharma, V.K.; Kieft, H.; Ouellet, T.; Zhang, L.; Hattori, J.; Liu, C.M.; van Lammeren, A.A.; Miki, B.L.; et al. Ectopic expression of BABY BOOM triggers a conversion from vegetative to embryonic growth. Plant Cell 2002, 4, 1737–1749. [Google Scholar] [CrossRef] [Green Version]
- Zuo, J.; Niu, Q.W.; Frugis, G.; Chua, N.H. The WUSCHEL gene promotes vegetative-to-embryonic transition in Arabidopsis. Plant J. 2002, 30, 349–359. [Google Scholar] [CrossRef] [PubMed]
- Gaj, M.D.; Zhang, S.; Harada, J.J.; Lemaux, P.G. Leafy cotyledon genes are essential for induction of somatic embryogenesis of Arabidopsis. Planta 2005, 222, 977–988. [Google Scholar] [CrossRef] [PubMed]
- Chen, X. Small RNAs and their roles in plant development. Annu. Rev. Cell Dev. Biol. 2009, 25, 21–44. [Google Scholar] [CrossRef] [Green Version]
- Yakovlev, I.A.; Fossdal, C.G.; Johnsen, Ø. MicroRNAs, the epigenetic memory and climatic adaptation in Norway spruce. New Phytol. 2010, 187, 1154–1169. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Chen, J.; Qiu, S.; Zhang, Y.; Wang, P.; Yang, L.; Lu, Y.; Shi, J. Deep sequencing and microarray hybridization identify conserved and species-specific microRNAs during somatic embryogenesis in hybrid yellow poplar. PLoS ONE 2012, 7, e43451. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.C.; Zhou, H.; Li, Y.; Chen, J.Y.; Yang, J.H.; Chen, Y.Q.; Qu, L.H. Rice embryogenic calli express a unique set of microRNAs, suggesting regulatory roles of microRNAs in plant post-embryogenic development. FEBS Lett. 2006, 580, 5111–5116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gou, J.Y.; Felippes, F.F.; Liu, C.J.; Weigel, D.; Wang, J.W. Negative regulation of anthocyanin biosynthesis in Arabidopsis by a miR156-targeted SPL transcription factor. Plant Cell 2011, 23, 1512–1522. [Google Scholar] [CrossRef] [Green Version]
- Tang, W.; Newton, R.J. Genome-wide expression analysis of genes involved in somatic embryogenesis. Somat. Embryog. 2005, 2, 69–83. [Google Scholar]
- Ye, C.Y.; Xu, H.; Shen, E.; Liu, Y.; Wang, Y.; Shen, Y.; Qiu, J.; Zhu, Q.H.; Fan, L. Genome-wide identification of non-coding RNAs interacted with microRNAs in soybean. Front. Plant Sci. 2014, 23, 743. [Google Scholar] [CrossRef]
- Jing, D.; Zhang, J.; Xia, Y.; Kong, L.; OuYang, F.; Zhang, S.; Zhang, H.; Wang, J. Proteomic analysis of stress-related proteins and metabolic pathways in Picea asperata somatic embryos during partial desiccation. Plant Biotechnol. J. 2017, 15, 27–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saunders, J.W.; Bingham, E.T. Production of Alfalfa Plants from Callus Tissue 1. Crop Sci. 1972, 12, 804–808. [Google Scholar] [CrossRef]
- Imin, N.; Nizamidin, M.; Daniher, D.; Nolan, K.E.; Rose, R.J.; Rolfe, B.G. Proteomic analysis of somatic embryogenesis in Medicago truncatula. Explant cultures grown under 6-benzylaminopurine and 1-naphthaleneacetic acid treatments. Plant Physiol. 2005, 137, 1250–1260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elhiti, M.; Stasolla, C.; Wang, A. Molecular regulation of plant somatic embryogenesis. Vitr. Cell. Dev. Biol.-Plant 2013, 49, 631–642. [Google Scholar] [CrossRef]
- Zhang, J.; Xue, B.; Gai, M.; Song, S.; Jia, N.; Sun, H. Small RNA and Transcriptome Sequencing Reveal a Potential miRNA-Mediated Interaction Network That Functions during Somatic Embryogenesis in Lilium pumilum DC. Fisch. Front Plant Sci. 2017, 8, 566. [Google Scholar] [CrossRef] [PubMed]
- Li, W.F.; Zhang, S.G.; Han, S.Y.; Wu, T.; Zhang, J.H.; Qi, L.W. Regulation of LaMYB33 by miR159 during maintenance of embryogenic potential and somatic embryo maturation in Larix kaempferi (Lamb.) Carr. Plant Cell Tissue Organ Cult. (PCTOC) 2013, 113, 131–136. [Google Scholar] [CrossRef]
- Reinert, J. Morphogenese und ihre Kontrolle an Gewebekulturen aus Carotten. Naturwissenschaften 1958, 45, 344–345. [Google Scholar] [CrossRef]
- Steward, F.C.; Mapes, M.O.; Smith, J. Growth and organized development of cultured cells. I. Growth and division of freely suspended cells. Am. J. Bot. 1958, 45, 693–703. [Google Scholar] [CrossRef]
- Galland, R.; Randoux, B.; Vasseur, J.; Hilbert, J.L. A glutathione S-transferase cDNA identified by mRNA differential display is upregulated during somatic embryogenesis in Cichorium. Biochim. Biophys. Acta 2001, 1522, 212–216. [Google Scholar] [CrossRef]
- Xu, H.; Gao, Y.; Wang, J. Transcriptomic analysis of rice (Oryza sativa) developing embryos using the RNA-Seq technique. PLoS ONE 2012, 7, e30646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldberg, R.B.; Barker, S.J.; Perez-Grau, L. Regulation of gene expression during plant embryogenesis. Cell 1989, 56, 149–160. [Google Scholar] [CrossRef]
- Bratzel, F.; López-Torrejón, G.; Koch, M.; Del Pozo, J.C.; Calonje, M. Keeping cell identity in Arabidopsis requires PRC1 RING-finger homologs that catalyze H2A monoubiquitination. Curr. Biol. 2010, 20, 1853–1859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hecht, V.; Vielle-Calzada, J.P.; Hartog, M.V.; Schmidt, E.D.; Boutilier, K.; Grossniklaus, U.; de Vries, S.C. The Arabidopsis SOMATIC EMBRYOGENESIS RECEPTOR KINASE 1 gene is expressed in developing ovules and embryos and enhances embryogenic competence in culture. Plant Physiol. 2001, 127, 803–816. [Google Scholar] [CrossRef] [PubMed]
- Puigderrajols, P.; Jofré, A.; Mir, G.; Pla, M.; Verdaguer, D.; Huguet, G.; Molinas, M. Developmentally and stress-induced small heat shock proteins in cork oak somatic embryos. J. Exp. Bot. 2002, 53, 1445–1452. [Google Scholar]
- Jiménez, V.M. Involvement of plant hormones and plant growth regulators on in vitro somatic embryogenesis. Plant Growth Regul. 2005, 47, 91–110. [Google Scholar] [CrossRef]
- Mashiguchi, K.; Tanaka, K.; Sakai, T.; Sugawara, S.; Kawaide, H.; Natsume, M.; Hanada, A.; Yaeno, T.; Shirasu, K.; Yao, H.; et al. The main auxin biosynthesis pathway in Arabidopsis. Proc. Natl. Acad. Sci. USA 2011, 108, 18512–18517. [Google Scholar] [CrossRef] [Green Version]
- Swarup, R.; Friml, J.; Marchant, A.; Ljung, K.; Sandberg, G.; Palme, K.; Bennett, M. Localization of the auxin permease AUX1 suggests two functionally distinct hormone transport pathways operate in the Arabidopsis root apex. Genes Dev. 2001, 15, 2648–2653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dharmasiri, N.; Dharmasiri, S.; Estelle, M. The F-box protein TIR1 is an auxin receptor. Nature 2005, 435, 441–445. [Google Scholar] [CrossRef]
- Zenk, M.H.; Müller, G. In vivo destruction of exogenously applied indolyl-3-acetic acid as influenced by naturally occurring phenolic acids. Nature 1963, 200, 761–763. [Google Scholar] [CrossRef]
- Hagen, G.; Guilfoyle, T. Auxin-responsive gene expression: Genes, promoters and regulatory factors. Plant Mol. Biol. 2002, 49, 373–385. [Google Scholar] [CrossRef]
- Abe, H.; Uchiyama, M.; Sato, R. Isolation and identification of native auxins in marine algae. Agric. Biol. Chem. 1972, 36, 2259–2260. [Google Scholar] [CrossRef]
- Wang, H.; Jones, B.; Li, Z.; Frasse, P.; Delalande, C.; Regad, F.; Chaabouni, S.; Latché, A.; Pech, J.C.; Bouzayen, M. The tomato Aux/IAA transcription factor IAA9 is involved in fruit development and leaf morphogenesis. Plant Cell 2005, 17, 2676–2692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez-Rizzo, S.; Crespi, M.; Frugier, F. The Medicago truncatula CRE1 cytokinin receptor regulates lateral root development and early symbiotic interaction with Sinorhizobium meliloti. Plant Cell 2006, 18, 2680–2693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tun, N.N.; Livaja, M.; Kieber, J.J.; Scherer, G.F. Zeatin-induced nitric oxide (NO) biosynthesis in Arabidopsis thaliana mutants of NO biosynthesis and of two-component signaling genes. New Phytol. 2008, 178, 515–531. [Google Scholar] [CrossRef] [PubMed]
- Argueso, C.T.; Ferreira, F.J.; Kieber, J.J. Environmental perception avenues: The interaction of cytokinin and environmental response pathways. Plant Cell Environ. 2009, 32, 1147–1160. [Google Scholar] [CrossRef] [PubMed]
- Hwang, I.; Sheen, J. Two-component circuitry in Arabidopsis cytokinin signal transduction. Nature 2001, 413, 383–389. [Google Scholar] [CrossRef]
- Papon, N.; Vansiri, A.; Gantet, P.; Chénieux, J.C.; Rideau, M.; Crèche, J. Histidine-containing phosphotransfer domain extinction by RNA interference turns off a cytokinin signalling circuitry in Catharanthus roseus suspension cells. FEBS Lett. 2004, 558, 85–88. [Google Scholar] [CrossRef] [Green Version]
- Ferreira, F.J.; Kieber, J.J. Cytokinin signaling. Curr. Opin. Plant Biol. 2005, 8, 518–525. [Google Scholar] [CrossRef] [PubMed]
- To, J.P.; Haberer, G.; Ferreira, F.J.; Deruère, J.; Mason, M.G.; Schaller, G.E.; Alonso, J.M.; Ecker, J.R.; Kieber, J.J. Type-A Arabidopsis response regulators are partially redundant negative regulators of cytokinin signaling. Plant Cell. 2004, 16, 658–671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Lucas, M.; Davière, J.M.; Rodríguez-Falcón, M.; Pontin, M.; Iglesias-Pedraz, J.M.; Lorrain, S.; Fankhauser, C.; Blázquez, M.A.; Titarenko, E.; Prat, S. A molecular framework for light and gibberellin control of cell elongation. Nature 2008, 451, 480–484. [Google Scholar] [CrossRef]
- Schoonheim, P.J.; Costa Pereira, D.D.; De Boer, A.H. Dual role for 14-3-3 proteins and ABF transcription factors in gibberellic acid and abscisic acid signalling in barley (Hordeum vulgare) aleurone cells. Plant Cell Environ. 2009, 32, 439–447. [Google Scholar] [CrossRef] [PubMed]
- Spoel, S.H.; Koornneef, A.; Claessens, S.M.; Korzelius, J.P.; Van Pelt, J.A.; Mueller, M.J.; Buchala, A.J.; Métraux, J.P.; Brown, R.; Kazan, K.; et al. NPR1 modulates cross-talk between salicylate- and jasmonate-dependent defense pathways through a novel function in the cytosol. Plant Cell 2003, 15, 760–770. [Google Scholar] [CrossRef] [Green Version]
- Yoong, F.Y.; O’Brien, L.K.; Truco, M.J.; Huo, H.; Sideman, R.; Hayes, R.; Michelmore, R.W.; Bradford, K.J. Genetic Variation for Thermotolerance in Lettuce Seed Germination Is Associated with Temperature-Sensitive Regulation of ETHYLENE RESPONSE FACTOR1 (ERF1). Plant Physiol. 2016, 170, 472–488. [Google Scholar] [CrossRef] [Green Version]
- Fraga, H.P.; Vieira, L.D.; Puttkammer, C.C.; Dos Santos, H.P.; Garighan, J.A.; Guerra, M.P. Glutathione and abscisic acid supplementation influences somatic embryo maturation and hormone endogenous levels during somatic embryogenesis in Podocarpus lambertii Klotzsch ex Endl. Plant Sci. 2016, 253, 98–106. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.C.; Luan, S. ABA signal transduction at the crossroad of biotic and abiotic stress responses. Plant Cell Environ. 2012, 35, 53–60. [Google Scholar] [CrossRef]
- Soon, F.F.; Ng, L.M.; Zhou, X.E.; West, G.M.; Kovach, A.; Tan, M.H.; Suino-Powell, K.M.; He, Y.; Xu, Y.; Chalmers, M.J.; et al. Molecular mimicry regulates ABA signaling by SnRK2 kinases and PP2C phosphatases. Science 2012, 335, 85–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, G.; Park, M.Y.; Conway, S.R.; Wang, J.W.; Weigel, D.; Poethig, R.S. The sequential action of miR156 and miR172 regulates developmental timing in Arabidopsis. Cell 2009, 138, 750–759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lauter, N.; Kampani, A.; Carlson, S.; Goebel, M.; Moose, S.P. microRNA172 down-regulates glossy15 to promote vegetative phase change in maize. Proc. Natl. Acad. Sci. USA 2005, 102, 9412–9417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, Z.; Hossain, M.S.; Wang, J.; Valdés-López, O.; Liang, Y.; Libault, M.; Qiu, L.; Stacey, G. miR172 regulates soybean nodulation. Mol. Plant Microbe Interact. 2013, 26, 1371–1377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, L.; Kim, Y.; Dinh, T.T.; Chen, X. miR172 regulates stem cell fate and defines the inner boundary of APETALA3 and PISTILLATA expression domain in Arabidopsis floral meristems. Plant J. 2007, 51, 840–849. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Zhou, J.; Han, S.; Yang, W.; Li, W.; Wei, H.; Li, X.; Qi, L. Four abiotic stress-induced miRNA families differentially regulated in the embryogenic and non-embryogenic callus tissues of Larix leptolepis. Biochem. Biophys. Res. Commun. 2010, 398, 355–360. [Google Scholar] [CrossRef]
- Glazińska, P.; Zienkiewicz, A.; Wojciechowski, W.; Kopcewicz, J. The putative miR172 target gene InAPETALA2-like is involved in the photoperiodic flower induction of Ipomoea nil. J. Plant Physiol. 2009, 166, 1801–1813. [Google Scholar] [CrossRef]
- Wang, J.W.; Czech, B.; Weigel, D. miR156-regulated SPL transcription factors define an endogenous flowering pathway in Arabidopsis thaliana. Cell 2009, 138, 738–749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.W.; Schwab, R.; Czech, B.; Mica, E.; Weigel, D. Dual effects of miR156-targeted SPL genes and CYP78A5/KLUH on plastochron length and organ size in Arabidopsis thaliana. Plant Cell 2008, 20, 1231–1243. [Google Scholar] [CrossRef] [Green Version]
- Wu, G.; Poethig, R.S. Temporal regulation of shoot development in Arabidopsis thaliana by miR156 and its target SPL3. Development 2006, 133, 3539–3547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, H.; Hu, F.; Wang, R.; Zhou, X.; Sze, S.H.; Liou, L.W.; Barefoot, A.; Dickman, M.; Zhang, X. Arabidopsis Argonaute10 specifically sequesters miR166/165 to regulate shoot apical meristem development. Cell 2011, 145, 242–256. [Google Scholar] [CrossRef] [Green Version]
- Miyashima, S.; Honda, M.; Hashimoto, K.; Tatematsu, K.; Hashimoto, T.; Sato-Nara, K.; Okada, K.; Nakajima, K. A comprehensive expression analysis of the Arabidopsis MICRORNA165/6 gene family during embryogenesis reveals a conserved role in meristem specification and a non-cell-autonomous function. Plant Cell Physiol. 2013, 54, 375–384. [Google Scholar] [CrossRef] [Green Version]
- Jung, J.H.; Park, C.M. MIR166/165 genes exhibit dynamic expression patterns in regulating shoot apical meristem and floral development in Arabidopsis. Planta 2007, 225, 1327–1338. [Google Scholar] [CrossRef]
- Li, W.F.; Zhang, S.G.; Han, S.Y.; Wu, T.; Zhang, J.H.; Qi, L.W. The post-transcriptional regulation of LaSCL6 by miR171 during maintenance of embryogenic potential in Larix kaempferi (Lamb.) Carr. Tree Genet. Genomes 2014, 10, 223–229. [Google Scholar] [CrossRef]
- Ma, Z.; Hu, X.; Cai, W.; Huang, W.; Zhou, X.; Luo, Q.; Yang, H.; Wang, J.; Huang, J. Arabidopsis miR171-targeted scarecrow-like proteins bind to GT cis-elements and mediate gibberellin-regulated chlorophyll biosynthesis under light conditions. PLoS Genet. 2014, 10, e1004519. [Google Scholar]
- Huang, W.; Peng, S.; Xian, Z.; Lin, D.; Hu, G.; Yang, L.; Ren, M.; Li, Z. Overexpression of a tomato miR171 target gene SlGRAS24 impacts multiple agronomical traits via regulating gibberellin and auxin homeostasis. Plant Biotechnol. J. 2017, 15, 472–488. [Google Scholar] [CrossRef] [PubMed]
- Marin, E.; Jouannet, V.; Herz, A.; Lokerse, A.S.; Weijers, D.; Vaucheret, H.; Nussaume, L.; Crespi, M.D.; Maizel, A. miR390, Arabidopsis TAS3 tasiRNAs, and their AUXIN RESPONSE FACTOR targets define an autoregulatory network quantitatively regulating lateral root growth. Plant Cell 2010, 22, 1104–1117. [Google Scholar] [PubMed] [Green Version]
- Lu, C.; Tej, S.S.; Luo, S.; Haudenschild, C.D.; Meyers, B.C.; Green, P.J. Elucidation of the small RNA component of the transcriptome. Science 2005, 309, 1567–1569. [Google Scholar] [PubMed] [Green Version]
- German, M.A.; Pillay, M.; Jeong, D.H.; Hetawal, A.; Luo, S.; Janardhanan, P.; Kannan, V.; Rymarquis, L.A.; Nobuta, K.; German, R.; et al. Global identification of microRNA-target RNA pairs by parallel analysis of RNA ends. Nat. Biotechnol. 2008, 26, 941–946. [Google Scholar]
- Liszkay, A.; van der Zalm, E.; Schopfer, P. Production of reactive oxygen intermediates (O(2)(.-), H(2)O(2), and (.)OH) by maize roots and their role in wall loosening and elongation growth. Plant Physiol. 2004, 136, 3114–3123. [Google Scholar] [CrossRef] [Green Version]
- Saldanha, A.J. Java Treeview—Extensible visualization of microarray data. Bioinformatics 2004, 20, 3246–3248. [Google Scholar] [PubMed] [Green Version]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Gene ontology analysis for RNA-seq: Accounting for selection bias. Genome Biol. 2010, 11, R14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, J.; Li, G.; Wang, B.M.; Liu, W.; Nan, T.G.; Zhai, Z.X.; Li, Z.H.; Li, Q.X. Development of a monoclonal antibody-based enzyme-linked immunosorbent assay for the analysis of glycyrrhizic acid. Anal. Bioanal. Chem. 2006, 386, 1735–1740. [Google Scholar]
- Jin, M.; Ren, Y.; Chen, X. Determination of 6-benzyladenine in bean sprout by LC–ESI-IT-MS–MS. Chromatographia 2007, 66, 407–410. [Google Scholar] [CrossRef]
- Goidin, D.; Mamessier, A.; Staquet, M.J.; Schmitt, D.; Berthier-Vergnes, O. Ribosomal 18S RNA prevails over glyceraldehyde-3-phosphate dehydrogenase and beta-actin genes as internal standard for quantitative comparison of mRNA levels in invasive and noninvasive human melanoma cell subpopulations. Anal. Biochem. 2001, 295, 17–21. [Google Scholar] [CrossRef] [Green Version]
- Miyagishi, M.; Taira, K. U6 promoter-driven siRNAs with four uridine 3′ overhangs efficiently suppress targeted gene expression in mammalian cells. Nat. Biotechnol. 2002, 20, 497–500. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Xu, J.; Liu, Y.; Zhao, X.; Deng, X.; Guo, L.; Gu, J. A novel bud mutation that confers abnormal patterns of lycopene accumulation in sweet orange fruit (Citrus sinensis L. Osbeck). J. Exp. Bot. 2007, 58, 4161–4171. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Raw Reads | Clean Reads | Clean Bases | Error (%) | Q20 (%) | Q30 (%) | GC (%) |
|---|---|---|---|---|---|---|---|
| ICE_1 | 52,384,258 | 51,607,148 | 7.74G | 0.01 | 97.75 | 94.00 | 41.28 |
| ICE_2 | 58,306,722 | 56,696,602 | 8.5G | 0.02 | 97.20 | 92.69 | 41.30 |
| ICE_3 | 50,810,168 | 50,103,162 | 7.52G | 0.01 | 97.86 | 94.21 | 41.34 |
| NEC_1 | 57,200,884 | 55,844,904 | 8.38G | 0.01 | 97.89 | 94.30 | 41.60 |
| NEC_2 | 60,219,472 | 56,035,448 | 8.41G | 0.02 | 96.97 | 92.05 | 41.86 |
| NEC_3 | 53,763,200 | 52,838,680 | 7.93G | 0.01 | 97.97 | 94.50 | 41.60 |
| EC_1 | 52,977,216 | 51,478,324 | 7.72G | 0.01 | 97.97 | 94.51 | 41.45 |
| EC_2 | 55,361,562 | 54,341,900 | 8.15G | 0.01 | 98.02 | 94.63 | 41.39 |
| EC_3 | 57,268,222 | 55,969,446 | 8.4G | 0.01 | 97.64 | 93.76 | 41.79 |
| CE_1 | 63,997,186 | 62,745,116 | 9.41G | 0.02 | 97.47 | 93.47 | 41.93 |
| CE_2 | 50,846,076 | 49,993,036 | 7.5G | 0.02 | 97.49 | 93.49 | 41.95 |
| CE_3 | 55,045,608 | 54,018,626 | 8.1G | 0.02 | 97.44 | 93.39 | 41.65 |
| Sample | ICE | NEC | EC | CE |
|---|---|---|---|---|
| total reads | 14,669,757 (100.00%) | 14,696,331 (100.00%) | 11,817,247 (100.00%) | 10,282,021 (100.00%) |
| N% > 10% | 18 (0.00%) | 25 (0.00%) | 0 (0.00%) | 0 (0.00%) |
| low quality | 753,071 (5.13%) | 1,954,044 (13.30%) | 488,075 (4.13%) | 225,087 (2.19%) |
| 5 adapter contamine | 23,386 (0.16%) | 16,804 (0.11%) | 9703 (0.08%) | 11,016 (0.11%) |
| 3 adapter null or insert null | 649,842 (4.43%) | 316,352 (2.15%) | 212,613 (1.80%) | 189,171 (1.84%) |
| With ployA/T/G/C | 39,873 (0.27%) | 55,440 (0.38%) | 35,208 (0.30%) | 32,043 (0.31%) |
| clean reads | 13,203,567 (90.01%) | 12,353,666 (84.06%) | 11,071,648 (93.69%) | 9,824,704 (95.55%) |
| Types | ICE | NEC | EC | CE |
|---|---|---|---|---|
| known miRNA | 740,026 (12.40%) | 302,053 (5.92%) | 299,136 (6.74%) | 479,121 (11.25%) |
| novel miRNA | 132,783 (2.23%) | 63,563 (1.25%) | 34,189 (0.77%) | 46,934 (1.10%) |
| rRNA | 432,250 (7.24%) | 121,355 (2.38%) | 252,580 (5.69%) | 149,802 (3.52%) |
| tRNA | 3 (0.00%) | 0 | 1 (0.00%) | 0 |
| snRNA | 3988 (0.07%) | 2290 (0.04%) | 5984 (0.13%) | 2902 (0.07%) |
| snoRNA | 9607 (0.16%) | 23,276 (0.46%) | 22,688 (0.51%) | 15,722 (0.37%) |
| TAS | 19,322 (0.32%) | 31,369 (0.61%) | 25,992 (0.59%) | 132,783 (0.46%) |
| other | 4,629,365 (77.58%) | 4,560,608 (89.34%) | 25,992 (0.59%) | 19,645 (0.46%) |
| total | 5,967,344 | 5,104,514 | 4,436,671 | 4,257,086 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yuan, J.; Chao, Y.; Han, L. Uncovering a Phenomenon of Active Hormone Transcriptional Regulation during Early Somatic Embryogenesis in Medicago sativa. Int. J. Mol. Sci. 2022, 23, 8633. https://doi.org/10.3390/ijms23158633
Yuan J, Chao Y, Han L. Uncovering a Phenomenon of Active Hormone Transcriptional Regulation during Early Somatic Embryogenesis in Medicago sativa. International Journal of Molecular Sciences. 2022; 23(15):8633. https://doi.org/10.3390/ijms23158633
Chicago/Turabian StyleYuan, Jianbo, Yuehui Chao, and Liebao Han. 2022. "Uncovering a Phenomenon of Active Hormone Transcriptional Regulation during Early Somatic Embryogenesis in Medicago sativa" International Journal of Molecular Sciences 23, no. 15: 8633. https://doi.org/10.3390/ijms23158633
APA StyleYuan, J., Chao, Y., & Han, L. (2022). Uncovering a Phenomenon of Active Hormone Transcriptional Regulation during Early Somatic Embryogenesis in Medicago sativa. International Journal of Molecular Sciences, 23(15), 8633. https://doi.org/10.3390/ijms23158633