
Four Cholesterol-Recognition Motifs in the Pore-Forming and Translocation Domains of Adenylate Cyclase Toxin Are Essential for Invasion of Eukaryotic Cells and Lysis of Erythrocytes


Abstract
:1. Introduction
2. Results
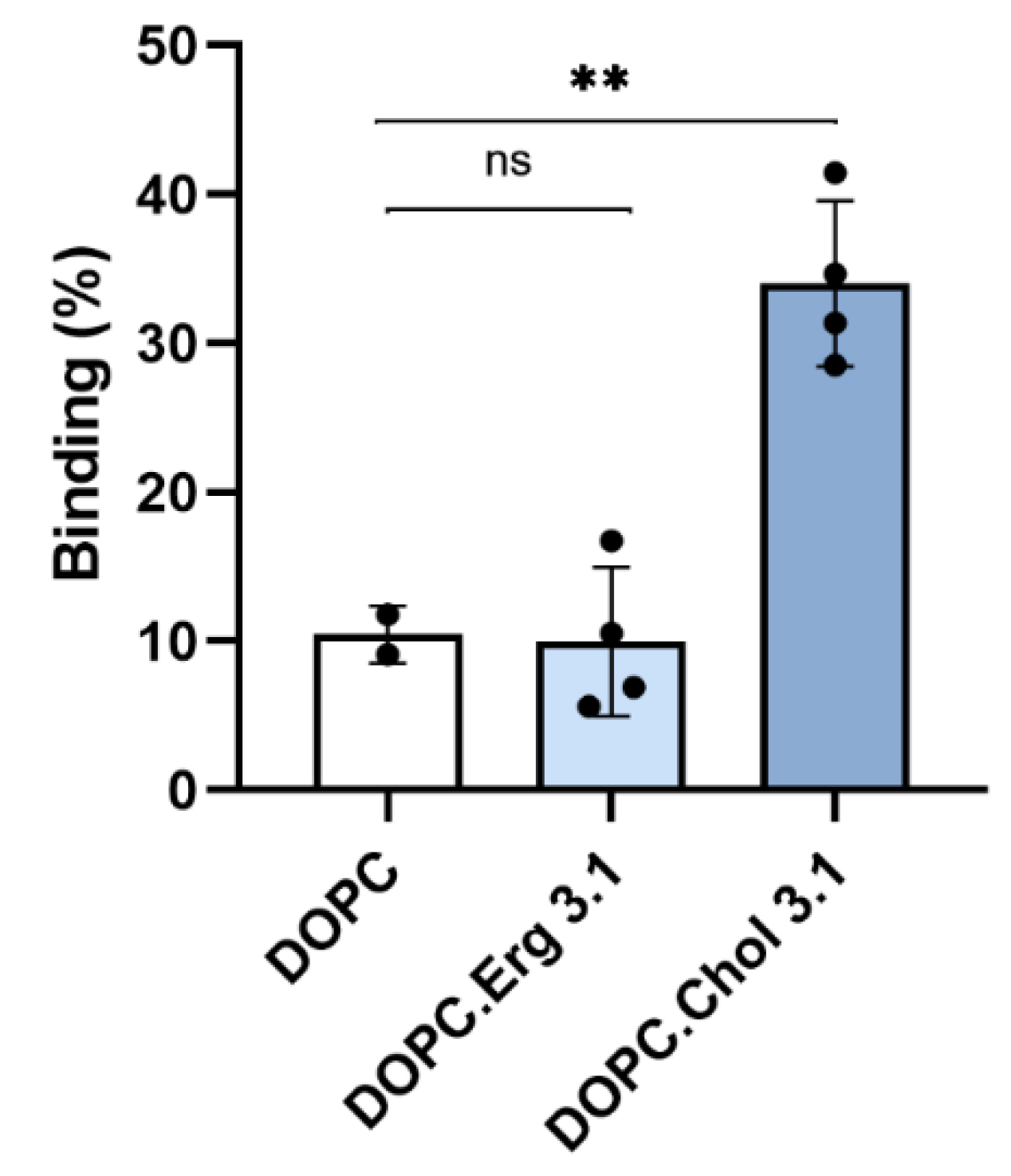
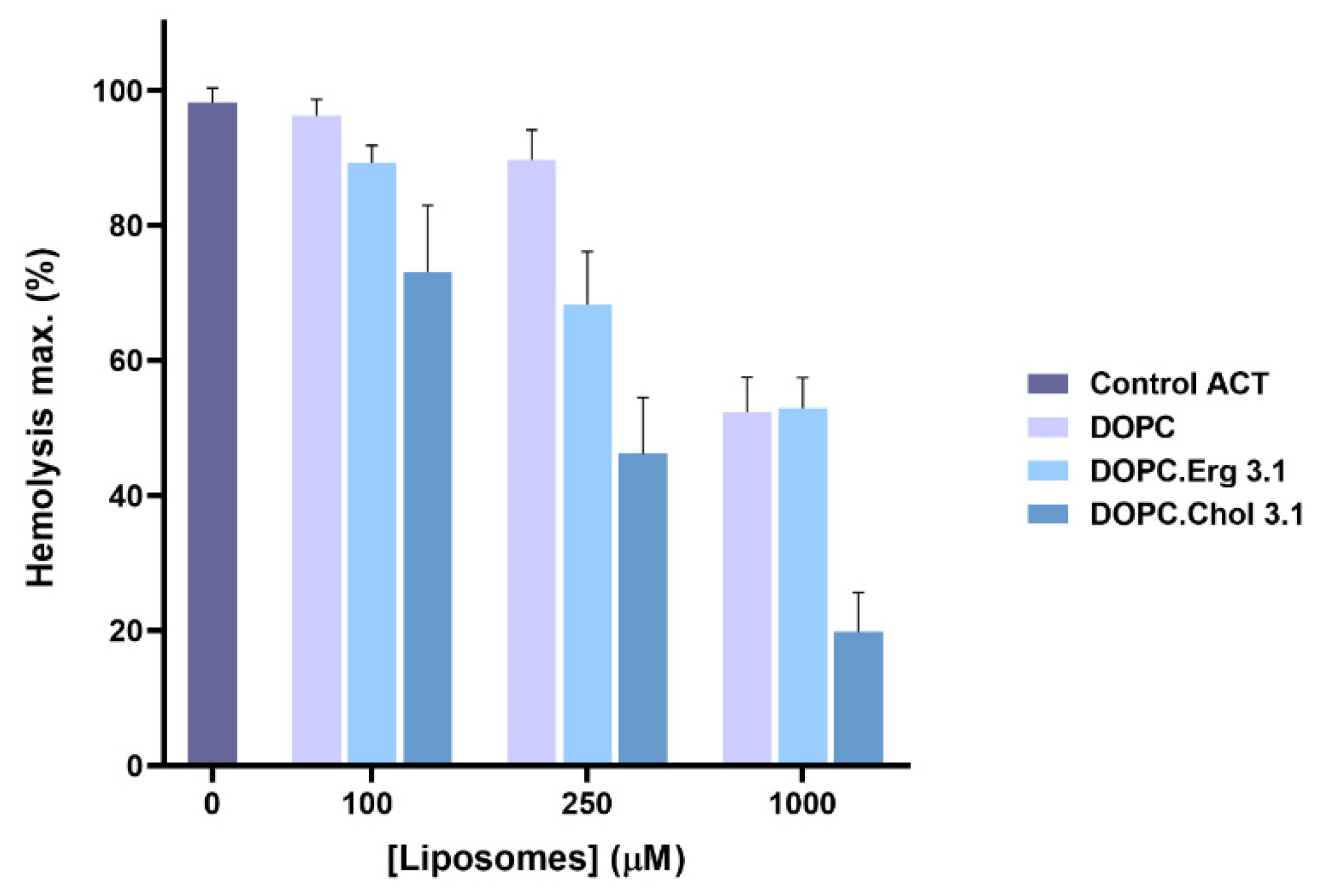
2.1. Specificity in the Interaction of ACT with Cholesterol
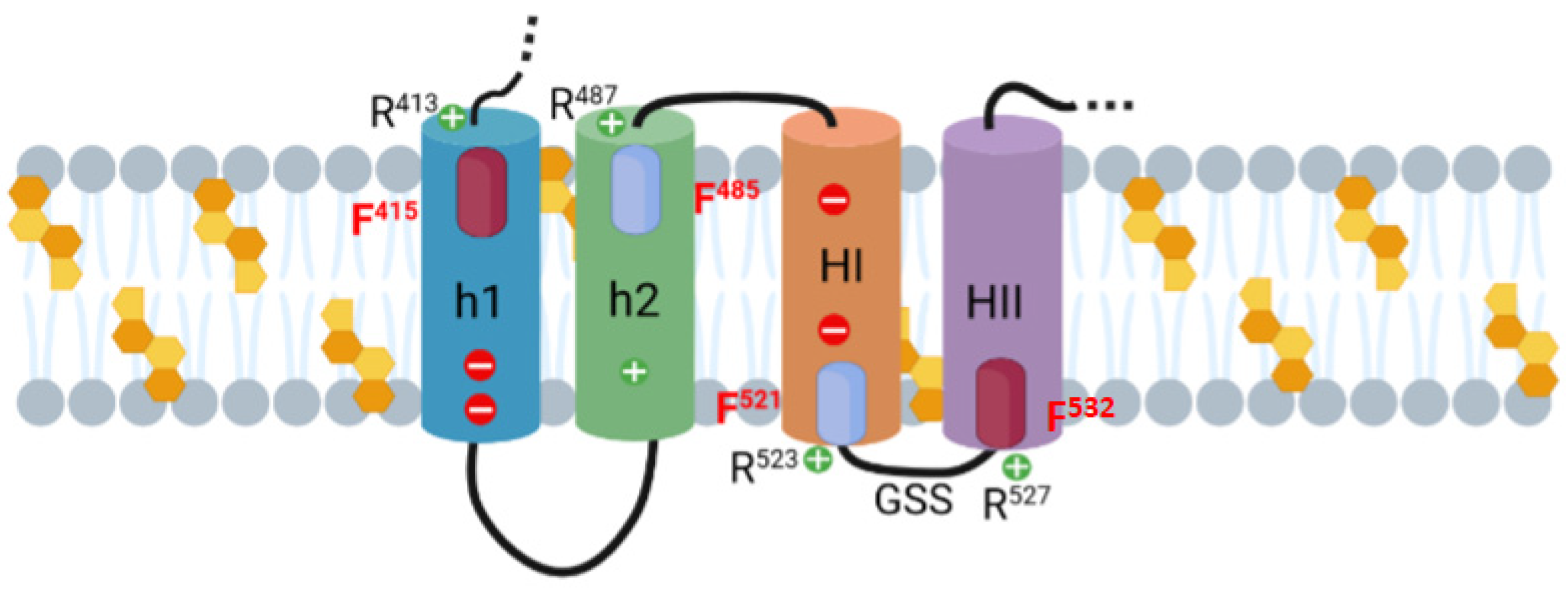
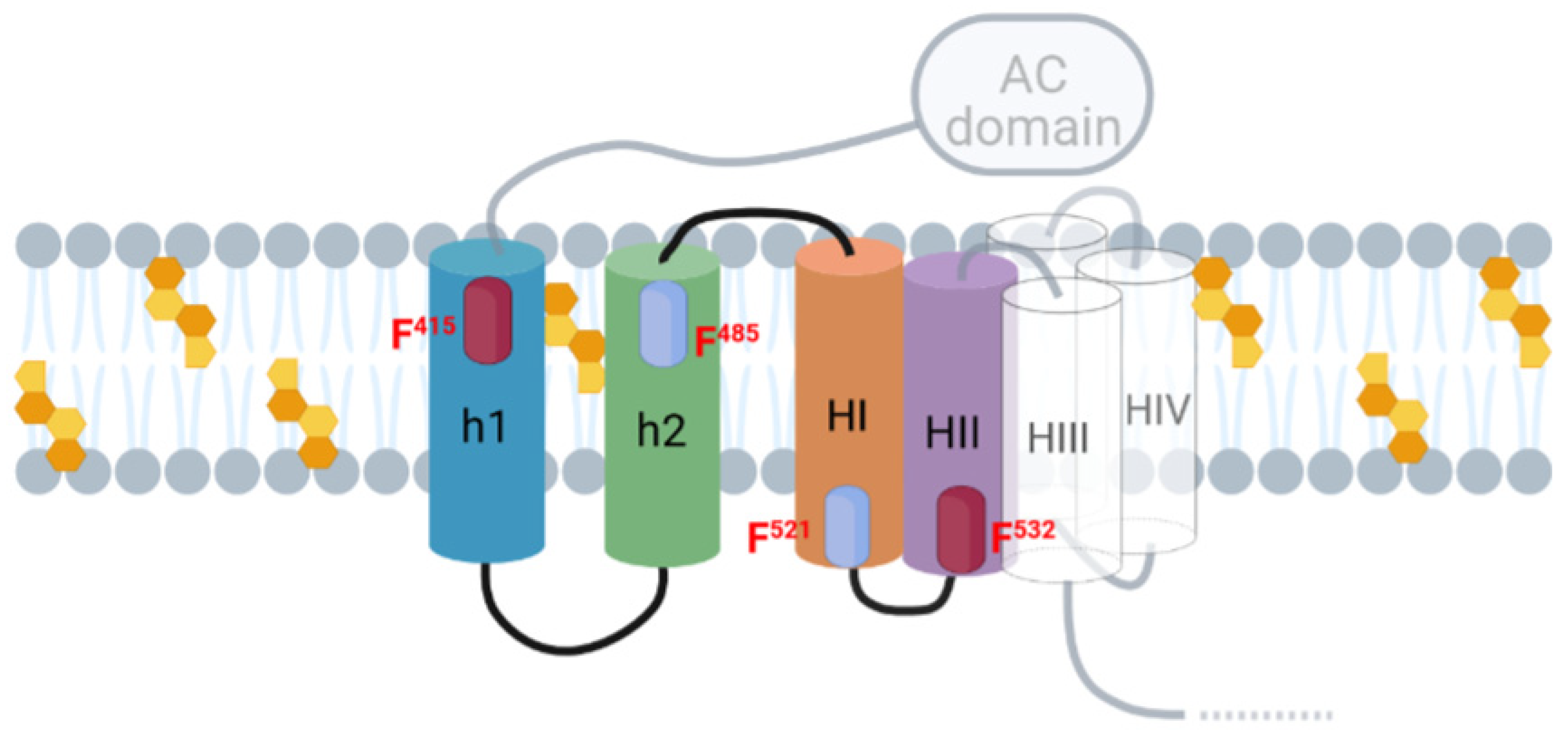
2.2. Potential Cholesterol-Recognition Motifs Can Be Identified in the Sequence of the Membrane-Interacting Translocation Region and Hydrophobic Domain of ACT
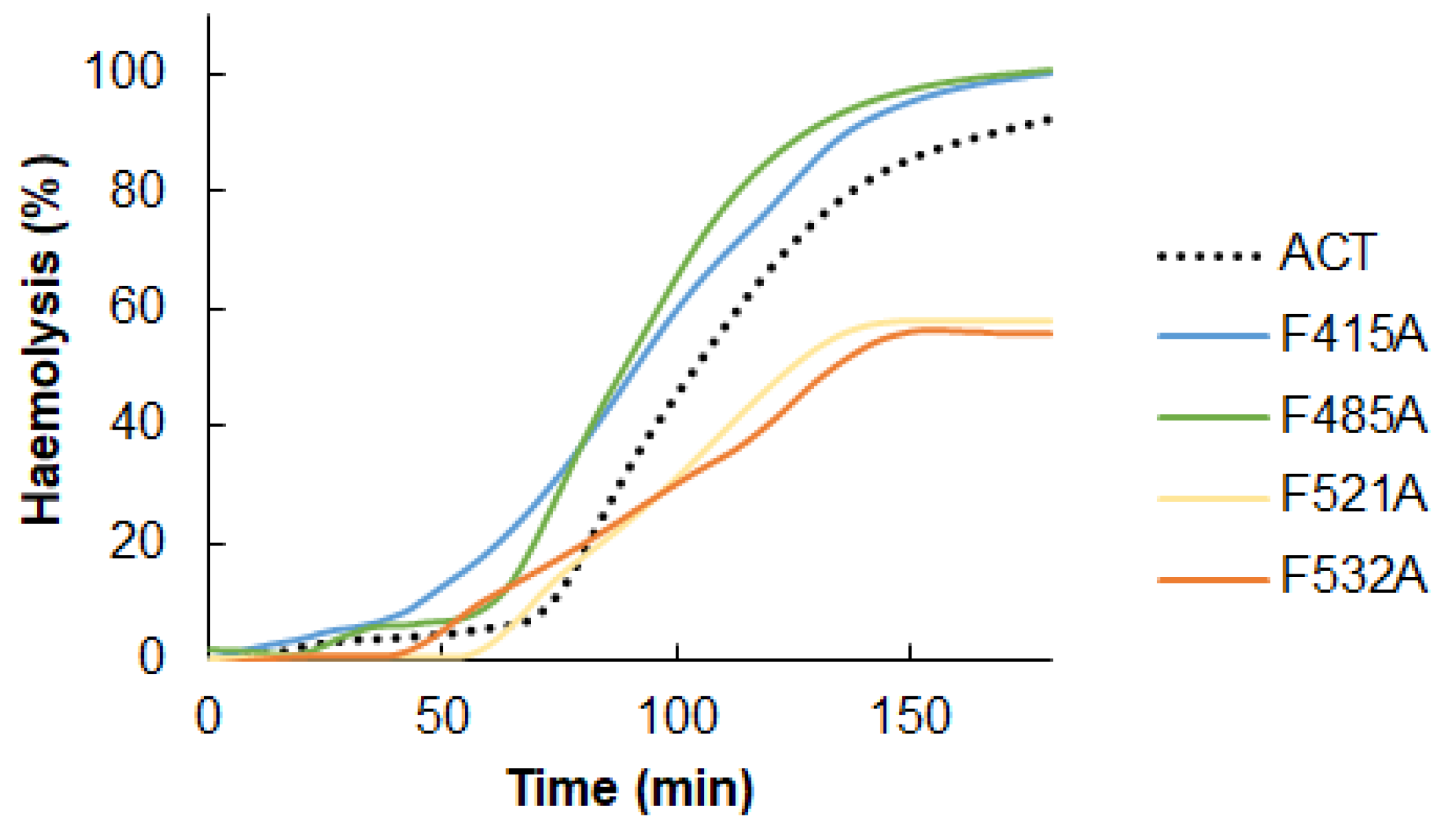
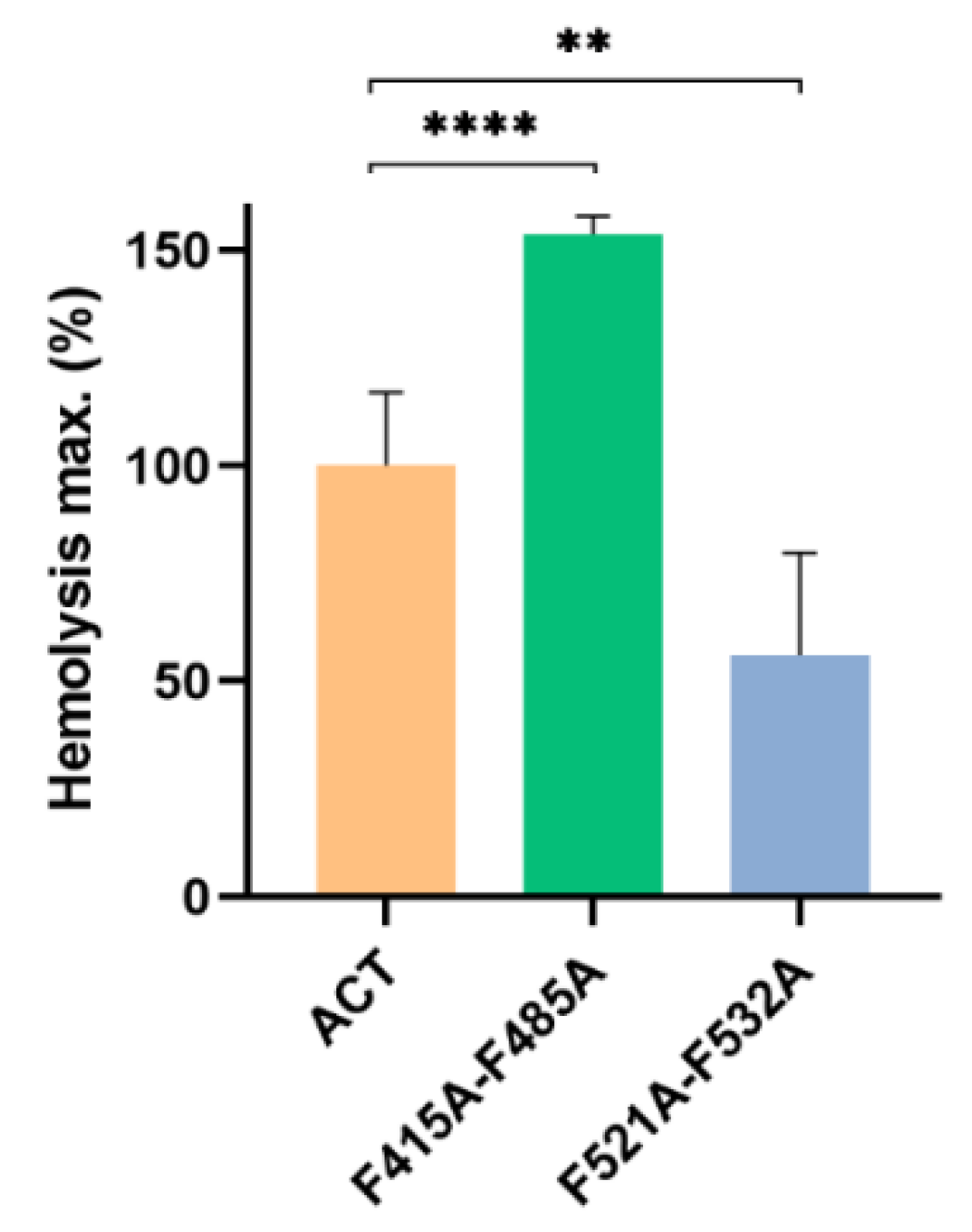
2.3. Point Mutation of the Central Phe Residue in the CRAC and CARC Motifs Have a Differentiated Effect on the ACT-Induced Haemolysis
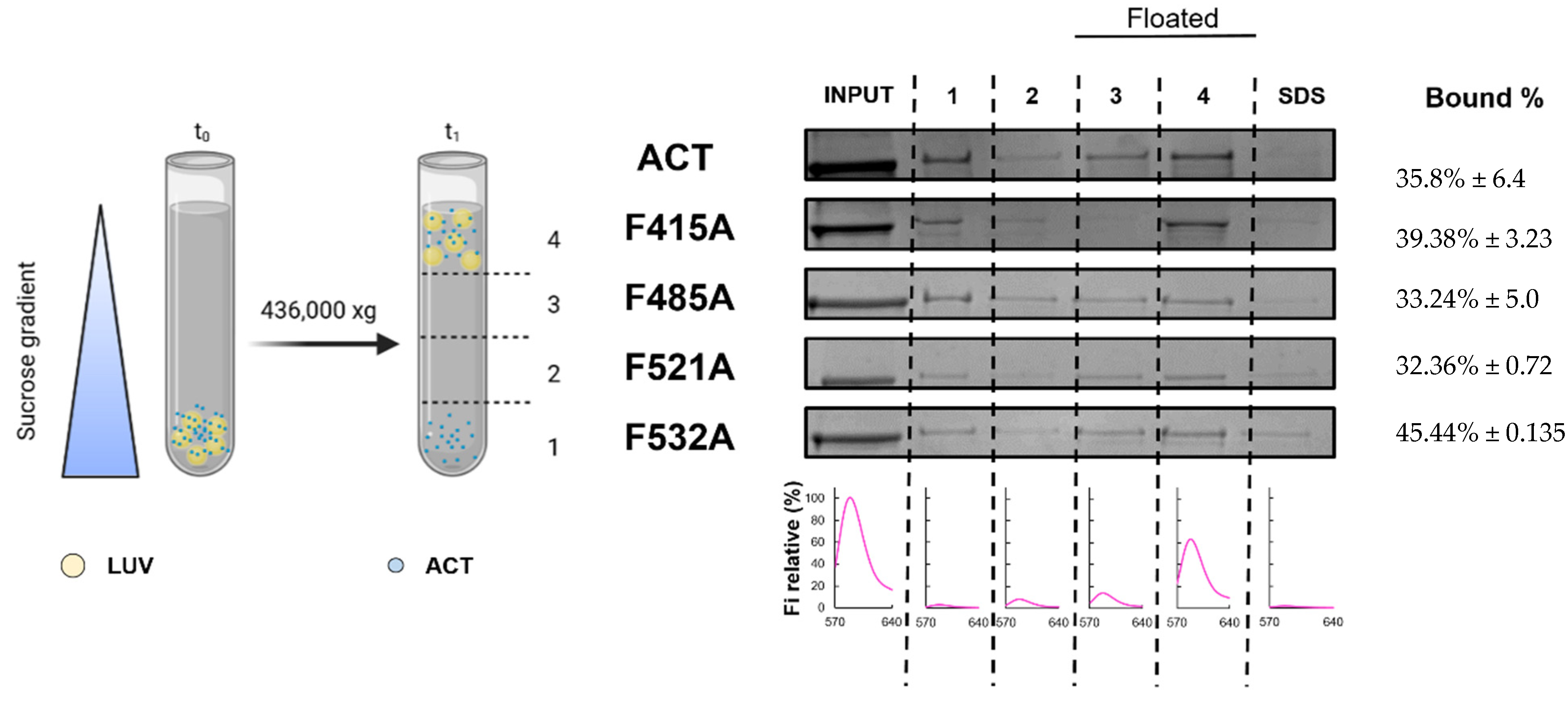
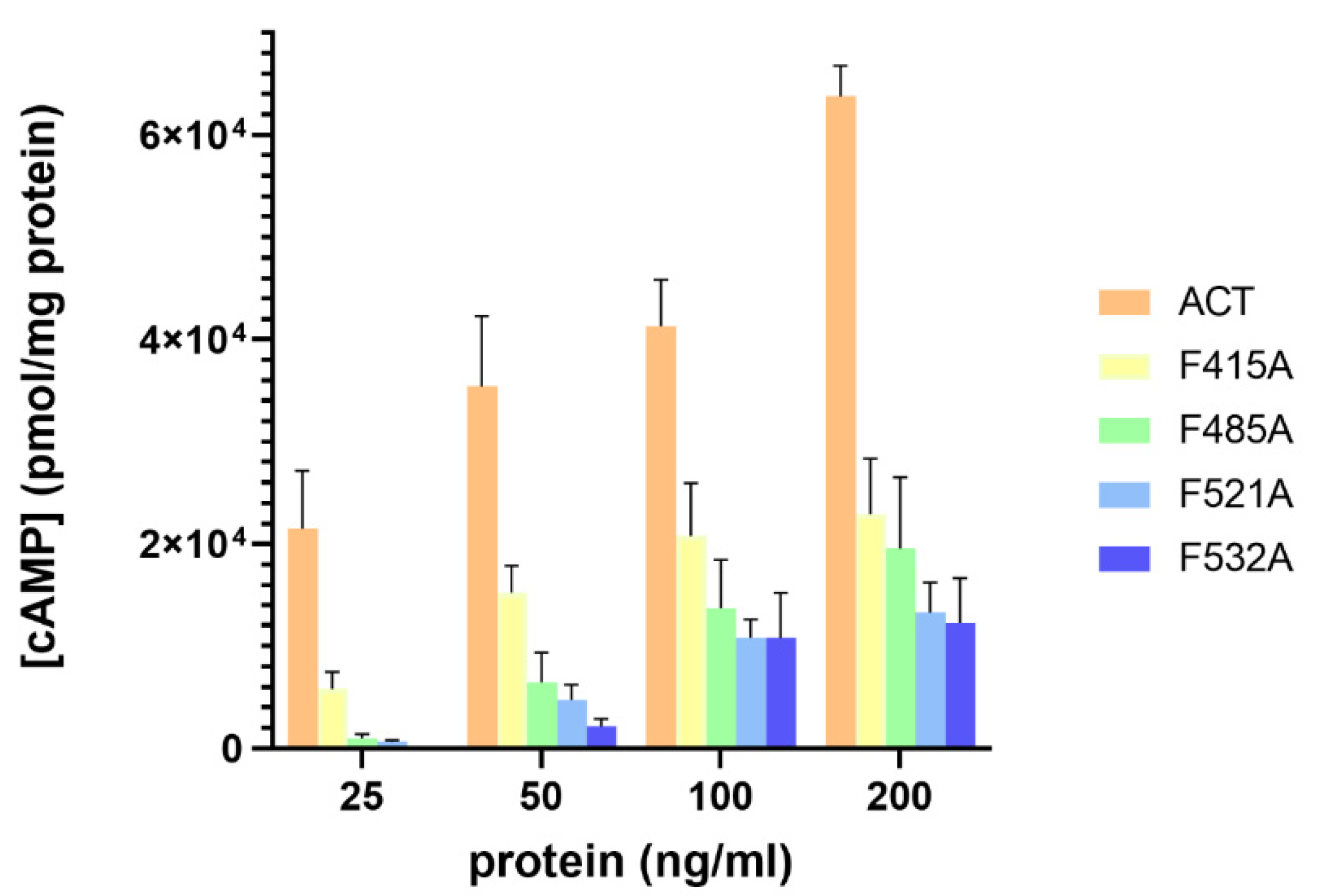
2.4. Substitutions by Ala of the Central Phe in 415, 485, 521 and 532 Positions, in the Respective Cholesterol-Recognition Motifs of ACT, Inhibit Prominently AC Domain Translocation
3. Discussion
4. Materials and Methods
4.1. Expression and Purification of Intact ACT
4.2. Construction, Expression and Purification of the ACT Mutants F415A, F485A, F521A and F532A
4.3. Haemolysis Assay
4.4. Cell Culture
4.5. Measurement of cAMP
4.6. Measurement of ACT or Mutant Toxins Binding to Lipid Membranes Determined by Flotation Assays
Author Contributions
Funding
Conflicts of Interest
References
- Parker, M.W.; Feil, S.C. Pore-forming protein toxins: From structure to function. Prog. Biophys. Mol. Biol. 2005, 88, 91–142. [Google Scholar] [CrossRef] [PubMed]
- Dal Peraro, M.; van der Goot, F.G. Pore-forming toxins: Ancient, but never really out of fashion. Nat. Rev. Microbiol. 2016, 14, 77–92. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, R.J.C.; Dalla Serra, M.; Froelich, C.J.; Wallace, M.I.; Anderluh, G. Membrane pore formation at protein-lipid interfaces. Trends Biochem. Sci. 2014, 39, 510–516. [Google Scholar] [CrossRef] [PubMed]
- Tilley, S.J.; Saibil, H.R. The mechanism of pore formation by bacterial toxins. Curr. Opin. Struct. Biol. 2006, 16, 230–236. [Google Scholar] [CrossRef] [PubMed]
- Janmey, P.A.; Kinnunen, P.K. Biophysical properties of lipids and dynamic membranes. Trends Cell Biol. 2006, 16, 538–546. [Google Scholar] [CrossRef]
- Escriba, P.V.; Gonzalez-Ros, J.M.; Goñi, F.M.; Kinnunen, P.K.; Vigh, L.; Sanchez-Magraner, L.; Barcelo-Coblijn, G. Membranes: A meeting point for lipids, proteins and therapies. J. Cell Mol. Med. 2008, 12, 829–875. [Google Scholar] [CrossRef] [Green Version]
- Salas-Estrada, L.A.; Leioatts, N.; Romo, T.D.; Grossfield, A. Lipids Alter Rhodopsin Function via Ligand-like and Solvent-like Interactions. Biophys. J. 2018, 114, 355–367. [Google Scholar] [CrossRef] [Green Version]
- Sych, T.; Levental, K.R.; Sezgin, E. Lipid-Protein Interactions in Plasma Membrane Organization and Function. Annu. Rev. Biophys. 2022, 51, 135–156. [Google Scholar] [CrossRef]
- Renard, K.; Byrne, B. Insights into the Role of Membrane Lipids in the Structure, Function and Regulation of Integral Membrane Proteins. Int. J. Mol. Sci. 2021, 22, 9026. [Google Scholar] [CrossRef]
- Laganowsky, A.; Reading, E.; Allison, T.M.; Ulmschneider, M.B.; Degiacomi, M.T.; Baldwin, A.J.; Robinson, C.V. Membrane Proteins Bind Lipids Selectively to Modulate Their Structure and Function. Nature 2014, 510, 172–175. [Google Scholar] [CrossRef] [Green Version]
- Carbonetti, N.H. Immunomodulation in the pathogenesis of Bordetella pertussis infection and disease. Curr. Opin. Pharmacol. 2007, 7, 272–278. [Google Scholar] [CrossRef] [PubMed]
- Melvin, J.A.; Scheller, E.V.; Miller, J.F.; Cotter, P.A. Bordetella pertussis pathogenesis: Current and future challenges. Nat. Rev. Microbiol. 2014, 12, 274–288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mattoo, S.; Cherry, J.D. Molecular pathogenesis, epidemiology, and clinical manifestations of respiratory infections due to Bordetella pertussis and other Bordetella subspecies. Clin. Microbiol. Rev. 2005, 18, 326–382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Welch, R.A. RTX toxin structure and function: A story of numerous anomalies and few analogies in toxin biology. Curr. Top. Microbiol. Immunol. 2001, 257, 85–111. [Google Scholar] [PubMed]
- Vojtova, J.; Kamanova, J.; Sebo, P. Bordetella adenylate cyclase toxin: A swift saboteur of host defense. Curr. Opin. Microbiol. 2006, 9, 69–75. [Google Scholar] [CrossRef]
- Confer, D.L.; Eaton, J.W. Phagocyte impotence caused by an invasive bacterial adenylate cyclase. Science 1982, 217, 948–950. [Google Scholar] [CrossRef]
- Wolff, J.; Cook, G.H.; Goldhammer, A.R.; Berkowitz, S.A. Calmodulin activates prokaryotic adenylate cyclase. Proc. Natl. Acad. Sci. USA 1980, 77, 3841–3844. [Google Scholar] [CrossRef] [Green Version]
- Ehrmann, I.E.; Gray, M.C.; Gordon, V.M.; Gray, L.S.; Hewlett, E.L. Hemolytic activity of adenylate cyclase toxin from Bordetella pertussis. FEBS Lett. 1991, 278, 79–83. [Google Scholar]
- Benz, R.; Maier, E.; Ladant, D.; Ullmann, A.; Sebo, P. Adenylate cyclase toxin (CyaA) of Bordetella pertussis. Evidence for the formation of small ion-permeable channels and comparison with HlyA of Escherichia coli. J. Biol. Chem. 1994, 269, 27231–27239. [Google Scholar] [CrossRef]
- González-Bullón, D.; Uribe, K.B.; Largo, E.; Guembelzu, G.; García-Arribas, A.B.; Martín, C.; Ostolaza, H. Membrane Permeabilization by Bordetella Adenylate Cyclase Toxin Involves Pores of Tunable Size. Biomolecules 2019, 9, 183. [Google Scholar] [CrossRef] [Green Version]
- Osickova, A.; Osicka, R.; Maier, E.; Benz, R.; Sebo, P. An amphipathic alpha-helix including glutamates 509 and 516 is crucial for membrane translocation of adenylate cyclase toxin and modulates formation and cation selectivity of its membrane channels. J. Biol. Chem. 1999, 274, 37644–37650. [Google Scholar] [CrossRef]
- Basler, M.; Knapp, O.; Masin, J.; Fiser, R.; Maier, E.; Benz, R.; Sebo, P.; Osicka, R. Segments crucial for membrane translocation and pore-forming activity of Bordetella adenylate cyclase toxin. J. Biol. Chem. 2007, 282, 12419–12429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Powthongchin, B.; Angsuthanasombat, C. Effects on haemolytic activity of single proline substitutions in the Bordetella pertussis CyaA pore-forming fragment. Arch. Microbiol. 2009, 191, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Juntapremjit, S.; Thamwiriyasati, N.; Kurehong, C.; Prangkio, P.; Shank, L.; Powthongchin, B.; Angsuthanasombat, C. Functional importance of the Gly cluster in transmembrane helix 2 of the Bordetella pertussis CyaA-hemolysin: Implications for toxin oligomerization and pore formation. Toxicon 2015, 106, 14–19. [Google Scholar] [CrossRef]
- Masin, J.; Osickova, A.; Sukova, A.; Fiser, R.; Halada, P.; Bumba, L.; Linhartova, I.; Osicka, R.; Sebo, P. Negatively charged residues of the segment linking the enzyme and cytolysin moieties restrict the membrane-permeabilizing capacity of adenylate cyclase toxin. Sci. Rep. 2016, 6, 29137. [Google Scholar] [CrossRef] [Green Version]
- Prangkio, P.; Juntapremjit, S.; Koehler, M.; Hinterdorfer, P.; Angsuthanasombt, C. Contributions of the hydrophobic Helix 2 of the Bordetella pertussis CyaA-hemolysin to membrane permeabilization. Protein Pept. Lett. 2018, 25, 236–243. [Google Scholar] [CrossRef]
- Roderova, J.; Osickova, A.; Sukova, A.; Mikusova, G.; Fiser, R.; Sebo, P.; Osicka, R.; Masin, J. Residues 529 to 549 participate in membrane penetration and pore-forming activity of the Bordetella adenylate cyclase toxin. Sci. Rep. 2019, 9, 5758. [Google Scholar] [CrossRef]
- Karst, J.C.; Barker, R.; Devi, U.; Swann, M.J.; Davi, M.; Roser, S.J.; Ladant, D.; Chenal, A. Identification of a region that assists membrane insertion and translocation of the catalytic domain of Bordetella pertussis CyaA toxin. J. Biol. Chem. 2012, 287, 9200–9212. [Google Scholar] [CrossRef] [Green Version]
- Sukova, A.; Bumba, L.; Srb, P.; Ververka, V.; Stanek, O.; Holulova, J.; Chmelik, J.; Sebo, P.; Masin, J. Negative charge of the AC-to-Hly linking segment modulates calcium-dependent membrane activities of Bordetella adenylate cyclase toxin. Biochimica et Biophys. Acta Biomembr. 2020, 1862, 18310–18316. [Google Scholar] [CrossRef]
- Bumba, L.; Masin, J.; Fiser, R.; Sebo, P. Bordetella adenylate cyclase toxin mobilizes its beta2 integrin receptor into lipid rafts to accomplish translocation across target cell membrane in two steps. PLoS Pathog. 2010, 9, e1000901. [Google Scholar]
- González-Bullón, D.; Uribe, K.B.; Amuategi, J.; Martín, C.; Ostolaza, H. Cholesterol stimulates the lytic activity of Adenylate Cyclase Toxin on lipid membranes by promoting toxin oligomerization and formation of pores with a greater effective size. FEBS J. 2021, 288, 6795–6814. [Google Scholar] [CrossRef] [PubMed]
- Bakás, L.; Ostolaza, H.; Vaz, W.L.; Goñi, F.M. Reversible adsorption and nonreversible insertion of Escherichia coli alpha-hemolysin into lipid bilayers. Biophys. J. 1996, 7, 1869–1876. [Google Scholar] [CrossRef] [Green Version]
- Brown, A.C.; Koufos, E.; Balashova, N.V.; Boesze-Battaglia, K.; Lally, E.T. Inhibition of LtxA toxicity by blocking cholesterol binding with peptides. Mol. Oral. Microbiol. 2016, 31, 94–105. [Google Scholar] [CrossRef] [PubMed]
- Vazquez, R.F.; Maté, S.M.; Bakás, L.S.; Fernández, M.M.; Malchiodi, E.L.; Herlax, V.S. Novel evidence for the specific interaction between cholesterol and alpha-haemolysin of Escherichia coli. Biochem. J. 2014, 458, 481–489. [Google Scholar] [CrossRef] [PubMed]
- Osickova, A.; Balashova, N.; Masin, J.; Sulc, M.; Roderova, J.; Wald, T.; Brown, A.C.; Koufos, E.; Chang, E.H.; Giannakakis, A.; et al. Cytotoxic activity of Kingella kingae RtxA toxin depends on post-translational acylation of lysine residues and cholesterol binding. Emer. Microbes Infect. 2018, 7, 178. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Papadopoulos, V. Peripheral-type benzodiazepine receptor function in cholesterol transport. Identification of a putative cholesterol recognition/interaction amino acid sequence and consensus pattern. Endocrinology 1998, 139, 4991–4997. [Google Scholar] [CrossRef]
- Fantini, J.; Barrantes, F.J. How cholesterol interacts with membrane proteins: An exploration of cholesterol-binding sites including CRAC, CARC and tilted domains. Front. Physiol. 2013, 4, 31–41. [Google Scholar] [CrossRef] [Green Version]
- Fantini, J.; Di Scala, C.; Evans, L.S.; Williamson, P.T.F.; Barrantes, F.J. A mirror code for protein-cholesterol interactions in the two leaflets of biological membranes. Sci. Rep. 2016, 6, 21907. [Google Scholar] [CrossRef] [Green Version]
- Baier, C.J.; Fantini, J.; Barrantes, F.J. Disclosure of cholesterol recognition motifs in transmembrane domains of the human nicotinic acetylcholine receptor. Sci. Rep. 2011, 1, 69. [Google Scholar] [CrossRef] [Green Version]
- Kumar, G.A.; Sarkar, P.; Stepniewski, T.M.; Jafurulla, M.; Singh, S.L.; Selent, J.; Chattopadhyay, A. A molecular sensor for cholesterol in human serotonin1A receptor. Sci. Adv. 2021, 7, eabh2922. [Google Scholar] [CrossRef]
- Aisenbrey, C.; Rifi, O.; Bechinger, B. Structure, membrane topology and influence of cholesterol of the membrane proximal region: Transmembrane helical anchor sequence of gp41 from HIV. Sci. Rep. 2020, 10, 22278. [Google Scholar] [CrossRef] [PubMed]
- Luz-Madrigal, A.; Asanov, A.; Camacho-Zarco, A.R.; Sampieri, A.; Vaca, L. A cholesterol recognition amino acid consensus domain in GP64 fusion protein facilitates anchoring of Baculovirus to mammalian cells. J. Virol. 2013, 87, 11894–11907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, J.; Buboltz, J.T.; Feigenson, G.W. Maximum solubility of cholesterol in phosphatidylcholine and phosphatidylethanolamine bilayers. Biochim. Biophys. Acta 1999, 1417, 89–100. [Google Scholar] [CrossRef] [Green Version]
- Masin, J.; Roderova, J.; Osickova, A.; Novak, P.; Bumba, L.; Fiser, R.; Sebo, P.; Osicka, R. The conserved tyrosine residue 940 plays a key structural role in membrane interaction of Bordetella adenylate cyclase toxin. Sci. Rep. 2017, 7, 9330. [Google Scholar] [CrossRef] [PubMed]
- Lashuel, H.A.; Lansbury, P.T., Jr. Are amyloid diseases caused by protein aggregates that mimic bacterial pore-forming toxins? Q. Rev. Biophys. 2006, 39, 167–201. [Google Scholar] [CrossRef] [PubMed]
- Rosado, C.J.; Buckle, A.L.; Law, R.H.P.; Butcher, R.E.; Kan, W.T.; Bird, C.H.; Ung, K.; Browne, K.A.; Baran, K.; Bashtannyk-Puhalovich, T.A.; et al. A common fold mediates vertebrate defense and bacterial attack. Science 2007, 317, 1548–1551. [Google Scholar] [CrossRef] [Green Version]
- Nishio, M.; Umezawa, Y.; Fantini, J.; Weiss, M.S.; Pinak, C. CH-[small pi] hydrogen bonds in biological macromolecules. Phys. Chem. Chem. Phys. 2014, 16, 12648–12683. [Google Scholar] [CrossRef]
- Strandberg, E.; Killian, J.A. Snorkeling of lysine side chains in transmembrane helices: How easy can it get? FEBS Lett. 2003, 544, 69–73. [Google Scholar] [CrossRef] [Green Version]
- Subrini, O.; Sotomayor-Pérez, A.C.; Hessel, A.; Spiaczka-Karst, J.; Selwa, E.; Sapay, N.; Veneziano, R.; Pansieri, J.; Chopineau, J.; Ladant, D.; et al. Characterization of a membrane-active peptide from the Bordetella pertussis CyaA toxin. J. Biol. Chem. 2013, 288, 32585–32598. [Google Scholar] [CrossRef] [Green Version]
- Gray, M.C.; Lee, S.J.; Gray, L.S.; Zaretzky, F.R.; Otero, A.S.; Hewlett, E.L. Translocation specific conformation of Adenylate Cyclase Toxin from Bordetella pertussis inhibits toxin-mediated hemolysis. J. Bacteriol. 2001, 183, 5904–5910. [Google Scholar] [CrossRef] [Green Version]
- Otero, A.S.; Yi, X.B.; Gray, M.C.; Szabo, G.; Hewlett, E.L. Membrane depolarization prevents cell invasion by Bordetella pertussis adenylate cyclase toxin. J. Biol. Chem. 1995, 270, 9695–9697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurehong, C.; Kanchanawarin, C.; Powthongchin, B.; Katzenmeier, G.; Angsuthanasombat, C. Membrane-pore forming characteristics of the Bordetella pertussis CyaA-hemolysin domain. Toxins 2015, 7, 1486–1496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Etxaniz, A.; González-Bullón, D.; Alonso, M.T.; Martín, C.; Ostolaza, H. Irreversible vs. Reparairable Membrane Poration: Differences in Permeabilization elicited by Bordetella Adenylate Cyclase Toxin and its Hemolysin Domain in Macrophages. FEBS J. 2020, 287, 1798–1815. [Google Scholar] [CrossRef]
- Cannella, S.E.; Ntsogo Enguéné, V.Y.; Davi, M.; Malosse, C.; Sotomayor Pérez, A.C.; Chamot-Rooke, J.; Vachette, P.; Durand, D.; Ladant, D.; Chenal, A. Stability, structural and functional properties of a monomeric, calcium-loaded adenylate cyclase toxin, CyaA, from Bordetella Pertussis. Sci. Rep. 2017, 7, 42065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hope, M.J.; Bally, M.B.; Webb, G.; Cullis, P.R. Production of large unilamellar vesicles by a rapid extrusion procedure. Characterization of size distribution, trapped volume and ability to maintain a membrane potential. Biochim. Biophys. Acta 1985, 812, 55–65. [Google Scholar] [CrossRef]
- Fiske, C.H.; Subbarow, Y. The colorimetric determination of phosphorus. J. Biol. Chem. 1925, 66, 375–400. [Google Scholar] [CrossRef]
- Yethon, J.A.; Epand, R.F.; Leber, B.; Epand, R.M.; Andrews, D.W. Interaction with a membrane surface triggers a reversible conformational change in Bax normally associated with induction of apoptosis. J. Biol. Chem. 2003, 278, 48935–48941. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pattern | Amino Acids | Sequence | |
|---|---|---|---|
| [L/V]-X(1,5)-Y-X(1,5)-[R/K] | HYDROPHOBIC DOMAIN | 626–638 | LVQQSHYADQLDK |
| 653–661 | LLAQLYRDK | ||
| 721–728 | LANDYARK | ||
| 732–741 | LGGPQAYFEK | ||
| [L/V]-X(1,5)-F-X(1,5)-[R/K] | TRANSLOCATION REGION | 481–487 | LMTQFGR |
| HYDROPHOBIC DOMAIN | 518–527 | VSGFFRGSSR | |
| [R/K]-X(1,5)-F-X(1,5)-[L/V] | TRANSLOCATION REGION | 413–420 | RSFSLGEV |
| HYDROPHOBIC DOMAIN | 527–534 | RWAGGFGV |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Amuategi, J.; Alonso, R.; Ostolaza, H. Four Cholesterol-Recognition Motifs in the Pore-Forming and Translocation Domains of Adenylate Cyclase Toxin Are Essential for Invasion of Eukaryotic Cells and Lysis of Erythrocytes. Int. J. Mol. Sci. 2022, 23, 8703. https://doi.org/10.3390/ijms23158703
Amuategi J, Alonso R, Ostolaza H. Four Cholesterol-Recognition Motifs in the Pore-Forming and Translocation Domains of Adenylate Cyclase Toxin Are Essential for Invasion of Eukaryotic Cells and Lysis of Erythrocytes. International Journal of Molecular Sciences. 2022; 23(15):8703. https://doi.org/10.3390/ijms23158703
Chicago/Turabian StyleAmuategi, Jone, Rocío Alonso, and Helena Ostolaza. 2022. "Four Cholesterol-Recognition Motifs in the Pore-Forming and Translocation Domains of Adenylate Cyclase Toxin Are Essential for Invasion of Eukaryotic Cells and Lysis of Erythrocytes" International Journal of Molecular Sciences 23, no. 15: 8703. https://doi.org/10.3390/ijms23158703
APA StyleAmuategi, J., Alonso, R., & Ostolaza, H. (2022). Four Cholesterol-Recognition Motifs in the Pore-Forming and Translocation Domains of Adenylate Cyclase Toxin Are Essential for Invasion of Eukaryotic Cells and Lysis of Erythrocytes. International Journal of Molecular Sciences, 23(15), 8703. https://doi.org/10.3390/ijms23158703