
Systematic Review: Drug Repositioning for Congenital Disorders of Glycosylation (CDG)

 , , , , , ,
, , , , , ,  ,
,  ,
,  ,
,
Abstract
:1. Introduction
2. Methods
2.1. Literature Analysis for Drug Repositioning in CDG
- (a)
- Only English-written manuscripts were included;
- (b)
- Articles reporting biomarkers, in vitro and/or in vivo models, compassionate use or clinical trials of therapies in CDG related to drug repositioning, were included;
- (c)
- Only articles reporting CDG with therapies related to drug repositioning, under development (compassionate use, clinical research) or already approved therapies were included;
- (d)
- Reviews were excluded, although we have included some for contextualization purposes.
2.2. Stakeholders’ Views on AI for Drug Development in CDG
3. Results
3.1. Literature Analysis
3.2. Disease Models
3.3. Biomarkers
3.4. Drug Repositioning
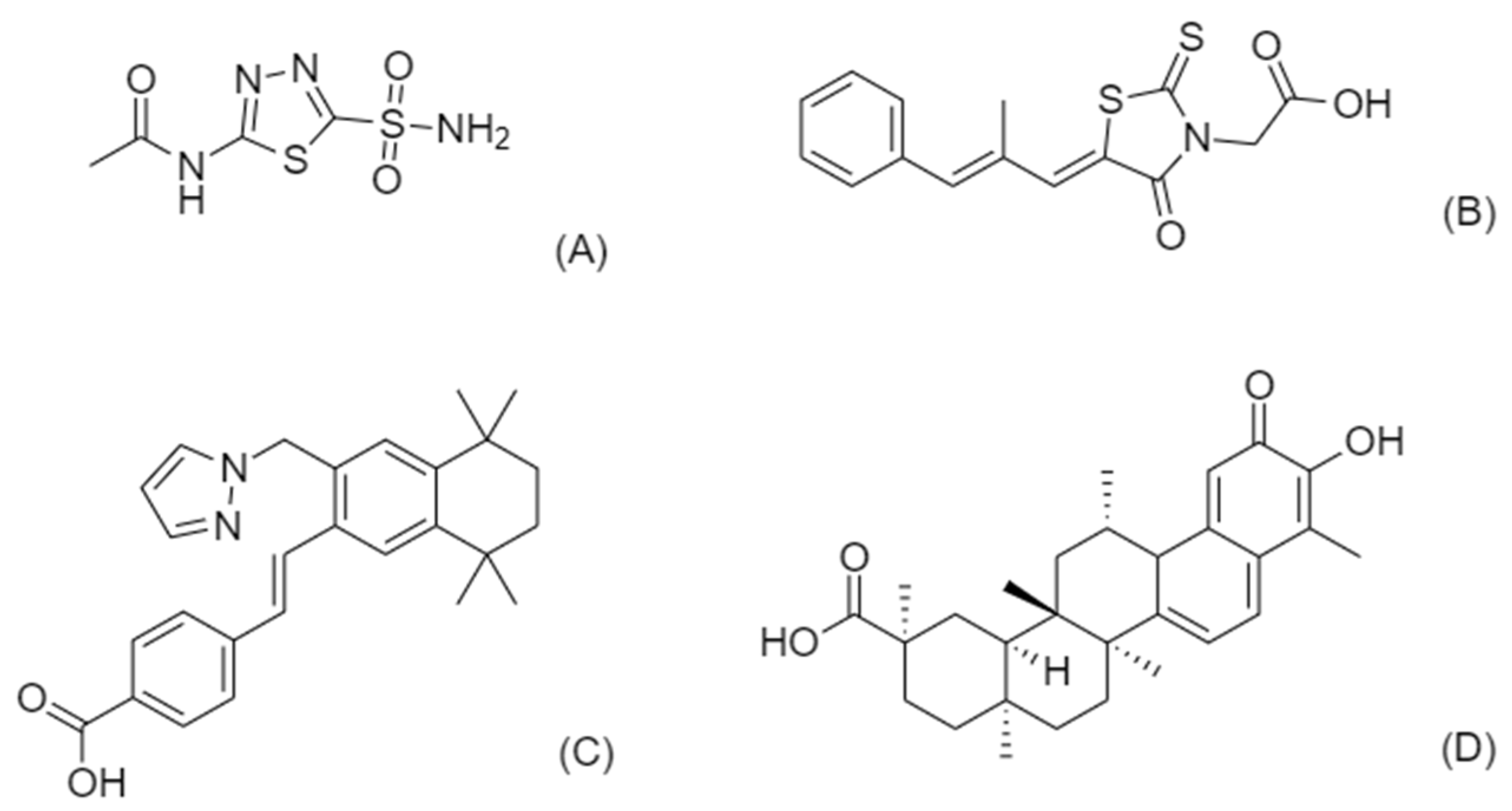
3.4.1. Celastrol for PMM2-CDG
3.4.2. Acetazolamide for PMM2-CDG
3.4.3. Epalrestat for PMM2-CDG
3.4.4. Palovarotene for EXT1/EXT2-CDG
3.5. Stakeholders’ Views on AI for Drug Development in CDG
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pushpakom, S.; Iorio, F.; Eyers, P.A.; Escott, K.J.; Hopper, S.; Wells, A.; Doig, A.; Guilliams, T.; Latimer, J.; McNamee, C.; et al. Drug Repurposing: Progress, Challenges and Recommendations. Nat. Rev. Drug Discov. 2018, 18, 41–58. [Google Scholar] [CrossRef] [PubMed]
- Juárez-López, D.; Schcolnik-Cabrera, A. Drug Repurposing: Considerations to Surpass While Re-Directing Old Compounds for New Treatments. Arch. Med. Res. 2021, 52, 243–251. [Google Scholar] [CrossRef] [PubMed]
- Xue, H.; Li, J.; Xie, H.; Wang, Y. Review of Drug Repositioning Approaches and Resources. Int. J. Biol. Sci. 2018, 14, 1232–1244. [Google Scholar] [CrossRef] [Green Version]
- Melnikova, I. Rare Diseases and Orphan Drugs. Nat. Rev. Drug Discov. 2012, 11, 267–268. [Google Scholar] [CrossRef]
- Richter, T.; Nestler-Parr, S.; Babela, R.; Khan, Z.M.; Tesoro, T.; Molsen, E.; Hughes, D.A. Rare Disease Terminology and Definitions—A Systematic Global Review: Report of the ISPOR Rare Disease Special Interest Group. Value Health 2015, 18, 906–914. [Google Scholar] [CrossRef] [Green Version]
- Lamoreaux, K. Rare-X The Power of Being Counted—A More Accurate Count of Rare Diseases and Steps to Getting Counted. 2022. Available online: https://rare-x.org/case-studies/the-power-of-being-counted/ (accessed on 24 March 2022).
- Sharma, A.; Jacob, A.; Tandon, M.; Kumar, D. Orphan Drug: Development Trends and Strategies. J. Pharm. Bioallied Sci. 2010, 2, 290–299. [Google Scholar] [CrossRef]
- Hechtelt Jonker, A.; Hivert, V.; Gabaldo, M.; Batista, L.; O’Connor, D.; Aartsma-Rus, A.; Day, S.; Sakushima, K.; Ardigo, D. Boosting Delivery of Rare Disease Therapies: The IRDiRC Orphan Drug Development Guidebook. Nat. Rev. Drug Discov. 2020, 19, 495–496. [Google Scholar] [CrossRef] [PubMed]
- Roessler, H.I.; Knoers, N.V.; van Haelst, M.M.; van Haaften, G. Drug Repurposing for Rare Diseases. Trends Pharmacol. Sci. 2021, 42, 255–267. [Google Scholar] [CrossRef] [PubMed]
- Fetro, C.; Scherman, D. Drug Repurposing in Rare Diseases: Myths and Reality. Therapies 2020, 75, 157–160. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, J.; Lehne, M.; Schepers, J.; Prasser, F.; Thun, S. The Use of Machine Learning in Rare Diseases: A Scoping Review. Orphanet J. Rare Dis. 2020, 15, 145. [Google Scholar] [CrossRef]
- Elbadawi, M.; Gaisford, S.; Basit, A.W. Advanced Machine-Learning Techniques in Drug Discovery. Drug Discov. Today 2021, 26, 769–777. [Google Scholar] [CrossRef]
- Patel, V.; Shah, M. A Comprehensive Study on Artificial Intelligence and Machine Learning in Drug Discovery and Drug Development. Intell. Med. 2021. [Google Scholar] [CrossRef]
- Brasil, S.; Pascoal, C.; Francisco, R.; Ferreira, V.D.R.; Videira, P.A.; Valadão, G. Artificial Intelligence (AI) in Rare Diseases: Is the Future Brighter? Genes 2019, 10, 978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toh, T.S.; Dondelinger, F.; Wang, D. Looking beyond the Hype: Applied AI and Machine Learning in Translational Medicine. eBioMedicine 2019, 47, 607–615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delavan, B.; Roberts, R.; Huang, R.; Bao, W.; Tong, W.; Liu, Z. Computational Drug Repositioning for Rare Diseases in the Era of Precision Medicine. Drug Discov. Today 2018, 23, 382–394. [Google Scholar] [CrossRef]
- Zhu, L.; Roberts, R.; Huang, R.; Zhao, J.; Xia, M.; Delavan, B.; Mikailov, M.; Tong, W.; Liu, Z. Drug Repositioning for Noonan and LEOPARD Syndromes by Integrating Transcriptomics With a Structure-Based Approach. Front. Pharmacol. 2020, 11, 927. [Google Scholar] [CrossRef]
- Ekins, S.; Gerlach, J.; Zorn, K.M.; Antonio, B.M.; Lin, Z.; Gerlach, A. Repurposing Approved Drugs as Inhibitors of K v 7.1 and Na v 1.8 to Treat Pitt Hopkins Syndrome. Pharmacol. Res. 2019, 36, 137. [Google Scholar] [CrossRef]
- Battista, T.; Pascarella, G.; Staid, D.S.; Colotti, G.; Rosati, J.; Fiorillo, A.; Casamassa, A.; Vescovi, A.L.; Giabbai, B.; Semrau, M.S.; et al. Known Drugs Identified by Structure-Based Virtual Screening Are Able to Bind Sigma-1 Receptor and Increase Growth of Huntington Disease Patient-Derived Cells. Int. J. Mol. Sci. 2021, 22, 1293. [Google Scholar] [CrossRef]
- Lee, Y.-s.; Krishnan, A.; Oughtred, R.; Rust, J.; Chang, C.S.; Ryu, J.; Kristensen, V.N.; Dolinski, K.; Theesfeld, C.L.; Troyanskaya, O.G. A Computational Framework for Genome-Wide Characterization of the Human Disease Landscape. Cell Syst. 2019, 8, 152–162. [Google Scholar] [CrossRef] [Green Version]
- Bottomley, S.S.; Fleming, M.D. Sideroblastic Anemia: Diagnosis and Management. Hematol. Oncol. Clin. N. Am. 2014, 28, 653–670. [Google Scholar] [CrossRef]
- Whittle, A.M.; Feyler, S.; Bowen, D.T. Durable Second Complete Remissions with Oral Melphalan in Hypocellular Acute Myeloid Leukemia and Refractory Anemia with Excess Blast with Normal Karyotype Relapsing after Intensive Chemotherapy. Leuk. Res. Rep. 2013, 2, 9–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esteban-Medina, M.; Peña-Chilet, M.; Loucera, C.; Dopazo, J. Exploring the Druggable Space around the Fanconi Anemia Pathway Using Machine Learning and Mechanistic Models. BMC Bioinform. 2019, 20, 370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bellomo, F.; de Leo, E.; Taranta, A.; Giaquinto, L.; di Giovamberardino, G.; Montefusco, S.; Rega, L.R.; Pastore, A.; Medina, D.L.; di Bernardo, D.; et al. Drug Repurposing in Rare Diseases: An Integrative Study of Drug Screening and Transcriptomic Analysis in Nephropathic Cystinosis. Int. J. Mol. Sci. 2021, 22, 12829. [Google Scholar] [CrossRef] [PubMed]
- Chang, I.J.; He, M.; Lam, C.T. Congenital Disorders of Glycosylation. Ann. Transl. Med. 2018, 6, 477. [Google Scholar] [CrossRef]
- Francisco, R.; Brasil, S.; Pascoal, C.; Jaeken, J.; Liddle, M.; Videira, P.A.; dos Reis Ferreira, V. The Road to Successful People-Centric Research in Rare Diseases: The Web-Based Case Study of the Immunology and Congenital Disorders of Glycosylation Questionnaire (ImmunoCDGQ). Orphanet J. Rare Dis. 2022, 17, 134. [Google Scholar] [CrossRef] [PubMed]
- Pajusalu, S.; Vals, M.-A.; Mihkla, L.; Šamarina, U.; Kahre, T.; Õunap, K. The Estimated Prevalence of N-Linked Congenital Disorders of Glycosylation Across Various Populations Based on Allele Frequencies in General Population Databases. Front. Genet. 2021, 12, 719437. [Google Scholar] [CrossRef]
- Brasil, S.; Pascoal, C.; Francisco, R.; Marques-da-Silva, D.; Andreotti, G.; Videira, P.A.; Morava, E.; Jaeken, J.; dos Reis Ferreira, V. CDG Therapies: From Bench to Bedside. Int. J. Mol. Sci. 2018, 19, 1304. [Google Scholar] [CrossRef] [Green Version]
- Péanne, R.; de Lonlay, P.; Foulquier, F.; Kornak, U.; Lefeber, D.J.; Morava, E.; Pérez, B.; Seta, N.; Thiel, C.; van Schaftingen, E.; et al. Congenital Disorders of Glycosylation (CDG): Quo Vadis? Eur. J. Med. Genet. 2018, 61, 643–663. [Google Scholar] [CrossRef]
- Boyer, S.W.; Johnsen, C.; Morava, E. Nutrition Interventions in Congenital Disorders of Glycosylation. Trends Mol. Med. 2022, 28, 463–481. [Google Scholar] [CrossRef]
- Kara, B.; Ayhan, Ö.; Gökçay, G.; Başboğaoğlu, N.; Tolun, A. Adult Phenotype and Further Phenotypic Variability in SRD5A3-CDG. BMC Med. Genet. 2014, 15, 10. [Google Scholar] [CrossRef] [Green Version]
- Kamarus Jaman, N.; Rehsi, P.; Henderson, R.H.; Löbel, U.; Mankad, K.; Grunewald, S. SRD5A3-CDG: Emerging Phenotypic Features of an Ultrarare CDG Subtype. Front. Genet. 2021, 12, 737094. [Google Scholar] [CrossRef] [PubMed]
- Westphal, V.; Kjaergaard, S.; Schollen, E.; Martens, K.; Grunewald, S.; Schwartz, M.; Matthijs, G.; Freeze, H.H. A Frequent Mild Mutation in ALG6 May Exacerbate the Clinical Severity of Patients with Congenital Disorder of Glycosylation La (CDG-La) Caused by Phosphomannomutase Deficiency. Hum. Mol. Genet. 2002, 11, 599–604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bortot, B.; Cosentini, D.; Faletra, F.; Biffi, S.; de Martino, E.; Carrozzi, M.; Severini, G.M. PMM2-CDG: Phenotype and Genotype in Four Affected Family Members. Gene 2013, 531, 506–509. [Google Scholar] [CrossRef] [PubMed]
- Citro, V.; Cimmaruta, C.; Monticelli, M.; Riccio, G.; Mele, B.H.; Cubellis, M.V.; Andreotti, G. The Analysis of Variants in the General Population Reveals That PMM2 Is Extremely Tolerant to Missense Mutations and That Diagnosis of PMM2-CDG Can Benefit from the Identification of Modifiers. Int. J. Mol. Sci. 2018, 19, 2218. [Google Scholar] [CrossRef] [Green Version]
- Park, J.H.; Marquardt, T. Treatment Options in Congenital Disorders of Glycosylation. Front. Genet. 2021, 12, 735348. [Google Scholar] [CrossRef]
- Monticelli, M.; Francisco, R.; Brasil, S.; Marques-da-Silva, D.; Rijoff, T.; Pascoal, C.; Jaeken, J.; Videira, P.A.; Ferreira, V.R. Stakeholders Views on Drug Development: The Congenital Disorders of Glycosylation Community Perspective. Orphanet J. Rare Dis. 2022; submitted. [Google Scholar]
- Page, M.J.; McKenzie, J.E.; Bossuyt, P.M.; Boutron, I.; Hoffmann, T.C.; Mulrow, C.D.; Shamseer, L.; Tetzlaff, J.M.; Akl, E.A.; Brennan, S.E.; et al. The PRISMA 2020 statement: An updated guideline for reporting systematic reviews. Syst. Rev. 2021, 11, 89. [Google Scholar] [CrossRef]
- Parrado, A.; Rubio, G.; Serrano, M.; de la Morena-Barrio, M.E.; Ibáñez-Micó, S.; Ruiz-Lafuente, N.; Schwartz-Albiez, R.; Esteve-Solé, A.; Alsina, L.; Corral, J.; et al. Dissecting the Transcriptional Program of Phosphomannomutase 2-Deficient Cells: Lymphoblastoide B Cell Lines as a Valuable Model for Congenital Disorders of Glycosylation Studies. Glycobiology 2022, 32, 84–100. [Google Scholar] [CrossRef]
- Lao, J.P.; Diprimio, N.; Prangley, M.; Sam, F.S.; Mast, J.D.; Perlstein, E.O. Yeast Models of Phosphomannomutase 2 Deficiency, a Congenital Disorder of Glycosylation. G3 Genes Genomes Genet. 2019, 9, 413–423. [Google Scholar] [CrossRef] [Green Version]
- Vignogna, R.C.; Allocca, M.; Monticelli, M.; Norris, J.W.; Steet, R.; Andreotti, G.; Perlstein, E.O.; Lang, G.I. Experimental Evolution of Phosphomannomutase-Deficient Yeast Reveals Compensatory 1 Mutations in a Phosphoglucomutase 2. eLife, 2022; submitted. [Google Scholar] [CrossRef]
- Inubushi, T.; Nozawa, S.; Matsumoto, K.; Irie, F.; Yamaguchi, Y. Aberrant Perichondrial BMP Signaling Mediates Multiple Osteochondromagenesis in Mice. JCI Insight 2017, 2, e90049. [Google Scholar] [CrossRef] [Green Version]
- Balakrishnan, B.; Verheijen, J.; Lupo, A.; Raymond, K.; Turgeon, C.T.; Yang, Y.; Carter, K.L.; Whitehead, K.J.; Kozicz, T.; Morava, E.; et al. A Novel Phosphoglucomutase-Deficient Mouse Model Reveals Aberrant Glycosylation and Early Embryonic Lethality HHS Public Access. J. Inherit. Metab. Dis. 2019, 42, 998–1007. [Google Scholar] [CrossRef]
- Gao, P.; Wang, F.; Huo, J.; Wan, D.; Zhang, J.; Niu, J.; Wu, J.; Yu, B.; Sun, T. ALG13 Deficiency Associated with Increased Seizure Susceptibility and Severity. Neuroscience 2019, 409, 204–221. [Google Scholar] [CrossRef] [PubMed]
- Neitzel, L.R.; Spencer, Z.T.; Nayak, A.; Cselenyi, C.S.; Benchabane, H.; Youngblood, C.A.Q.; Zouaoui, A.; Ng, V.; Stephens, L.; Hann, T.; et al. Developmental Regulation of Wnt Signaling by Nagk and the UDP-GlcNAc Salvage Pathway. Mech. Dev. 2019, 156, 20–31. [Google Scholar] [CrossRef] [PubMed]
- Inubushi, T.; Lemire, I.; Irie, F.; Yamaguchi, Y. Palovarotene Inhibits Osteochondroma Formation in a Mouse Model of Multiple Hereditary Exostoses. J. Bone Marrow Res. 2018, 33, 658–666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iyer, S.; Sam, F.S.; DiPrimio, N.; Preston, G.; Verheijen, J.; Murthy, K.; Parton, Z.; Tsang, H.; Lao, J.; Morava, E.; et al. Repurposing the Aldose Reductase Inhibitor and Diabetic Neuropathy Drug Epalrestat for the Congenital Disorder of Glycosylation PMM2-CDG. Dis. Models Mech. 2019, 12, dmm040584. [Google Scholar] [CrossRef] [Green Version]
- Kantautas, K.; Sappani Foundation, Brampton, ON, Canada. Personal communication, 2022.
- Gücüm, S.; Sakson, R.; Hoffmann, M.; Grote, V.; Becker, C.; Pakari, K.; Beedgen, L.; Thiel, C.; Rapp, E.; Ruppert, T.; et al. A Patient-Based Medaka Alg2 Mutant as a Model for Hypo-N-Glycosylation. Development 2021, 148, dev199385. [Google Scholar] [CrossRef] [PubMed]
- Ng, B.G.; Xu, G.; Chandy, N.; Steyermark, J.; Shinde, D.N.; Radtke, K.; Raymond, K.; Lebrilla, C.B.; AlAsmari, A.; Suchy, S.F.; et al. Biallelic Mutations in FUT8 Cause a Congenital Disorder of Glycosylation with Defective Fucosylation. Am. J. Hum. Genet. 2018, 102, 188–195. [Google Scholar] [CrossRef] [Green Version]
- Matsuda-Lennikov, M.; Biancalana, M.; Zou, J.; Ravell, J.C.; Zheng, L.; Kanellopoulou, C.; Jiang, P.; Notarangelo, G.; Jing, H.; Masutani, E.; et al. Magnesium Transporter 1 (MAGT1) Deficiency Causes Selective Defects in N-Linked Glycosylation and Expression of Immune-Response Genes. J. Biol. Chem. 2019, 294, 13638–13656. [Google Scholar] [CrossRef]
- Gallo, G.L.; Valko, A.; Aramburu, S.I.; Etchegaray, E.; Völker, C.; Parodi, A.J.; D’Alessio, C. Abrogation of Glucosidase I–Mediated Glycoprotein Deglucosylation Results in a Sick Phenotype in Fission Yeasts: Model for the Human MOGS-CDG Disorder. J. Biol. Chem. 2018, 293, 19957–19973. [Google Scholar] [CrossRef] [Green Version]
- DeRossi, C.; Bambino, K.; Morrison, J.; Sakarin, I.; Villacorta-Martin, C.; Zhang, C.; Ellis, J.L.; Fiel, M.I.; Ybanez, M.; Lee, Y.A.; et al. Mannose Phosphate Isomerase and Mannose Regulate Hepatic Stellate Cell Activation and Fibrosis in Zebrafish and Humans. Hepatology 2019, 70, 2107–2122. [Google Scholar] [CrossRef]
- Klaver, E.J.; Dukes-Rimsky, L.; Kumar, B.; Xia, Z.-J.; Dang, T.; Lehrman, M.A.; Angel, P.; Drake, R.R.; Freeze, H.H.; Steet, R.; et al. Protease-Dependent Defects in N-Cadherin Processing Drive PMM2-CDG Pathogenesis. JCI Insight 2021, 6, e149217. [Google Scholar] [CrossRef]
- Taubenschmid, J.; Stadlmann, J.; Jost, M.; Klokk, T.I.; Rillahan, C.D.; Leibbrandt, A.; Mechtler, K.; Paulson, J.C.; Jude, J.; Zuber, J.; et al. A Vital Sugar Code for Ricin Toxicity. Nat. Publ. Group 2017, 27, 1351–1364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cataldi, M.P.; Lu, P.; Blaeser, A.; Lu, Q.L. Ribitol Restores Functionally Glycosylated α-Dystroglycan and Improves Muscle Function in Dystrophic FKRP-Mutant Mice. Nat. Commun. 2018, 9, 3448. [Google Scholar] [CrossRef] [PubMed]
- Cataldi, M.P.; Blaeser, A.; Lu, P.; Leroy, V.; Lu, Q.L. ISPD Overexpression Enhances Ribitol-Induced Glycosylation of α-Dystroglycan in Dystrophic FKRP Mutant Mice. Mol. Ther. Methods Clin. Dev. 2019, 17, 271–280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sinha, S.; Mundy, C.; Sgariglia, F.; Ibrahim, M.M.; Billings, P.C.; Carroll, K.; Koyama, E.; Jones, K.B.; Pacifici, M. Unsuspected Osteochondroma-like Outgrowths in the Cranial Base of Hereditary Multiple Exostoses Patients and Modeling and Treatment with a BMP Antagonist in Mice. PLOS Genet. 2017, 13, e1006742. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Sun, X.; Yu, F.; Perle, M.A.; Araten, D.; Boeke, J.D. Application of Counter-Selectable Marker PIGA in Engineering Designer Deletion Cell Lines and Characterization of CRISPR Deletion Efficiency. Nucleic Acids Res. 2021, 49, 2642–2654. [Google Scholar] [CrossRef]
- Kandasamy, L.C.; Tsukamoto, M.; Banov, V.; Tsetsegee, S.; Nagasawa, Y.; Kato, M.; Matsumoto, N.; Takeda, J.; Itohara, S.; Ogawa, S.; et al. Limb-Clasping, Cognitive Deficit and Increased Vulnerability to Kainic Acid-Induced Seizures in Neuronal Glycosylphosphatidylinositol Deficiency Mouse Models. Hum. Mol. Genet. 2021, 30, 758–770. [Google Scholar] [CrossRef]
- del Caño-Ochoa, F.; Ng, B.G.; Abedalthagafi, M.; Almannai, M.; Cohn, R.D.; Costain, G.; Elpeleg, O.; Houlden, H.; Karimiani, E.G.; Liu, P.; et al. Cell-Based Analysis of CAD Variants Identifies Individuals Likely to Benefit from Uridine Therapy. Genet. Med. 2020, 22, 1598–1605. [Google Scholar] [CrossRef]
- Liu, Z.; Wang, Y.; Yang, F.; Yang, Q.; Mo, X.; Burstein, E.; Jia, D.; Cai, X.-T.; Tu, Y. GMPPB-Congenital Disorders of Glycosylation Associate with Decreased Enzymatic Activity of GMPPB. Mol. Biomed. 2021, 2, 13. [Google Scholar] [CrossRef]
- Willems, A.P.; Sun, L.; Schulz, M.A.; Tian, W.; Ashikov, A.; van Scherpenzeel, M.; Hermans, E.; Clausen, H.; Yang, Z.; Lefeber, D.J. Activity of N-Acylneuraminate-9-Phosphatase (NANP) Is Not Essential for de Novo Sialic Acid Biosynthesis. Biochim. Biophys. Acta (BBA)-Gen. Subj. 2019, 1863, 1471–1479. [Google Scholar] [CrossRef]
- Sumya, F.T.; Pokrovskaya, I.D.; Lupashin, V. Development and Initial Characterization of Cellular Models for COG Complex-Related CDG-II Diseases. Front. Genet. 2021, 12, dmm040584. [Google Scholar] [CrossRef]
- Ishii, M.; Lupashin, V.V.; Nakano, A. Detailed Analysis of the Interaction of Yeast COG Complex. Cell Struct. Funct. 2018, 43, 119–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fári, K.F.; Sándor, S.; Takács, T.; Dániel, D.; Ungár, U.; Sinka, R. The Role of Acroblast Formation during Drosophila Spermatogenesis. Biol. Open 2016, 5, 1102–1110. [Google Scholar] [CrossRef] [Green Version]
- Frappaolo, A.; Sechi, S.; Kumagai, T.; Robinson, S.; Fraschini, R.; Karimpour-Ghahnavieh, A.; Belloni, G.; Piergentili, R.; Tiemeyer, K.H.; Tiemeyer, M.; et al. COG7 Deficiency in Drosophila Generates Multifaceted Developmental, Behavioral and Protein Glycosylation Phenotypes. J. Cell Sci. 2017, 130, 3637–3649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, W.; Vann, D.R.; Doulias, P.T.; Wang, T.; Landesberg, G.; Li, X.; Ricciotti, E.; Scalia, R.; He, M.; Hand, N.J.; et al. Hepatic Metal Ion Transporter ZIP8 Regulates Manganese Homeostasis and Manganese-Dependent Enzyme Activity. J. Clin. Investig. 2017, 127, 2407–2417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Medina-Cano, D.; Ucuncu, E.; Nguyen, L.S.; Nicouleau, M.; Lipecka, J.; Bizot, J.-C.; Thiel, C.; Ois Foulquier, F.; Lefort, N.; Faivre-Sarrailh, C.; et al. High N-Glycan Multiplicity Is Critical for Neuronal Adhesion and Sensitizes the Developing Cerebellum to N-Glycosylation Defect. eLife 2018, 7, e38309. [Google Scholar] [CrossRef]
- Indellicato, R.; Domenighini, R.; Malagolini, N.; Cereda, A.; Mamoli, D.; Pezzani, L.; Iascone, M.; dall’Olio, F.; Trinchera, M. A Novel Nonsense and Inactivating Variant of ST3GAL3 in Two Infant Siblings Suffering Severe Epilepsy and Expressing Circulating CA19.9. Glycobiology 2020, 30, 95–104. [Google Scholar] [CrossRef]
- Indellicato, R.; Parini, R.; Domenighini, R.; Malagolini, N.; Iascone, M.; Gasperini, S.; Masera, N.; Dall’olio, F.; Trinchera, M. Total Loss of GM3 Synthase Activity by a Normally Processed Enzyme in a Novel Variant and in All ST3GAL5 Variants Reported to Cause a Distinct Congenital Disorder of Glycosylation. Glycobiology 2019, 29, 229–241. [Google Scholar] [CrossRef]
- Park, J.H.; Mealer, R.G.; Abdallah, F.E.; Hoffmann, S.; Grüneberg, M.; Biskup, S.; Fobker, M.; Haven, J.; Mangels, U.; Reunert, J.; et al. N-Glycome Analysis Detects Dysglycosylation Missed by Conventional Methods in SLC39A8 Deficiency. J. Inherit. Metab. Dis. 2020, 43, 1370–1381. [Google Scholar] [CrossRef]
- Bruneel, A.; Fenaille, F. Editorial Commentary Integrating Mass Spectrometry-Based Plasma (or Serum) Protein N-Glycan Profiling into the Clinical Practice? Increased Clinical Sensitivity and Specificity of Plasma Protein N-Glycan Profiling for Diagnosing Congenital Disorders of Glycosylation by Use of Flow Injection-Electrospray Ionization-Quadrupole Time-of-Flight Mass Spectrometry. Clin. Ann. Transl. Med. 2019, 7, 653–663. [Google Scholar] [CrossRef]
- Chen, J.; Li, X.; Edmondson, A.; Meyers, G.D.; Izumi, K.; Ackermann, A.M.; Morava, E.; Ficicioglu, C.; Bennett, M.J.; He, M. Increased Clinical Sensitivity and Specificity of Plasma Protein N-Glycan Profiling for Diagnosing Congenital Disorders of Glycosylation by Use of Flow Injection-Electrospray Ionization-Quadrupole Time-of-Flight Mass Spectrometry. Clin. Chem. 2019, 65, 653–663. [Google Scholar] [CrossRef]
- Abu Bakar, N.; Ashikov, A.; Brum, J.M.; Smeets, R.; Kersten, M.; Huijben, K.; Keng, W.T.; Speck-Martins, C.E.; de Carvalho, D.R.; de Rizzo, I.M.P.O.; et al. Synergistic Use of Glycomics and Single-Molecule Molecular Inversion Probes for Identification of Congenital Disorders of Glycosylation Type-1. J. Inherit. Metab. Dis. 2022, 45, 769–781. [Google Scholar] [CrossRef] [PubMed]
- Ligezka, A.N.; Radenkovic, S.; Saraswat, M.; Garapati, K.; Ranatunga, W.; Krzysciak, W.; Yanaihara, H.; Preston, G.; Brucker, W.; McGovern, R.M.; et al. Sorbitol Is a Severity Biomarker for PMM2-CDG with Therapeutic Implications. Ann. Neurol. 2021, 90, 887–900. [Google Scholar] [CrossRef] [PubMed]
- Perales-Clemente, E.; Liedtke, K.; Studinski, A.; Radenkovic, S.; Gavrilov, D.; Oglesbee, D.; Matern, D.; Rinaldo, P.; Tortorelli, S.; Morava, E.; et al. A New D-Galactose Treatment Monitoring Index for PGM1-CDG. J. Inherit. Metab. Dis. 2021, 44, 1263–1271. [Google Scholar] [CrossRef] [PubMed]
- Cascão, R.; Fonseca, J.E.; Moita, L.F. Celastrol: A Spectrum of Treatment Opportunities in Chronic Diseases. Front. Med. 2017, 4, 69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boridy, S.; Le, P.U.; Petrecca, K.; Maysinger, D. Celastrol Targets Proteostasis and Acts Synergistically with a Heat-Shock Protein 90 Inhibitor to Kill Human Glioblastoma Cells. Cell Death Dis. 2014, 5, e1216. [Google Scholar] [CrossRef] [PubMed]
- Balch, W.E.; Morimoto, R.I.; Dillin, A.; Kelly, J.W. Adapting Proteostasis for Disease Intervention. Science 2008, 319, 916–919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seemann, S.; Ernst, M.; Cimmaruta, C.; Struckmann, S.; Cozma, C.; Koczan, D.; Knospe, A.-M.; Haake, L.R.; Citro, V.; Bräuer, A.U.; et al. Proteostasis Regulators Modulate Proteasomal Activity and Gene Expression to Attenuate Multiple Phenotypes in Fabry Disease. Biochem. J. 2020, 477, 359–380. [Google Scholar] [CrossRef]
- Brusa, I.; Sondo, E.; Falchi, F.; Pedemonte, N.; Roberti, M.; Cavalli, A. Proteostasis Regulators in Cystic Fibrosis: Current Development and Future Perspectives. J. Med. Chem. 2022, 65, 5212–5243. [Google Scholar] [CrossRef]
- Vilas, A.; Yuste-Checa, P.; Gallego, D.; Desviat, L.R.; Ugarte, M.; Pérez-Cerda, C.; Gámez, A.; Pérez, B. Proteostasis Regulators as Potential Rescuers of PMM2 Activity. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2020, 1866, 165777. [Google Scholar] [CrossRef]
- Mu, T.-W.; Sek Tong Ong, D.; Wang, Y.-J.; Balch, W.E.; Yates, J.R.; Segatori, L.; Kelly, J.W. Proteostasis Regulators and Pharmacologic Chaperones Synergize to Correct Protein Misfolding Diseases. Cell 2008, 134, 769–781. [Google Scholar] [CrossRef] [Green Version]
- Gámez, A.; Serrano, M.; Gallego, D.; Vilas, A.; Pérez, B. New and Potential Strategies for the Treatment of PMM2-CDG. Biochim. Biophys. Acta (BBA)-Gen. Subj. 2020, 1864, 129686. [Google Scholar] [CrossRef] [PubMed]
- Banderali, G.; Salvatici, E.; Rovelli, V.; Jaeken, J. PMM2- CDG and Nephrotic Syndrome: A Case Report. Clin. Case Rep. 2022, 10, e05347. [Google Scholar] [CrossRef]
- Izquierdo-Serra, M.; Martínez-Monseny, A.F.; López, L.; Carrillo-García, J.; Edo, A.; Darío Ortigoza-Escobar, J.; García, Ó.; Cancho-Candela, R.; Llanos Carrasco-Marina, M.; Gutiérrez-Solana, L.G.; et al. Stroke-Like Episodes and Cerebellar Syndrome in Phosphomannomutase Deficiency (PMM2-CDG): Evidence for Hypoglycosylation-Driven Channelopathy. Int. J. Mol. Sci 2018, 19, 619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andreotti, G.; de Vaca, I.C.; Poziello, A.; Monti, M.C.; Guallar, V.; Cubellis, M.V. Conformational Response to Ligand Binding in Phosphomannomutase2: Insights into inborn glycosylation disorder. J. Biol. Chem. 2014, 289, 34900–34910. [Google Scholar] [CrossRef] [Green Version]
- Bain, P.G.; O’Brien, M.D.; Keevil, S.F.; Porter, D.A. Familial Periodic Cerebellar Ataxia: A Problem of Cerebellar Intracellular PH Homeostasis. Ann. Neurol. 1992, 31, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Fine, A.L.; Wirrell, E.C.; Wong-Kisiel, L.C.; Nickels, K.C. Acetazolamide for Electrical Status Epilepticus in Slow-Wave Sleep. Epilepsia 2015, 56, 134–138. [Google Scholar] [CrossRef] [Green Version]
- Hong, E.H.; Ahn, S.J.; Lim, H.W.; Lee, B.R. The Effect of Oral Acetazolamide on Cystoid Macular Edema in Hydroxychloroquine Retinopathy: A Case Report. BMC Ophthalmol. 2017, 17, 124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salman, M.S. Epidemiology of Cerebellar Diseases and Therapeutic Approaches. Cerebellum 2018, 17, 4–11. [Google Scholar] [CrossRef]
- Martínez-Monseny, A.F.; Bolasell, M.; Callejón-Póo, L.; Cuadras, D.; Freniche, V.; Itzep, D.C.; Gassiot, S.; Arango, P.; Casas-Alba, D.; de La Mo-rena, E.; et al. AZATAX: Acetazolamide Safety and Efficacy in Cerebellar Syndrome in PMM2 Congenital Disorder of Glycosylation (PMM2-CDG) and the CDG Spanish Consortium. Ann. Neurol. 2019, 85, 740–751. [Google Scholar] [CrossRef]
- Kawai, T.; Takei, I.; Tokui, M.; Funae, O.; Miyamoto, K.; Tabata, M.; Hirata, T.; Saruta, T.; Shimada, A.; Itoh, H. Effects of Epalrestat, an Aldose Reductase Inhibitor, on Diabetic Peripheral Neuropathy in Patients with Type 2 Diabetes, in Relation to Suppression of Nɛ-Carboxymethyl Lysine. J. Diabetes Its Complicat. 2010, 24, 424–432. [Google Scholar] [CrossRef]
- Monticelli, M.; Liguori, L.; Allocca, M.; Andreotti, G.; Cubellis, M.V. Bisphosphate Stabilizes Pathological Phophomannomutase2 Mutants In Vitro and Represents a Lead Compound to Develop Pharmacological Chaperones for the Most Common Disorder of Glycosylation, PMM2-CDG. Int. J. Mol. Sci. 2019, 20, 4164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delgado, M.A.; Martinez-Domenech, G.; Sarrión, P.; Urreizti, R.; Zecchini, L.; Robledo, H.H.; Segura, F.; Dodelson De Kremer, R.; Balcells, S.; Grinberg, D.; et al. A Broad Spectrum of Genomic Changes in Latinamerican Patients with EXT1/ EXT2-CDG. Sci. Rep. 2014, 4, 6407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huegel, J.; Mundy, C.; Sgariglia, F.; Nygren, P.; Billings, P.C.; Yamaguchi, Y.; Koyama, E.; Pacifici, M. Perichondrium Phenotype and Border Function Are Regulated by Ext1 and Heparan Sulfate in Developing Long Bones: A Mechanism Likely Deranged in Hereditary Multiple Exostoses. Dev. Biol. 2013, 377, 100–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shore, E.M.; Kaplan, F.S. Insights from a Rare Genetic Disorder of Extra-Skeletal Bone Formation, Fibrodysplasia Ossificans Progressiva (FOP). Bone 2008, 43, 427–433. [Google Scholar] [CrossRef] [Green Version]
- Stolk, J.; Stockley, R.A.; Stoel, B.C.; Cooper, B.G.; Piitulainen, E.; Seersholm, N.; Chapman, K.R.; Burdon, J.G.W.; Decramer, M.; Abboud, R.T.; et al. Randomised Controlled Trial for Emphysema with a Selective Agonist of the C-Type Retinoic Acid Receptor. Eur. Respir. J. 2012, 40, 306–312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DiMasi, J.A.; Feldman, L.; Seckler, A.; Wilson, A. Trends in Risks Associated With New Drug Development: Success Rates for Investigational Drugs. Clin. Pharm. Ther. 2010, 87, 272–277. [Google Scholar] [CrossRef]
- Wouters, O.J.; Mckee, M.; Luyten, J. Estimated Research and Development Investment Neededto Bring a New Medicine to Market, 2009–2018. JAMA|Orig. Investig. 2020, 323, 844–853. [Google Scholar] [CrossRef]
- Sernadela, P.; González-Castro, L.; Carta, C.; van der Horst, E.; Lopes, P.; Kaliyaperumal, R.; Thompson, M.; Thompson, R.; Queralt-Rosinach, N.; Lopez, E.; et al. Linked Registries: Connecting Rare Diseases Patient Registries through a Semantic Web Layer. BioMed Res. Int. 2017, 2017, 8327980. [Google Scholar] [CrossRef] [Green Version]
- Oprea, T.I.; Bauman, J.E.; Bologa, C.G.; Buranda, T.; Chigaev, A.; Edwards, B.S.; Jarvik, J.W.; Gresham, H.D.; Haynes, M.K.; Hjelle, B.; et al. Drug Repurposing from an Academic Perspective. Drug Discov. Today Ther. Strateg. 2011, 8, 61–69. [Google Scholar] [CrossRef] [Green Version]
- Augustine, E.F.; Adams, H.R.; Mink, J.W. Clinical Trials in Rare Disease: Challenges and Opportunities. J. Child Neurol. 2013, 28, 1142–1150. [Google Scholar] [CrossRef]
- Murillo-Cuesta, S.; Varela-Nieto, I.; Artuch, R.; Asensio, F.; de La Villa, P.; Dierssen, M.; Antonio Enríquez, J.; Fillat, C.; Fourcade, S.; Ibáñez, B.; et al. The Value of Mouse Models of Rare Diseases: A Spanish Experience. Front. Genet. 2020, 11, 583932. [Google Scholar] [CrossRef]
- Bender, A.; Cortés-Ciriano, I. Artificial Intelligence in Drug Discovery: What Is Realistic, What Are Illusions? Part 1: Ways to Make an Impact, and Why We Are Not There Yet. Drug Discov. Today 2021, 26, 511–524. [Google Scholar] [CrossRef] [PubMed]
- Jiménez-Luna, J.; Grisoni, F.; Schneider, G. Drug Discovery with Explainable Artificial Intelligence. Nat. Mach. Intell. 2020, 2, 573–584. [Google Scholar] [CrossRef]
- Sakate, R.; Kimura, T. Drug Repositioning Trends in Rare and Intractable Diseases. Drug Discov. Today 2022, 27, 1789–1795. [Google Scholar] [CrossRef] [PubMed]
- Challa, A.P.; Zaleski, N.M.; Jerome, R.N.; Lavieri, R.R.; Shirey-Rice, J.K.; Barnado, A.; Lindsell, C.J.; Aronoff, D.M.; Crofford, L.J.; Harris, R.C.; et al. Human and Machine Intelligence Together Drive Drug Repurposing in Rare Diseases. Front. Genet. 2021, 12, 1374. [Google Scholar] [CrossRef] [PubMed]
- Modelis Projects—Rapidly Avancing Programs. Available online: https://modelis.ca/projects/ (accessed on 24 June 2022).




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene (Gene ID) | Protein | Disorder |
|---|---|---|
| ALG2 (85365) | alpha-1,3/1,6-mannosyltransferase | ALG2-CDG |
| ALG13 (79868) | UDP-N-acetylglucosaminyltransferase (subunit) | ALG13-CDG |
| B4GALT1 (2683) | beta-1,4-galactosyltransferase 1 | B4GALT1-CDG |
| CAD (790) | carbamoyl-phosphate synthetase 2, aspartate transcarbamylase, and dihydroorotase (enzyme complex) | CAD-CDG |
| COG4 (25839) | component of oligomeric golgi complex 4 | COG4-CDG |
| COG5 (10466) | component of oligomeric golgi complex 5 | COG5-CDG |
| COG7 (91949) | component of oligomeric golgi complex 7 | COG7-CDG |
| DPAGT1 (1798) | dolichyl-phosphate N-acetylglucosaminephosphotransferase 1 | DPAGT1-CDG |
| EXT1 (2131) | exostosin glycosyltransferase 1 | EXT1-CDG |
| EXT2 (2132) | exostosin glycosyltransferase 2 | EXT2-CDG |
| FUT8 (2530) | fucosyltransferase 8 | FUT8-CDG |
| GMPPB (29925) | GDP-mannose pyrophosphorylase B | GMPPB-CDG |
| GNE (10020) | glucosamine (UDP-N-acetyl)-2-epimerase/N-acetylmannosamine kinase | GNE-CDG |
| MAGT1 (84061) | magnesium transporter 1 | MAGT1-CDG |
| MOGS (7841) | mannosyl-oligosaccharide glucosidase | MOGS-CDG |
| MPI (4351) | mannose phosphate isomerase | MPI-CDG |
| NANS (54187) | N-acetylneuraminate synthase | NANS-CDG |
| PGM1 (5236) | phosphoglucomutase 1 | PGM1-CDG |
| PGM3 (5238) | phosphoglucomutase 3 | PGM3-CDG |
| PIGA (5277) | phosphatidylinositol glycan anchor biosynthesis class A | PIGA-CDG |
| PMM2 (5373) | phosphomannomutase 2 | PMM2-CDG |
| SLC35C1 (55343) | solute carrier family 35 member C1 | SLC35C1-CDG |
| SLC39A8 (64116) | solute carrier family 39 member 8 | SLC39A8-CDG |
| SRD5A3 (79644) | steroid 5 alpha-reductase 3 | SRD5A3-CDG |
| ST3GAL3 (6487) | ST3 beta-galactoside alpha-2,3-sialyltransferase 3 | ST3GAL3-CDG |
| ST3GAL4 (6484) | ST3 beta-galactoside alpha-2,3-sialyltransferase 4 | ST3GAL4-CDG |
| ST3GAL5 (8869) | ST3 beta-galactoside alpha-2,3-sialyltransferase 5 | ST3GAL5-CDG |
| Defect | CDG | Cell/Organism | Model | Major Findings/Phenotype | Reference |
|---|---|---|---|---|---|
| N-linked glycosylation | ALG2-CDG | Oryzias latipes (medaka) | Alg2+/p.G336 Alg2p.G336/p.G336 | Modelling ALG2-CDG patient phenotypes, in terms of morphology (facial skeleton and neuronal defects) and hypo-N-glycosylation (especially affecting rod photoreceptors) | [49] |
| ALG13-CDG | Mus musculus (mouse) | Alg13 KO |
| [44] | |
| DPAGT1-CDG | Xenopus laevis | Dpagt1 KO (mRNA) |
| [45] | |
| Danio rerio (zebrafish) | Dpagt1 KO (mRNA) | Inhibition of eye formation | |||
| FUT8-CDG | Mouse | Fut8−/− | High mortality rate after birth due to respiratory defects and severe growth retardation | [50] | |
| MAGT1-CDG | Jurkat cell line | Magt1−/− | Selective deficiency of N-glycoproteins and glycosylation defects in immune-response proteins such as CD28 | [51] | |
| Human embryonic kidney (HEK) 293T cell line | Magt1 KO Magt1/Tusc3 KO |
| |||
| MOGS-CDG | Schizosaccharomyces pombe (yeast) | Δgls1-S |
| [52] | |
| MPI-CDG | TWNT-4 and LX-2 a human hepatic stellate cells | Mpi KD (siRNA) |
| [53] | |
| PMM2-CDG | Caenorhabditis elegans | Pmm2F125L/F125L |
| [47] | |
| Saccharomyces cerevisiae (yeast) | Sec53Δ Sec53E146K (E139K) Sec53V238M (V231M) Sec53F126L (F119L) Sec53E100K (E93A) Sec53R148H (R141H) | Drug repurposing screen revealed three novel chemical modifiers that subdued growth defects in SEC53 protein variants | [40] | ||
| Zebrafish | Pmm2 KD (MO) Mmp2 KD (MO) Mmp9 KD (MO) Furina KD (MO) | Reducing proconvertase activity restores matrix metalloproteinase (mmp) activity and improves N-cadherin processing | [54] | ||
| EBV-transformed lymphoblastoid B cell lines (B-LCL) from 13 patients | Carbonic anhydrase 2 is proposed as a cellular biomarker for CDG | [39] | |||
| O-linked glycosylation | B4GALT1-CDG | Mouse embryonic stem cells (mESCs) | B4Galt1 KO | Enhanced resistance to ricin | [55] |
| CRPP-CDG | Mouse | FKRPP448L/P448L |
| [56,57] | |
| EXT1/EXT2-CDG | Mouse | Col2a1-Ext1CKO stochastic KO |
| [42] | |
| Fsp1-Ext1CKO (perichondrium-targeted Ext1–conditional KO) | Development of multiple osteochondromas | ||||
| Ext1f/f Agr-CreER |
| [58] | |||
| Ext1f/f Col2-CreER |
| ||||
| GPI-biosynthesis | PIGA-CDG | Human male colon cancer cell line (HCT116) | PigaΔ | NR | [59] |
| Mouse | a,b In-M-cko a,c Ex-M-cko Th-H-cko |
| [60] | ||
| Multiple and other glycosylation pathways | CAD-CDG/ Enzyme complex (ATase, CPSase, ATCase and DHOase) | Human U20S cells | CAD KO (homozygous c.70delG frameshift (p.Ala24-Profs*27) within exon 1 using CRISPR/Cas9) | No expression of CAD protein | [61] |
| GMPPB-CDG | Zebrafish | Gmppb KD (MO) | Gmppb involvement in neuronal and muscle development | [62] | |
| GNE-CDG | Chinese hamster ovary (CHO) cell line | Gne KO |
| [63] | |
| COG4-CDG | RPE1 and HEK293T cell lines | Cog4 KO |
| [64] | |
| COG5-8 | S. cerevisiae | Cog5-8Δ (cog5-8::kanMX6) |
| [65] | |
| COG5-CDG | Drosophila melanogaster | P element insertion mutations in the Cog5 (fws) subunit | Impairment of spermatocyte cytokinesis, acroblast structure and elongation and individualization of differentiating spermatids | [66] | |
| COG7-CDG | D. melanogaster | Cog7z4495/z5797 |
| [67] | |
| NANS-CDG | CHO cell line | Nans KO | CMP-sialic acid reduction | [63] | |
| PGM1-CDG | Mouse | Pgm2−/− | Embryonic lethality | [43] | |
| Pgm2+/− |
| ||||
| PGM3-CDG | D. melanogaster | DPgm3 KO (RNAi) |
| [45] | |
| Xenopus laevis | Pgm3 (mRNA) | Posteriorization of embryos | |||
| Pgm3 KO (MO) | Anteriorization of embryos | ||||
| Zebrafish | Pgm3 (mRNA) | Inhibition of eye formation | |||
| SLC35C1-CDG | mESCs (haploid state) | Slc35c1−/− | Lack of fucosylated structures | [55] | |
| Mouse intestinal organoids | Improved ricin resistance | ||||
| SLC39A8-CDG | Mouse | ZIP8-iKO (Slc39a8fl/fl UBC-CreERT2) |
| [68] | |
| ZIP8-LSKO (Slc39a8fl/fl Alb-Cre, a liver-specific KO) |
| ||||
| SRD5A3-CDG | Mouse | Cerebellar conditional KO En1-Cre; Srd5a3fl/- |
| [69] | |
| ST3GAL3-CDG | Mouse | St3gal3 KO | Minor hematologic abnormalities | [70] | |
| St3gal2/st3gal3 double KO | Lack of GD1a and GT1b gangliosides | ||||
| ST3GAL4-CDG | KBM7 ST3GAL4 KO-1 and KO-2 cells | ST3GAL4−/− |
| [55] | |
| ST3GAL5-CDG | HEK 293T | G342S- C195S a- G201A a- E355K-HaloTag-ST3GAL5 | a Complete loss of GM3 synthase activity | [71] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brasil, S.; Allocca, M.; Magrinho, S.C.M.; Santos, I.; Raposo, M.; Francisco, R.; Pascoal, C.; Martins, T.; Videira, P.A.; Pereira, F.; et al. Systematic Review: Drug Repositioning for Congenital Disorders of Glycosylation (CDG). Int. J. Mol. Sci. 2022, 23, 8725. https://doi.org/10.3390/ijms23158725
Brasil S, Allocca M, Magrinho SCM, Santos I, Raposo M, Francisco R, Pascoal C, Martins T, Videira PA, Pereira F, et al. Systematic Review: Drug Repositioning for Congenital Disorders of Glycosylation (CDG). International Journal of Molecular Sciences. 2022; 23(15):8725. https://doi.org/10.3390/ijms23158725
Chicago/Turabian StyleBrasil, Sandra, Mariateresa Allocca, Salvador C. M. Magrinho, Inês Santos, Madalena Raposo, Rita Francisco, Carlota Pascoal, Tiago Martins, Paula A. Videira, Florbela Pereira, and et al. 2022. "Systematic Review: Drug Repositioning for Congenital Disorders of Glycosylation (CDG)" International Journal of Molecular Sciences 23, no. 15: 8725. https://doi.org/10.3390/ijms23158725
APA StyleBrasil, S., Allocca, M., Magrinho, S. C. M., Santos, I., Raposo, M., Francisco, R., Pascoal, C., Martins, T., Videira, P. A., Pereira, F., Andreotti, G., Jaeken, J., Kantautas, K. A., Perlstein, E. O., & Ferreira, V. d. R. (2022). Systematic Review: Drug Repositioning for Congenital Disorders of Glycosylation (CDG). International Journal of Molecular Sciences, 23(15), 8725. https://doi.org/10.3390/ijms23158725