
Exploring Ligand Binding Domain Dynamics in the NRs Superfamily

 , , ,
, , , 
Abstract
:1. Introduction
2. Results
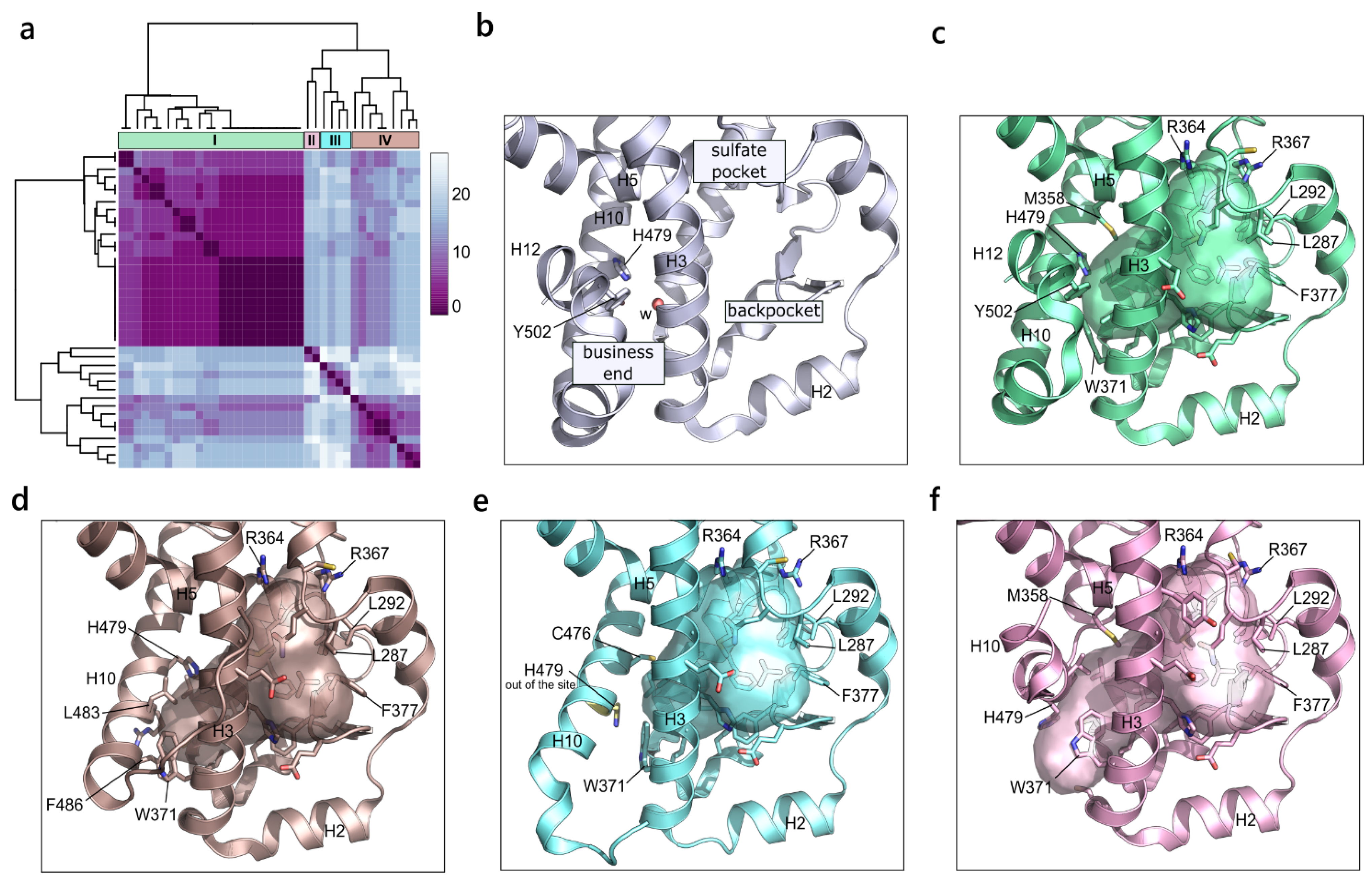
2.1. Intrafamily Variability
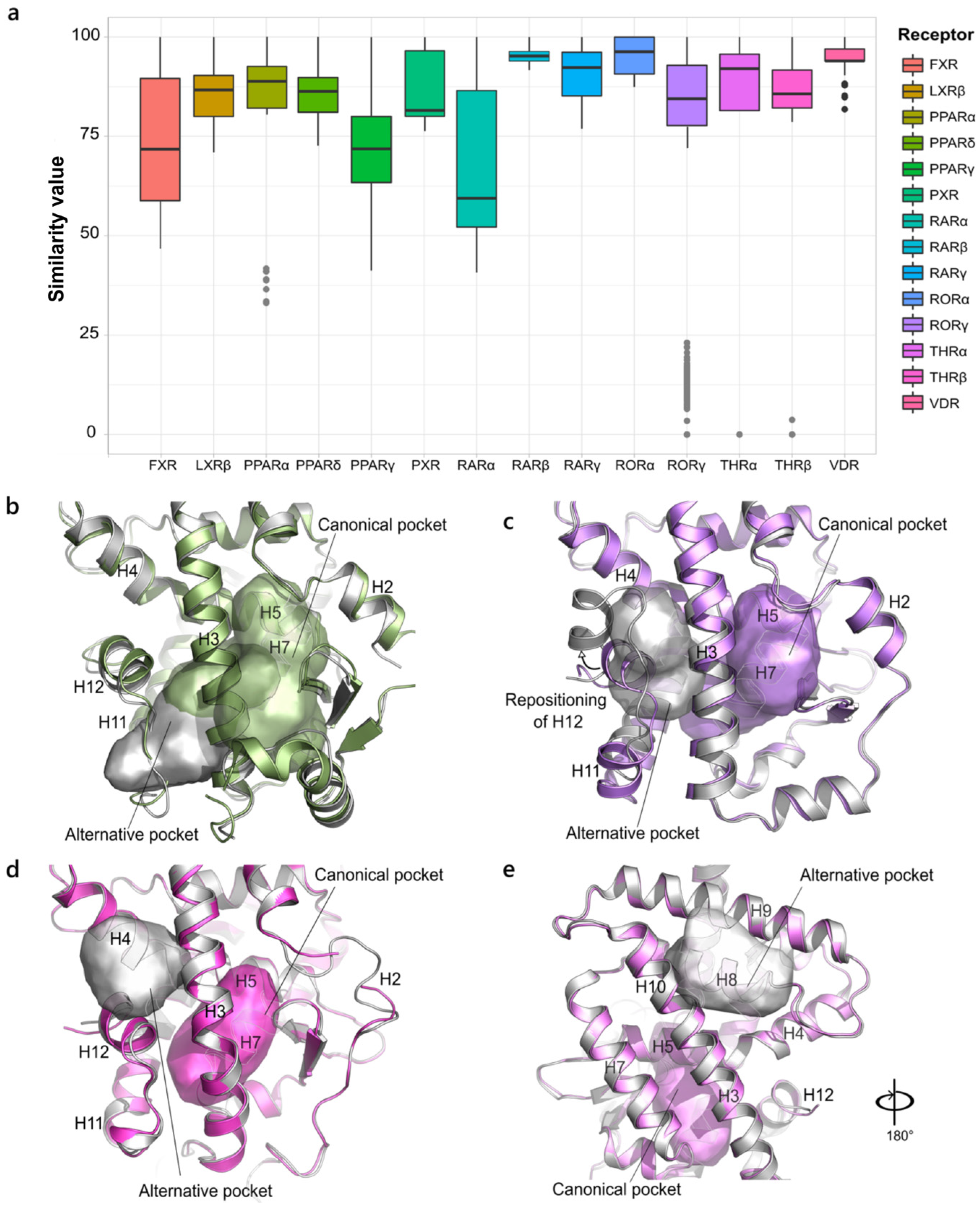
2.1.1. NR1. Thyroid Hormone Receptor-like Family
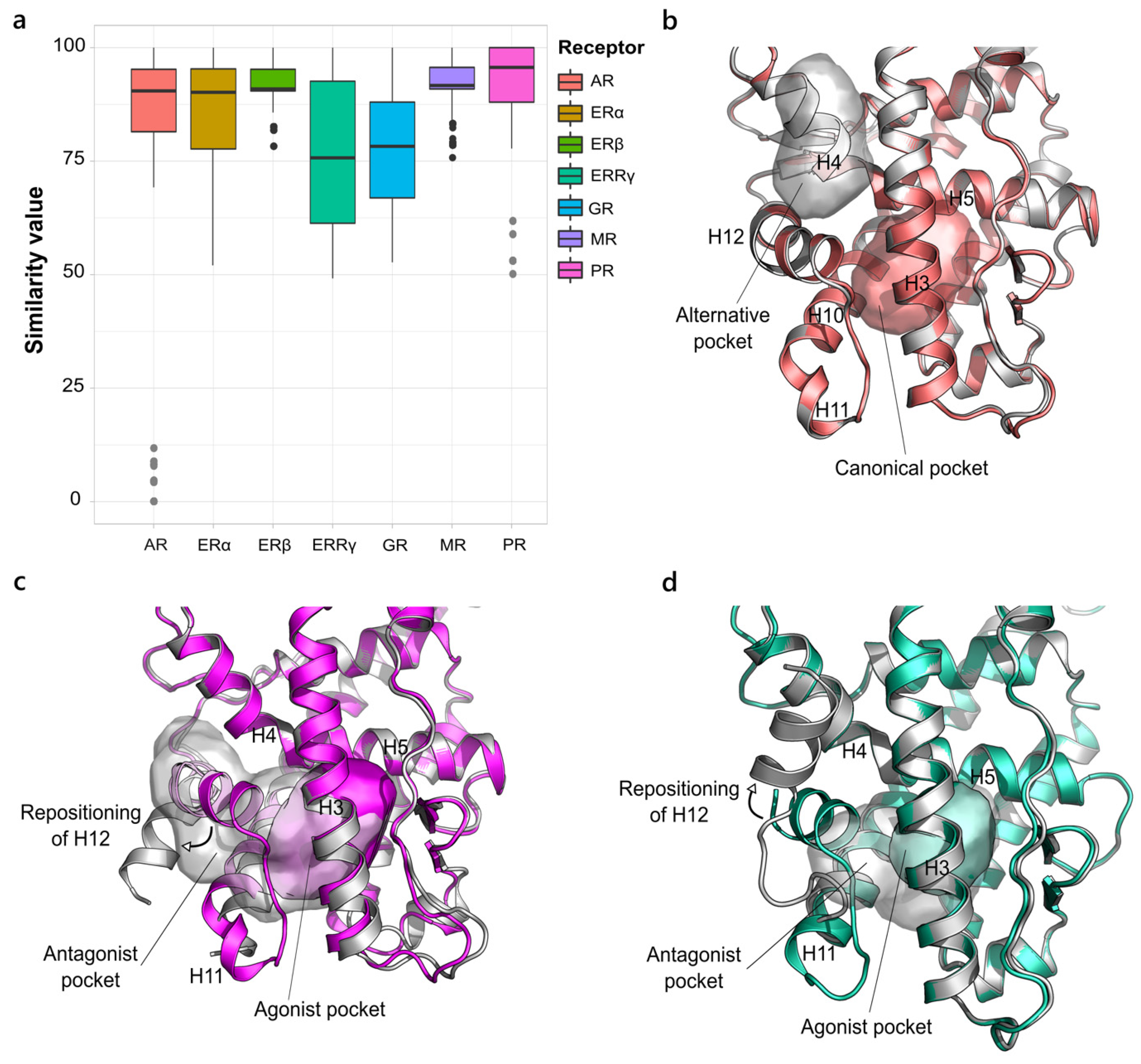
2.1.2. NR3. Estrogen Receptor-like Family
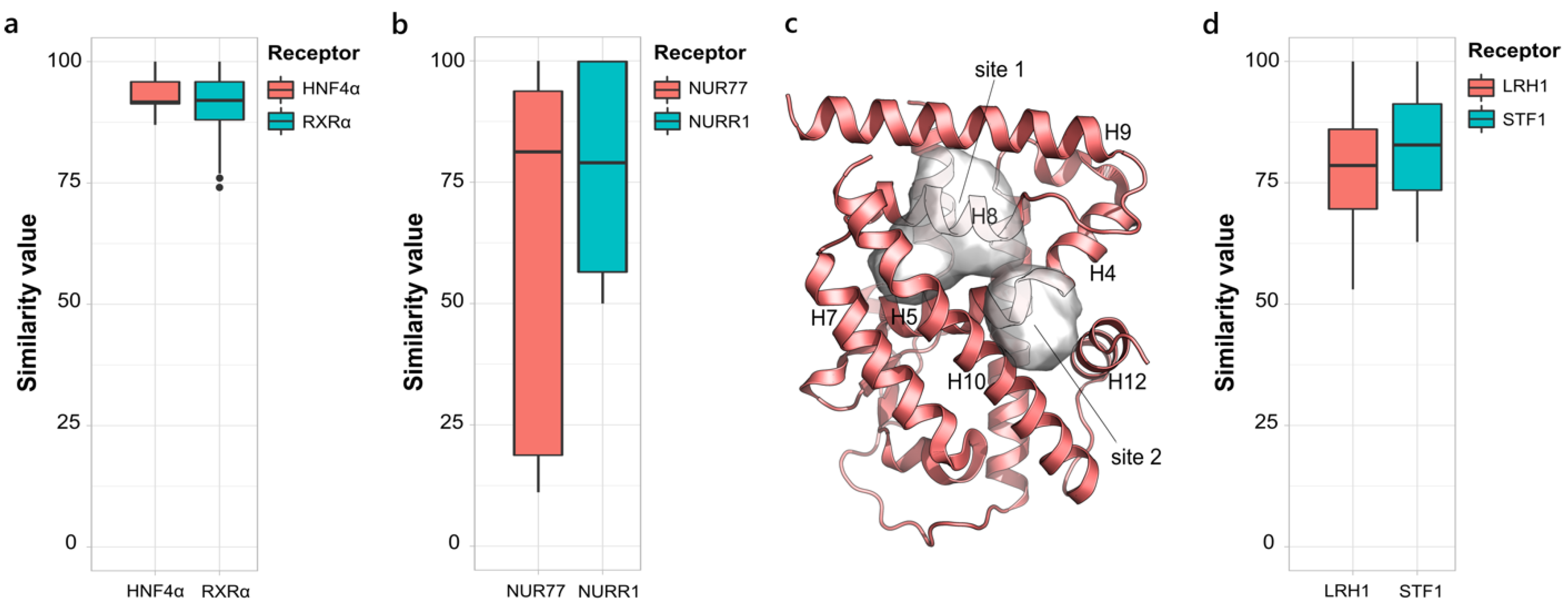
2.1.3. NR2, NR4 and NR5. Retinoid X Receptor-like, Nerve Growth Factor IB-like and Steroidogenic Factor-like Families
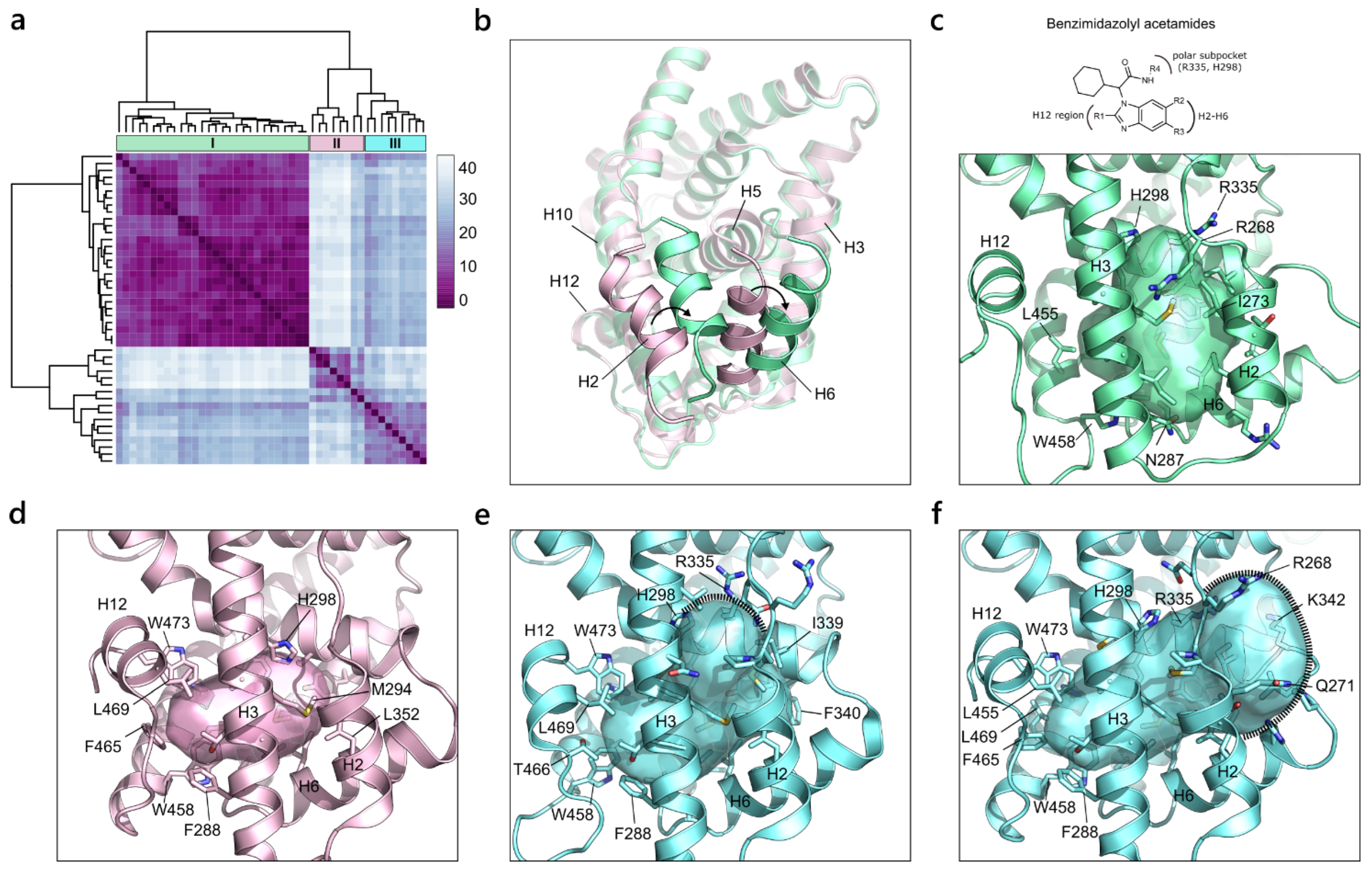
2.2. Intra-Sub-Family Variability
2.2.1. Androgen Receptor (NR3 Family)
2.2.2. Glucocorticoid Receptor (NR3 Family)
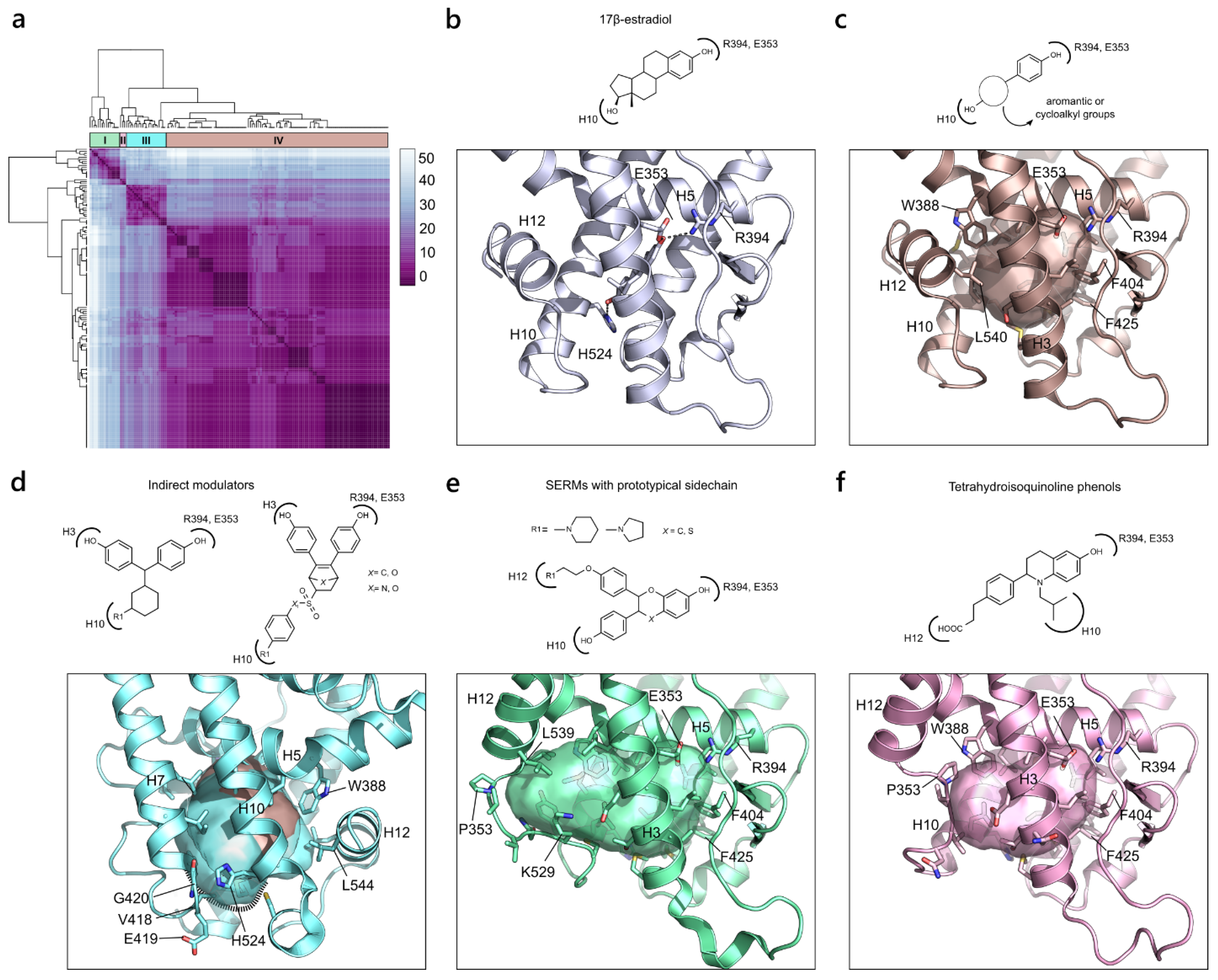
2.2.3. Estrogen Receptor α (NR3 Family)
2.2.4. Retinoic Acid-Related Orphan Receptor γ (NR1 Family)
2.2.5. Farnesoid X Receptor (NR1 Family)
2.3. Identification of Representative Structures
3. Discussion
4. Materials and Methods
4.1. Dataset Preparation
4.2. Pocket Detection
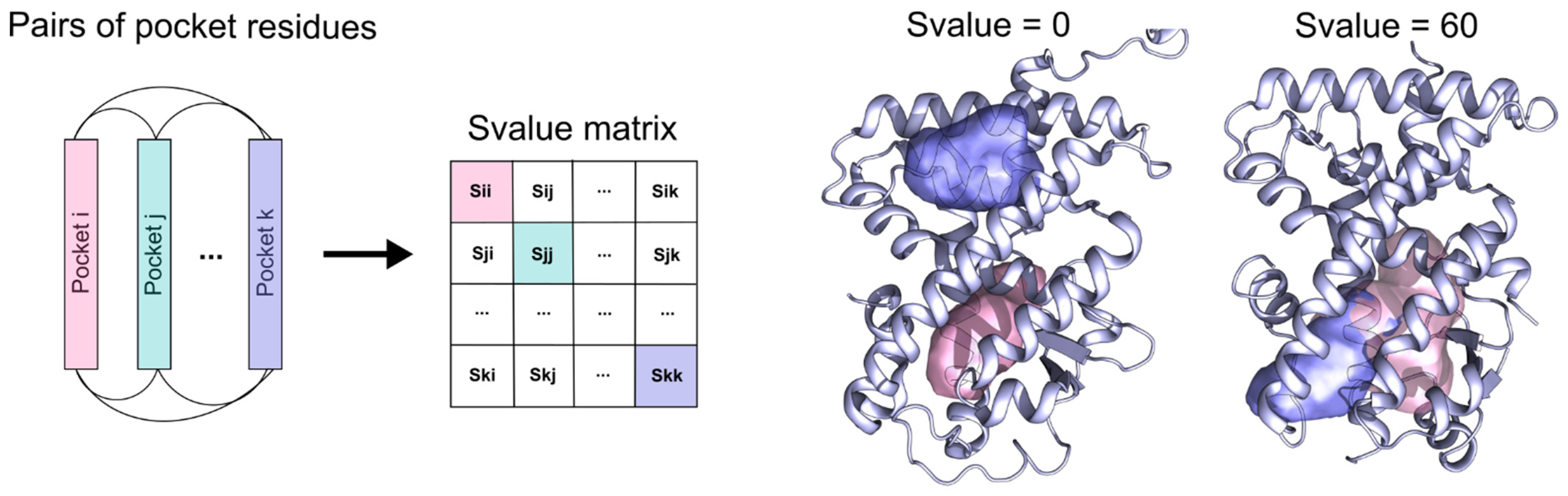
4.3. Pocket Comparison
4.4. Data Elaboration
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sever, R.; Glass, C.K. Signaling by Nuclear Receptors. Cold Spring Harb. Perspect. Biol. 2013, 5, a016709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazaira, G.I.; Zgajnar, N.R.; Lotufo, C.M.; Daneri-Becerra, C.; Sivils, J.C.; Soto, O.B.; Cox, M.B.; Galigniana, M.D. The Nuclear Receptor Field: A Historical Overview and Future Challenges. Nucl. Recept. Res. 2018, 5, 101320. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Zhou, S.; Gustafsson, J.-Å. Nuclear Receptors: Recent Drug Discovery for Cancer Therapies. Endocr. Rev. 2019, 40, 1207–1249. [Google Scholar] [CrossRef] [PubMed]
- Sherman, M.H.; Downes, M.; Evans, R.M. Nuclear Receptors as Modulators of the Tumor Microenvironment. Cancer Prev. Res. 2012, 5, 3–10. [Google Scholar] [CrossRef] [Green Version]
- Ahmadian, M.; Suh, J.M.; Hah, N.; Liddle, C.; Atkins, A.R.; Downes, M.; Evans, R.M. PPARγ Signaling and Metabolism: The Good, the Bad and the Future. Nat. Med. 2013, 19, 557–566. [Google Scholar] [CrossRef] [Green Version]
- Evans, R.M.; Mangelsdorf, D.J. Nuclear Receptors, RXR, and the Big Bang. Cell 2014, 157, 255–266. [Google Scholar] [CrossRef] [Green Version]
- De Bosscher, K.; Desmet, S.J.; Clarisse, D.; Estébanez-Perpiña, E.; Brunsveld, L. Nuclear Receptor Crosstalk—Defining the Mechanisms for Therapeutic Innovation. Nat. Rev. Endocrinol. 2020, 16, 363–377. [Google Scholar] [CrossRef]
- McKenna, N.J.; Cooney, A.J.; DeMayo, F.J.; Downes, M.; Glass, C.K.; Lanz, R.B.; Lazar, M.A.; Mangelsdorf, D.J.; Moore, D.D.; Qin, J.; et al. Minireview: Evolution of NURSA, the Nuclear Receptor Signaling Atlas. Mol. Endocrinol. 2009, 23, 740–746. [Google Scholar] [CrossRef]
- Folkertsma, S.; van Noort, P.I.; Brandt, R.F.J.; Bettler, E.; Vriend, G.; de Vlieg, J. The Nuclear Receptor Ligand-Binding Domain: A Family-Based Structure Analysis. Curr. Med. Chem. 2005, 12, 1001–1016. [Google Scholar] [CrossRef]
- Folkertsma, S.; van Noort, P.; Van Durme, J.; Joosten, H.-J.; Bettler, E.; Fleuren, W.; Oliveira, L.; Horn, F.; de Vlieg, J.; Vriend, G. A Family-Based Approach Reveals the Function of Residues in the Nuclear Receptor Ligand-Binding Domain. J. Mol. Biol. 2004, 341, 321–335. [Google Scholar] [CrossRef]
- Maity, A.; Majumdar, S.; Priya, P.; De, P.; Saha, S.; Ghosh Dastidar, S. Adaptability in Protein Structures: Structural Dynamics and Implications in Ligand Design. J. Biomol. Struct. Dyn. 2015, 33, 298–321. [Google Scholar] [CrossRef] [PubMed]
- Nagy, L.; Schwabe, J.W.R. Mechanism of the Nuclear Receptor Molecular Switch. Trends Biochem. Sci. 2004, 29, 317–324. [Google Scholar] [CrossRef] [PubMed]
- Renaud, J.-P.; Rochel, N.; Ruff, M.; Vivat, V.; Chambon, P.; Gronemeyer, H.; Moras, D. Crystal Structure of the RAR-γ Ligand-Binding Domain Bound to All-Trans Retinoic Acid. Nature 1995, 378, 681–689. [Google Scholar] [CrossRef] [PubMed]
- Kallenberger, B.C.; Love, J.D.; Chatterjee, V.K.K.; Schwabe, J.W.R. A Dynamic Mechanism of Nuclear Receptor Activation and Its Perturbation in a Human Disease. Nat. Struct Mol. Biol. 2003, 10, 136–140. [Google Scholar] [CrossRef]
- Gu, X. Helix 12 in the Human Estrogen Receptor (HER) Is Essential for the HER Function by Overcoming Nucleosome Repression in Yeast. J. Cell Biochem. 2002, 86, 224–238. [Google Scholar] [CrossRef]
- Watanabe, C.; Watanabe, H.; Tanaka, S. An Interpretation of Positional Displacement of the Helix12 in Nuclear Receptors: Preexistent Swing-up Motion Triggered by Ligand Binding. Biochim. Et Biophys. Acta (BBA)-Proteins Proteom. 2010, 1804, 1832–1840. [Google Scholar] [CrossRef]
- Kronenberger, T.; Keminer, O.; Wrenger, C.; Windshügel, B. Nuclear Receptor Modulators—Current Approaches and Future Perspectives; IntechOpen: London, UK, 2015; ISBN 978-953-51-2128-2. [Google Scholar]
- Hellal-Levy, C.; Fagart, J.; Souque, A.; Wurtz, J.M.; Moras, D.; Rafestin-Oblin, M.E. Crucial Role of the H11-H12 Loop in Stabilizing the Active Conformation of the Human Mineralocorticoid Receptor. Mol. Endocrinol. 2000, 14, 1210–1221. [Google Scholar] [CrossRef]
- Shizu, R.; Nishiguchi, H.; Tashiro, S.; Sato, T.; Sugawara, A.; Kanno, Y.; Hosaka, T.; Sasaki, T.; Yoshinari, K. Helix 12 Stabilization Contributes to Basal Transcriptional Activity of PXR. J. Biol. Chem. 2021, 297, 100978. [Google Scholar] [CrossRef]
- Souza, P.C.T.; Textor, L.C.; Melo, D.C.; Nascimento, A.S.; Skaf, M.S.; Polikarpov, I. An Alternative Conformation of ERβ Bound to Estradiol Reveals H12 in a Stable Antagonist Position. Sci. Rep. 2017, 7, 3509. [Google Scholar] [CrossRef]
- Siragusa, L.; Cross, S.; Baroni, M.; Goracci, L.; Cruciani, G. BioGPS: Navigating Biological Space to Predict Polypharmacology, off-Targeting, and Selectivity. Proteins Struct. Funct. Bioinform. 2015, 83, 517–532. [Google Scholar] [CrossRef]
- Yeturu, K.; Chandra, N. PocketMatch: A New Algorithm to Compare Binding Sites in Protein Structures. BMC Bioinform. 2008, 9, 543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kinoshita, K.; Murakami, Y.; Nakamura, H. EF-Seek: Prediction of the Functional Sites of Proteins by Searching for Similar Electrostatic Potential and Molecular Surface Shape. Nucleic Acids Res. 2007, 35, W398–W402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sael, L.; Kihara, D. Detecting Local Ligand-Binding Site Similarity in Nonhomologous Proteins by Surface Patch Comparison. Proteins Struct. Funct. Bioinform. 2012, 80, 1177–1195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kinoshita, K.; Furui, J.; Nakamura, H. Identification of Protein Functions from a Molecular Surface Database, EF-Site. J. Struct. Func. Genom. 2002, 2, 9–22. [Google Scholar] [CrossRef]
- Schmitt, S.; Kuhn, D.; Klebe, G. A New Method to Detect Related Function Among Proteins Independent of Sequence and Fold Homology. J. Mol. Biol. 2002, 323, 387–406. [Google Scholar] [CrossRef]
- Shulman-Peleg, A.; Nussinov, R.; Wolfson, H.J. SiteEngines: Recognition and Comparison of Binding Sites and Protein–Protein Interfaces. Nucleic Acids Res. 2005, 33, W337–W341. [Google Scholar] [CrossRef]
- Gao, M.; Skolnick, J. APoc: Large-Scale Identification of Similar Protein Pockets. Bioinformatics 2013, 29, 597–604. [Google Scholar] [CrossRef] [Green Version]
- Xie, L.; Bourne, P.E. Detecting Evolutionary Relationships across Existing Fold Space, Using Sequence Order-Independent Profile–Profile Alignments. Proc. Natl. Acad. Sci. USA 2008, 105, 5441–5446. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.S.; Im, W. G-LoSA for Prediction of Protein-Ligand Binding Sites and Structures. In Protein Function Prediction: Methods and Protocols; Kihara, D., Ed.; Methods in Molecular Biology; Springer: New York, NY, USA, 2017; pp. 97–108. ISBN 978-1-4939-7015-5. [Google Scholar]
- Capelli, D.; Cerchia, C.; Montanari, R.; Loiodice, F.; Tortorella, P.; Laghezza, A.; Cervoni, L.; Pochetti, G.; Lavecchia, A. Structural Basis for PPAR Partial or Full Activation Revealed by a Novel Ligand Binding Mode. Sci. Rep. 2016, 6, 34792. [Google Scholar] [CrossRef] [Green Version]
- Scheepstra, M.; Leysen, S.; van Almen, G.C.; Miller, J.R.; Piesvaux, J.; Kutilek, V.; van Eenennaam, H.; Zhang, H.; Barr, K.; Nagpal, S.; et al. Identification of an Allosteric Binding Site for RORγt Inhibition. Nat. Commun. 2015, 6, 8833. [Google Scholar] [CrossRef]
- Estébanez-Perpiñá, E.; Arnold, L.A.; Jouravel, N.; Togashi, M.; Blethrow, J.; Mar, E.; Nguyen, P.; Phillips, K.J.; Baxter, J.D.; Webb, P.; et al. Structural Insight into the Mode of Action of a Direct Inhibitor of Coregulator Binding to the Thyroid Hormone Receptor. Mol. Endocrinol. 2007, 21, 2919–2928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Souza, P.C.T.; Puhl, A.C.; Martínez, L.; Aparício, R.; Nascimento, A.S.; Figueira, A.C.M.; Nguyen, P.; Webb, P.; Skaf, M.S.; Polikarpov, I. Identification of a New Hormone-Binding Site on the Surface of Thyroid Hormone Receptor. Mol. Endocrinol. 2014, 28, 534–545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hughes, N.E.; Bleisch, T.J.; Jones, S.A.; Richardson, T.I.; Doti, R.A.; Wang, Y.; Stout, S.L.; Durst, G.L.; Chambers, M.G.; Oskins, J.L.; et al. Identification of Potent and Selective Retinoic Acid Receptor Gamma (RARγ) Antagonists for the Treatment of Osteoarthritis Pain Using Structure Based Drug Design. Bioorg. Med. Chem. Lett. 2016, 26, 3274–3277. [Google Scholar] [CrossRef] [Green Version]
- le Maire, A.; Teyssier, C.; Erb, C.; Grimaldi, M.; Alvarez, S.; de Lera, A.R.; Balaguer, P.; Gronemeyer, H.; Royer, C.A.; Germain, P.; et al. A Unique Secondary-Structure Switch Controls Constitutive Gene Repression by Retinoic Acid Receptor. Nat. Struct. Mol. Biol. 2010, 17, 801–807. [Google Scholar] [CrossRef] [PubMed]
- Fradera, X.; Vu, D.; Nimz, O.; Skene, R.; Hosfield, D.; Wynands, R.; Cooke, A.J.; Haunsø, A.; King, A.; Bennett, D.J.; et al. X-Ray Structures of the LXRalpha LBD in Its Homodimeric Form and Implications for Heterodimer Signaling. J. Mol. Biol. 2010, 399, 120–132. [Google Scholar] [CrossRef]
- Wu, C.-C.; Baiga, T.J.; Downes, M.; La Clair, J.J.; Atkins, A.R.; Richard, S.B.; Fan, W.; Stockley-Noel, T.A.; Bowman, M.E.; Noel, J.P.; et al. Structural Basis for Specific Ligation of the Peroxisome Proliferator-Activated Receptor δ. Proc. Natl. Acad. Sci. USA 2017, 114, E2563–E2570. [Google Scholar] [CrossRef] [Green Version]
- Lack, N.A.; Axerio-Cilies, P.; Tavassoli, P.; Han, F.Q.; Chan, K.H.; Feau, C.; LeBlanc, E.; Guns, E.T.; Guy, R.K.; Rennie, P.S.; et al. Targeting the Binding Function 3 (BF3) Site of the Human Androgen Receptor through Virtual Screening. J. Med. Chem. 2011, 54, 8563–8573. [Google Scholar] [CrossRef] [Green Version]
- Pike, A.C.W. Structure of the Ligand-Binding Domain of Oestrogen Receptor Beta in the Presence of a Partial Agonist and a Full Antagonist. EMBO J. 1999, 18, 4608–4618. [Google Scholar] [CrossRef] [Green Version]
- Nahoum, V.; Pérez, E.; Germain, P.; Rodríguez-Barrios, F.; Manzo, F.; Kammerer, S.; Lemaire, G.; Hirsch, O.; Royer, C.A.; Gronemeyer, H.; et al. Modulators of the Structural Dynamics of the Retinoid X Receptor to Reveal Receptor Function. Proc. Natl. Acad. Sci. USA 2007, 104, 17323–17328. [Google Scholar] [CrossRef] [Green Version]
- Scheepstra, M.; Andrei, S.A.; de Vries, R.M.J.M.; Meijer, F.A.; Ma, J.-N.; Burstein, E.S.; Olsson, R.; Ottmann, C.; Milroy, L.-G.; Brunsveld, L. Ligand Dependent Switch from RXR Homo- to RXR-NURR1 Heterodimerization. ACS Chem. Neurosci. 2017, 8, 2065–2077. [Google Scholar] [CrossRef]
- Martínez-González, J.; Badimon, L. The NR4A Subfamily of Nuclear Receptors: New Early Genes Regulated by Growth Factors in Vascular Cells. Cardiovasc. Res. 2005, 65, 609–618. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Wang, Y.; Hou, P.; Li, F.; Zhou, B.; Chen, H.; Bian, X.; Cai, Q.; Xing, Y.; He, J.; et al. Induction of Autophagic Death in Cancer Cells by Agonizing TR3 and Attenuating Akt2 Activity. Chem. Biol. 2015, 22, 1040–1051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Liu, Y.; Chen, H.; Li, F.; Wu, J.; Zhang, H.; He, J.; Xing, Y.; Chen, Y.; Wang, W.; et al. Impeding the Interaction between Nur77 and P38 Reduces LPS-Induced Inflammation. Nat. Chem. Biol. 2015, 11, 339–346. [Google Scholar] [CrossRef] [Green Version]
- Sablin, E.P.; Krylova, I.N.; Fletterick, R.J.; Ingraham, H.A. Structural Basis for Ligand-Independent Activation of the Orphan Nuclear Receptor LRH-1. Mol. Cell 2003, 11, 1575–1585. [Google Scholar] [CrossRef]
- Heinlein, C.A.; Chang, C. Androgen Receptor in Prostate Cancer. Endocr. Rev. 2004, 25, 276–308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cadilla, R.; Turnbull, P. Selective Androgen Receptor Modulators in Drug Discovery: Medicinal Chemistry and Therapeutic Potential. Curr. Top. Med. Chem. 2006, 6, 245–270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hara, T.; Miyazaki, J.I.; Araki, H.; Yamaoka, M.; Kanzaki, N.; Kusaka, M.; Miyamoto, M. Novel Mutations of Androgen Receptor: A Possible Mechanism of Bicalutamide Withdrawal Syndrome. Cancer Res. 2003, 63, 149–153. [Google Scholar]
- Tang, Z.-R.; Zhang, R.; Lian, Z.-X.; Deng, S.-L.; Yu, K. Estrogen-Receptor Expression and Function in Female Reproductive Disease. Cells 2019, 8, 1123. [Google Scholar] [CrossRef] [Green Version]
- Srinivasan, S.; Nwachukwu, J.C.; Bruno, N.E.; Dharmarajan, V.; Goswami, D.; Kastrati, I.; Novick, S.; Nowak, J.; Cavett, V.; Zhou, H.-B.; et al. Full Antagonism of the Estrogen Receptor without a Prototypical Ligand Side Chain. Nat. Chem. Biol. 2017, 13, 111–118. [Google Scholar] [CrossRef] [Green Version]
- Kallen, J.; Izaac, A.; Be, C.; Arista, L.; Orain, D.; Kaupmann, K.; Guntermann, C.; Hoegenauer, K.; Hintermann, S. Structural States of RORγt: X-Ray Elucidation of Molecular Mechanisms and Binding Interactions for Natural and Synthetic Compounds. ChemMedChem 2017, 12, 1014–1021. [Google Scholar] [CrossRef]
- Richter, H.G.F.; Benson, G.M.; Bleicher, K.H.; Blum, D.; Chaput, E.; Clemann, N.; Feng, S.; Gardes, C.; Grether, U.; Hartman, P.; et al. Optimization of a Novel Class of Benzimidazole-Based Farnesoid X Receptor (FXR) Agonists to Improve Physicochemical and ADME Properties. Bioorg. Med. Chem. Lett. 2011, 21, 1134–1140. [Google Scholar] [CrossRef]
- Williams, C.; Lin, C.-Y. Oestrogen Receptors in Breast Cancer: Basic Mechanisms and Clinical Implications. Ecancermedicalscience 2013, 7, 370. [Google Scholar] [CrossRef] [PubMed]
- Belachew, E.B.; Sewasew, D.T. Molecular Mechanisms of Endocrine Resistance in Estrogen-Receptor-Positive Breast Cancer. Front. Endocrinol. 2021, 12, 599586. [Google Scholar] [CrossRef] [PubMed]
- Feng, Q.; He, B. Androgen Receptor Signaling in the Development of Castration-Resistant Prostate Cancer. Front. Oncol. 2019, 9, 858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Y.; Bolton, E.C.; Jones, J.O. Androgens and Androgen Receptor Signaling in Prostate Tumorigenesis. J. Mol. Endocrinol. 2015, 54, R15–R29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Timmermans, S.; Souffriau, J.; Libert, C. A General Introduction to Glucocorticoid Biology. Front. Immunol. 2019, 10, 1545. [Google Scholar] [CrossRef] [Green Version]
- Jiang, L.; Zhang, H.; Xiao, D.; Wei, H.; Chen, Y. Farnesoid X Receptor (FXR): Structures and Ligands. Comput. Struct. Biotechnol. J. 2021, 19, 2148–2159. [Google Scholar] [CrossRef]
- Weikum, E.R.; Liu, X.; Ortlund, E.A. The Nuclear Receptor Superfamily: A Structural Perspective: The Nuclear Receptor Superfamily. Protein Sci. 2018, 27, 1876–1892. [Google Scholar] [CrossRef]
- Ecoeur, F.; Weiss, J.; Kaupmann, K.; Hintermann, S.; Orain, D.; Guntermann, C. Antagonizing Retinoic Acid-Related-Orphan Receptor Gamma Activity Blocks the T Helper 17/Interleukin-17 Pathway Leading to Attenuated Pro-Inflammatory Human Keratinocyte and Skin Responses. Front. Immunol. 2019, 10, 577. [Google Scholar] [CrossRef]
- Janani, C.; Ranjitha Kumari, B.D. PPAR Gamma Gene—A Review. Diabetes Metab. Syndr. 2015, 9, 46–50. [Google Scholar] [CrossRef]
- de Vink, P.J.; Koops, A.A.; D’Arrigo, G.; Cruciani, G.; Spyrakis, F.; Brunsveld, L. Cooperativity as Quantification and Optimization Paradigm for Nuclear Receptor Modulators. Chem. Sci. 2022, 13, 2744–2752. [Google Scholar] [CrossRef] [PubMed]
- Kongsbak, M.; Levring, T.; Geisler, C.; von Essen, M. The Vitamin D Receptor and T Cell Function. Front. Immunol. 2013, 4, 148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaveri, N.T.; Murphy, B.J. 2.25—Nuclear Hormone Receptors. In Comprehensive Medicinal Chemistry II; Taylor, J.B., Triggle, D.J., Eds.; Elsevier: Oxford, MS, USA, 2007; pp. 993–1036. ISBN 978-0-08-045044-5. [Google Scholar]
- Núñez, V.; Alameda, D.; Rico, D.; Mota, R.; Gonzalo, P.; Cedenilla, M.; Fischer, T.; Boscá, L.; Glass, C.K.; Arroyo, A.G.; et al. Retinoid X Receptor α Controls Innate Inflammatory Responses through the Up-Regulation of Chemokine Expression. Proc. Natl. Acad. Sci. USA 2010, 107, 10626–10631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, M.E.; Li, J.; Xu, H.E.; Melcher, K.; Yong, E. Androgen Receptor: Structure, Role in Prostate Cancer and Drug Discovery. Acta Pharmacol. Sin. 2015, 36, 3–23. [Google Scholar] [CrossRef] [Green Version]
- Schneider, M.; Pons, J.-L.; Labesse, G. Exploring the Conformational Space of a Receptor for Drug Design: An ERα Case Study. J. Mol. Graph. Model. 2021, 108, 107974. [Google Scholar] [CrossRef]
- Osguthorpe, D.J.; Sherman, W.; Hagler, A.T. Exploring Protein Flexibility: Incorporating Structural Ensembles From Crystal Structures and Simulation into Virtual Screening Protocols. J. Phys. Chem. B 2012, 116, 6952–6959. [Google Scholar] [CrossRef] [Green Version]
- Hou, T.-Y.; Weng, C.-F.; Leong, M.K. Insight Analysis of Promiscuous Estrogen Receptor α-Ligand Binding by a Novel Machine Learning Scheme. Chem. Res. Toxicol. 2018, 31, 799–813. [Google Scholar] [CrossRef]
- Baroni, M.; Cruciani, G.; Sciabola, S.; Perruccio, F.; Mason, J.S. A Common Reference Framework for Analyzing/Comparing Proteins and Ligands. Fingerprints for Ligands And Proteins (FLAP): Theory and Application. J. Chem. Inf. Model. 2007, 47, 279–294. [Google Scholar] [CrossRef]
- RStudio Team. RStudio: Integrated Development for R. RStudio; PBC: Boston, MA, USA, 2020; Available online: http://www.rstudio.com/ (accessed on 1 August 2022).



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Family | Sub-Family | Total LBD Structures | LBD Structures (R ≤ 2.5) | Final N° Liganded Pockets |
|---|---|---|---|---|
| NR1 | CAR | 2 | / | / |
| FXR | 81 | 61 | 45 | |
| LXRα | 7 | 4 | / | |
| LXRβ | 13 | 9 | 7 | |
| PPARα | 18 | 15 | 9 | |
| PPARδ | 43 | 34 | 17 | |
| PPARγ | 204 | 156 | 40 | |
| PXR | 23 | 7 | 4 | |
| RARα | 6 | 4 | 4 | |
| RARβ | 7 | 5 | 4 | |
| RARγ | 11 | 11 | 10 | |
| Rev-erba-A | 1 | / | / | |
| Rev-erba-B | 4 | 4 | / | |
| RORα | 3 | 3 | 3 | |
| RORβ * | / | / | / | |
| RORγ | 88 | 62 | 46 | |
| THRα | 8 | 8 | 9 | |
| THRβ | 18 | 9 | 10 | |
| VDR | 49 | 39 | 39 | |
| NR2 | COUP-TFI ** | / | / | / |
| COUP-TFII | 1 | 1 | / | |
| HNF4α | 6 | 3 | 3 | |
| HNF4γ | 1 | / | / | |
| PNR (E3) | 1 | / | / | |
| RXRα | 80 | 38 | 38 | |
| RXRβ | 6 | 6 | / | |
| RXRγ | 1 | 1 | / | |
| TLX (E1) | 1 | / | / | |
| TR2 (C1) * | / | / | / | |
| TR4 (C2) | 1 | / | / | |
| NR3 | AR | 75 | 68 | 70 |
| ERα | 274 | 227 | 148 | |
| ER2β | 32 | 27 | 27 | |
| ERRα | 4 | 4 | / | |
| ERRγ | 16 | 12 | 8 | |
| GR | 43 | 37 | 18 | |
| MR | 28 | 28 | 28 | |
| PR | 19 | 17 | 17 | |
| NR4 | NGFI-B | 15 | 12 | 5 |
| NOR1 * | / | / | / | |
| NURR1 | 4 | 4 | 3 | |
| NR5 | LRH-1 | 16 | 14 | 14 |
| SF-1 | 4 | 4 | 4 |
| Family | Sub-Family | Representative Structures |
|---|---|---|
| NR1 | FXR | 5Q1H, 5ICK, 5Q0W |
| LXRα | 3IPU | |
| LXRβ | 1PQ9, 3KFC | |
| PPARα | 2P54, 5HYK | |
| PPARδ | 3SP9, 5U3Z, 5U45 | |
| PPARγ | 3NOA, 2I4P, 1ZGY, 3TY0, 4E4Q, 6FZF | |
| PXR | 1NRL, 5A86 | |
| RARα | 1DKF, 3KMR, 3KMZ | |
| RARβ | 4JYI | |
| RARγ | 1FCX | |
| RORα | 1S0X | |
| RORγ | 3KYT, 4WQP, 5X8Q, 6Q2W | |
| THRα | 2H79 | |
| THRβ | 2J4A | |
| VDR | 2HB8 | |
| NR2 | HNF4α | 3FS1 |
| RXRα | 1FBY | |
| NR3 | AR | 1T63, 3B67 |
| ERα | 1L2I, 2AYR, 5DUH, 5FQP | |
| ER2β | 1U9E, 2GIU | |
| ERRγ | 2P7G, 2GPU | |
| GR | 6EL7, 4CSJ | |
| MR | 3VHU | |
| PR | 1A28, 2OVH | |
| NR4 | NURR1 | 6DDA, 5Y41 |
| NGFI-B | 4R38, 3V3Q | |
| NR5 | LRH-1 | 4PLE, 1YOK |
| SF-1 | 4QJR |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
D’Arrigo, G.; Autiero, I.; Gianquinto, E.; Siragusa, L.; Baroni, M.; Cruciani, G.; Spyrakis, F. Exploring Ligand Binding Domain Dynamics in the NRs Superfamily. Int. J. Mol. Sci. 2022, 23, 8732. https://doi.org/10.3390/ijms23158732
D’Arrigo G, Autiero I, Gianquinto E, Siragusa L, Baroni M, Cruciani G, Spyrakis F. Exploring Ligand Binding Domain Dynamics in the NRs Superfamily. International Journal of Molecular Sciences. 2022; 23(15):8732. https://doi.org/10.3390/ijms23158732
Chicago/Turabian StyleD’Arrigo, Giulia, Ida Autiero, Eleonora Gianquinto, Lydia Siragusa, Massimo Baroni, Gabriele Cruciani, and Francesca Spyrakis. 2022. "Exploring Ligand Binding Domain Dynamics in the NRs Superfamily" International Journal of Molecular Sciences 23, no. 15: 8732. https://doi.org/10.3390/ijms23158732
APA StyleD’Arrigo, G., Autiero, I., Gianquinto, E., Siragusa, L., Baroni, M., Cruciani, G., & Spyrakis, F. (2022). Exploring Ligand Binding Domain Dynamics in the NRs Superfamily. International Journal of Molecular Sciences, 23(15), 8732. https://doi.org/10.3390/ijms23158732