
Extracellular Vesicles in Myeloid Neoplasms


Abstract
:1. Introduction
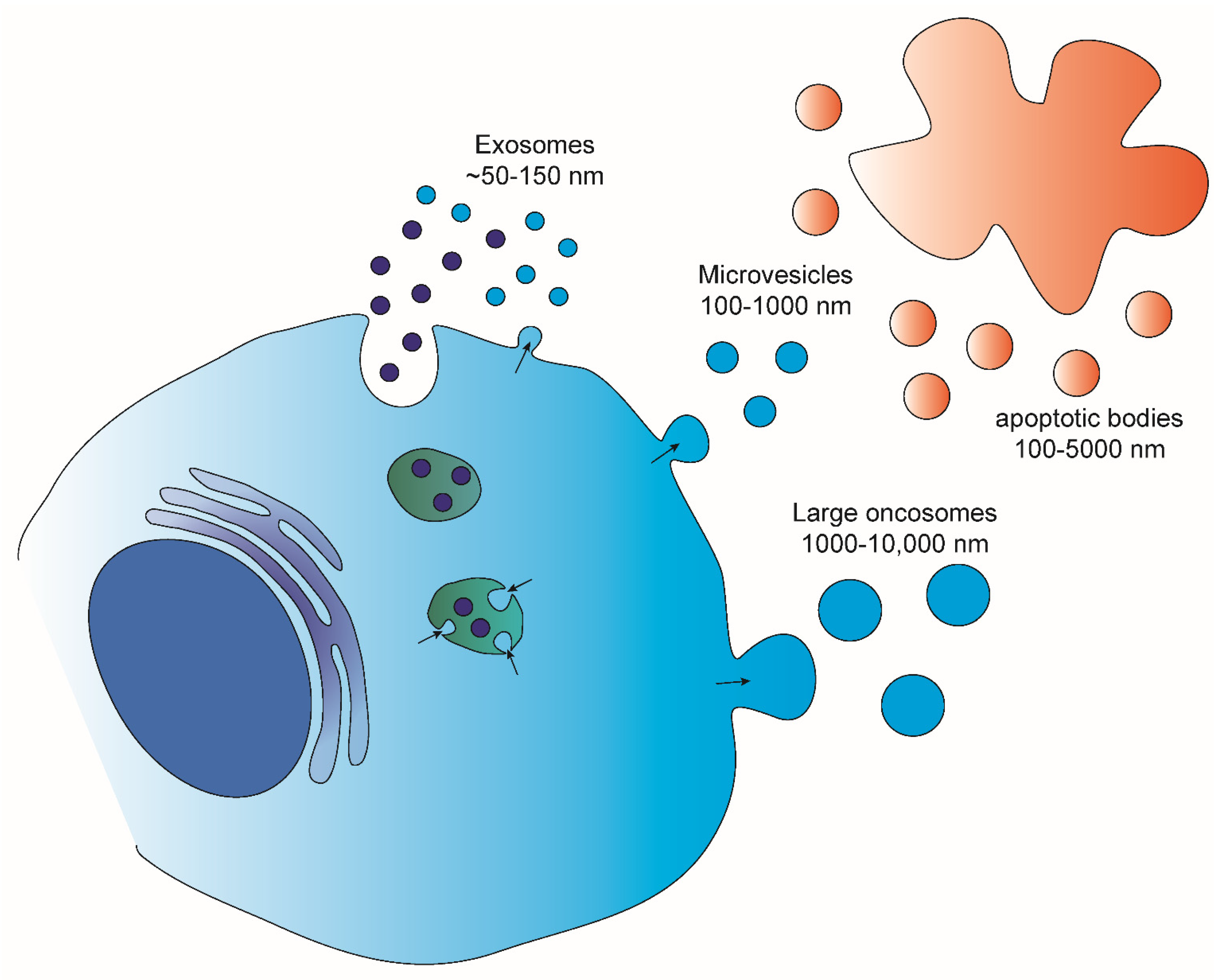
2. Extracellular Vesicles
2.1. Extracellular Vesicles in Philadelphia-Negative Myeloproliferative Neoplasms
2.2. Extracellular Vesicles in Essential Thrombocytosis
2.3. Extracellular Vesicles in Polycythemia Vera
2.4. Extracellular Vesicles in Primary Myelofibrosis
2.5. Extracellular Vesicles in Chronic Myeloid Leukemia
2.6. Extracellular Vesicles in Myelodysplastic Syndrome
2.7. Extracellular Vesicles in Acute Myeloid Leukemia

{kind=link}
| Disease | Extracellular Vesicle | Cargo | Association/Effect | References |
|---|---|---|---|---|
| ET | Platelet MPs | Platelet and endothelial markers (CD61, CD144) | Hypercoagulable state, increased thrombosis risk | [70] |
| ET | BM exo | circDAP3, circASXL1, circRUNX1 | Decreased exo number | [74] |
| PV | Platelet EVs | Protein diversity, specific DNA microbial signature | Increased EV number, pro-coagulation, inflammation | [76,77] |
| PMF | Platelet EVs, endothelial cell EVs, erythrocyte EVs | N/A | Increased EV number | [83] |
| PMF-TN | Plasma EVs | mRNA (miR-361-5p) | CD34+ cell survival | [85] |
| CML | CML-exo | TGFβ1 | Apoptosis inhibition | [97] |
| CML | CML-exo | Amphiregulin (AREG) | Enhanced CML proliferation via BM stroma, increased adhesion to BM stroma | [98] |
| CML | CML-exo | Ν/A | Polarization of MΦ to tumor-associated MΦ | [99] |
| CML | CML-exo | Ν/A | Induced angiogenic activity of EC | [102] |
| CML | BM stromal cell-exo | FGF2 | TKI resistance | [103] |
| MDS | Plasma EVs | miR-1237-3p, U33, hsa_piR_019420, miR-548av-5p | Poor survival | [117] |
| MDS | MDS EVs | N/A | Suppressed MSC differentiation to OB | [111] |
| MDS | MSC EVs | miR-10a, miR-132 | Altered EV protein expression, increased viability and clonogenicity of CD34+ cells | [118] |
| MDS | MSC EVs | miR-486-5p | Increased DNA damage and mutagenesis of HSC | [120] |
| AML | AML MVs | TGFβ1 | Suppressed NK function | [130] |
| AML | AML EVs | IGF-IR | Increased proliferation and VEGF expression in MSC | [131] |
| AML | AML EVs | UPR | ER stress in MSC and osteoprogenitors | [132] |
| AML | MSC exo | miR-7-5-p | Increased AML apoptosis, inhibition of PI3K/AKT/mTOR | [135] |
| AML | AML exo | miR-150, miR-155 | Suppressed HSPC differentiation and proliferation | [137] |
| AML | AML EVs | miR-1246 | Increased LT-HSC quiescence via Raptor | [138] |
| AML | AML exo | AML-related coding and non-coding RNAs | Reduced migration of pre-B cells | [131] |
| AML | AML exo | N/A | Suppressed osteogenesis and normal hematopoiesis via DKK1, leukemia development | [139] |
3. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016, 127, 2391–2405. [Google Scholar] [CrossRef]
- Lindsley, R.C.; Ebert, B.L. The biology and clinical impact of genetic lesions in myeloid malignancies. Blood 2013, 122, 3741–3748. [Google Scholar] [CrossRef] [Green Version]
- Korn, C.; Méndez-Ferrer, S. Myeloid malignancies and the microenvironment. Blood 2017, 129, 811–822. [Google Scholar] [CrossRef] [Green Version]
- Teodorescu, P.; Pasca, S.; Dima, D.; Tomuleasa, C.; Ghiaur, G. Targeting the Microenvironment in MDS: The Final Frontier. Front. Pharmacol. 2020, 11, 1044. [Google Scholar] [CrossRef]
- Wagner, P.N.; Shi, Q.; Salisbury-Ruf, C.T.; Zou, J.; Savona, M.R.; Fedoriw, Y.; Zinkel, S.S. Increased Ripk1-mediated bone marrow necroptosis leads to myelodysplasia and bone marrow failure in mice. Blood 2019, 133, 107–120. [Google Scholar] [CrossRef]
- Bhagat, T.D.; Chen, S.; Bartenstein, M.; Barlowe, A.T.; Von Ahrens, D.; Choudhary, G.S.; Tivnan, P.; Amin, E.; Marcondes, A.M.; Sanders, M.A.; et al. Epigenetically Aberrant Stroma in MDS Propagates Disease via Wnt/β-Catenin Activation. Cancer Res. 2017, 77, 4846–4857. [Google Scholar] [CrossRef] [Green Version]
- Doyle, L.M.; Wang, M.Z. Overview of Extracellular Vesicles, Their Origin, Composition, Purpose, and Methods for Exosome Isolation and Analysis. Cells 2019, 8, 727. [Google Scholar] [CrossRef] [Green Version]
- Cavallari, C.; Camussi, G.; Brizzi, M.F. Extracellular Vesicles in the Tumour Microenvironment: Eclectic Supervisors. Int. J. Mol. Sci. 2020, 21, 768. [Google Scholar] [CrossRef]
- Jafari, R.; Rahbarghazi, R.; Ahmadi, M.; Hassanpour, M.; Rezaie, J. Hypoxic exosomes orchestrate tumorigenesis: Molecular mechanisms and therapeutic implications. J. Transl. Med. 2020, 18, 474. [Google Scholar] [CrossRef]
- Becker, A.; Thakur, B.K.; Weiss, J.M.; Kim, H.S.; Peinado, H.; Lyden, D. Extracellular Vesicles in Cancer: Cell-to-Cell Mediators of Metastasis. Cancer Cell 2016, 30, 836–848. [Google Scholar] [CrossRef] [Green Version]
- Wu, Q.; Zhang, H.; Sun, S.; Wang, L.; Sun, S. Extracellular vesicles and immunogenic stress in cancer. Cell Death Dis. 2021, 12, 894. [Google Scholar] [CrossRef]
- Zhang, D.X.; Vu, L.T.; Ismail, N.N.; Le, M.T.N.; Grimson, A. Landscape of extracellular vesicles in the tumour microenvironment: Interactions with stromal cells and with non-cell components, and impacts on metabolic reprogramming, horizontal transfer of neoplastic traits, and the emergence of therapeutic resistance. Semin. Cancer Biol. 2021, 74, 24–44. [Google Scholar] [CrossRef]
- Yáñez-Mó, M.; Siljander, P.R.; Andreu, Z.; Zavec, A.B.; Borràs, F.E.; Buzas, E.I.; Buzas, K.; Casal, E.; Cappello, F.; Carvalho, J.; et al. Biological properties of extracellular vesicles and their physiological functions. J. Extracell. Vesicles 2015, 4, 27066. [Google Scholar] [CrossRef] [Green Version]
- Lötvall, J.; Hill, A.F.; Hochberg, F.; Buzás, E.I.; Di Vizio, D.; Gardiner, C.; Gho, Y.S.; Kurochkin, I.V.; Mathivanan, S.; Quesenberry, P.; et al. Minimal experimental requirements for definition of extracellular vesicles and their functions: A position statement from the International Society for Extracellular Vesicles. J. Extracell. Vesicles 2014, 3, 26913. [Google Scholar] [CrossRef]
- Théry, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G.K.; et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J. Extracell. Vesicles 2018, 7, 1535750. [Google Scholar] [CrossRef] [Green Version]
- Minciacchi, V.R.; Freeman, M.R.; Di Vizio, D. Extracellular vesicles in cancer: Exosomes, microvesicles and the emerging role of large oncosomes. Semin. Cell Dev. Biol. 2015, 40, 41–51. [Google Scholar] [CrossRef] [Green Version]
- van Niel, G.; D’Angelo, G.; Raposo, G. Shedding light on the cell biology of extracellular vesicles. Nat. Rev. Mol. Cell Biol. 2018, 19, 213–228. [Google Scholar] [CrossRef]
- O′Brien, K.; Breyne, K.; Ughetto, S.; Laurent, L.C.; Breakefield, X.O. RNA delivery by extracellular vesicles in mammalian cells and its applications. Nat. Rev. Mol. Cell Biol. 2020, 21, 585–606. [Google Scholar] [CrossRef]
- Hoshino, A.; Kim, H.S.; Bojmar, L.; Gyan, K.E.; Cioffi, M.; Hernandez, J.; Zambirinis, C.P.; Rodrigues, G.; Molina, H.; Heissel, S.; et al. Extracellular Vesicle and Particle Biomarkers Define Multiple Human Cancers. Cell 2020, 182, 1044–1061.e1018. [Google Scholar] [CrossRef]
- Möller, A.; Lobb, R.J. The evolving translational potential of small extracellular vesicles in cancer. Nat. Rev. Cancer 2020, 20, 697–709. [Google Scholar] [CrossRef]
- Peinado, H.; Alečković, M.; Lavotshkin, S.; Matei, I.; Costa-Silva, B.; Moreno-Bueno, G.; Hergueta-Redondo, M.; Williams, C.; García-Santos, G.; Ghajar, C.; et al. Melanoma exosomes educate bone marrow progenitor cells toward a pro-metastatic phenotype through MET. Nat. Med. 2012, 18, 883–891. [Google Scholar] [CrossRef] [Green Version]
- Chen, I.H.; Xue, L.; Hsu, C.C.; Paez, J.S.; Pan, L.; Andaluz, H.; Wendt, M.K.; Iliuk, A.B.; Zhu, J.K.; Tao, W.A. Phosphoproteins in extracellular vesicles as candidate markers for breast cancer. Proc. Natl. Acad. Sci. USA 2017, 114, 3175–3180. [Google Scholar] [CrossRef] [Green Version]
- Costa-Silva, B.; Aiello, N.M.; Ocean, A.J.; Singh, S.; Zhang, H.; Thakur, B.K.; Becker, A.; Hoshino, A.; Mark, M.T.; Molina, H.; et al. Pancreatic cancer exosomes initiate pre-metastatic niche formation in the liver. Nat. Cell Biol. 2015, 17, 816–826. [Google Scholar] [CrossRef]
- Fong, M.Y.; Zhou, W.; Liu, L.; Alontaga, A.Y.; Chandra, M.; Ashby, J.; Chow, A.; O’Connor, S.T.; Li, S.; Chin, A.R.; et al. Breast-cancer-secreted miR-122 reprograms glucose metabolism in premetastatic niche to promote metastasis. Nat. Cell Biol. 2015, 17, 183–194. [Google Scholar] [CrossRef] [Green Version]
- McKiernan, J.; Donovan, M.J.; O’Neill, V.; Bentink, S.; Noerholm, M.; Belzer, S.; Skog, J.; Kattan, M.W.; Partin, A.; Andriole, G.; et al. A Novel Urine Exosome Gene Expression Assay to Predict High-grade Prostate Cancer at Initial Biopsy. JAMA Oncol. 2016, 2, 882–889. [Google Scholar] [CrossRef] [Green Version]
- Mathieu, M.; Martin-Jaular, L.; Lavieu, G.; Théry, C. Specificities of secretion and uptake of exosomes and other extracellular vesicles for cell-to-cell communication. Nat. Cell Biol. 2019, 21, 9–17. [Google Scholar] [CrossRef]
- Barman, B.; Sung, B.H.; Krystofiak, E.; Ping, J.; Ramirez, M.; Millis, B.; Allen, R.; Prasad, N.; Chetyrkin, S.; Calcutt, M.W.; et al. VAP-A and its binding partner CERT drive biogenesis of RNA-containing extracellular vesicles at ER membrane contact sites. Dev. Cell 2022, 57, 974–994.e978. [Google Scholar] [CrossRef]
- Crivelli, S.M.; Giovagnoni, C.; Zhu, Z.; Tripathi, P.; Elsherbini, A.; Quadri, Z.; Pu, J.; Zhang, L.; Ferko, B.; Berkes, D.; et al. Function of ceramide transfer protein for biogenesis and sphingolipid composition of extracellular vesicles. J. Extracell. Vesicles 2022, 11, e12233. [Google Scholar] [CrossRef]
- Matsui, T.; Osaki, F.; Hiragi, S.; Sakamaki, Y.; Fukuda, M. ALIX and ceramide differentially control polarized small extracellular vesicle release from epithelial cells. EMBO Rep. 2021, 22, e51475. [Google Scholar] [CrossRef]
- Baietti, M.F.; Zhang, Z.; Mortier, E.; Melchior, A.; Degeest, G.; Geeraerts, A.; Ivarsson, Y.; Depoortere, F.; Coomans, C.; Vermeiren, E.; et al. Syndecan-syntenin-ALIX regulates the biogenesis of exosomes. Nat. Cell Biol. 2012, 14, 677–685. [Google Scholar] [CrossRef]
- Colombo, M.; Moita, C.; van Niel, G.; Kowal, J.; Vigneron, J.; Benaroch, P.; Manel, N.; Moita, L.F.; Théry, C.; Raposo, G. Analysis of ESCRT functions in exosome biogenesis, composition and secretion highlights the heterogeneity of extracellular vesicles. J. Cell Sci. 2013, 126, 5553–5565. [Google Scholar] [CrossRef] [Green Version]
- Ostrowski, M.; Carmo, N.B.; Krumeich, S.; Fanget, I.; Raposo, G.; Savina, A.; Moita, C.F.; Schauer, K.; Hume, A.N.; Freitas, R.P.; et al. Rab27a and Rab27b control different steps of the exosome secretion pathway. Nat. Cell Biol 2010, 12, 19–30. [Google Scholar] [CrossRef] [Green Version]
- Wei, D.; Zhan, W.; Gao, Y.; Huang, L.; Gong, R.; Wang, W.; Zhang, R.; Wu, Y.; Gao, S.; Kang, T. RAB31 marks and controls an ESCRT-independent exosome pathway. Cell Res. 2021, 31, 157–177. [Google Scholar] [CrossRef]
- Fei, X.; Li, Z.; Yang, D.; Kong, X.; Lu, X.; Shen, Y.; Li, X.; Xie, S.; Wang, J.; Zhao, Y.; et al. Neddylation of Coro1a determines the fate of multivesicular bodies and biogenesis of extracellular vesicles. J. Extracell. Vesicles 2021, 10, e12153. [Google Scholar] [CrossRef]
- Sinha, S.; Hoshino, D.; Hong, N.H.; Kirkbride, K.C.; Grega-Larson, N.E.; Seiki, M.; Tyska, M.J.; Weaver, A.M. Cortactin promotes exosome secretion by controlling branched actin dynamics. J. Cell Biol. 2016, 214, 197–213. [Google Scholar] [CrossRef] [Green Version]
- Dar, G.H.; Mendes, C.C.; Kuan, W.L.; Speciale, A.A.; Conceição, M.; Görgens, A.; Uliyakina, I.; Lobo, M.J.; Lim, W.F.; El Andaloussi, S.; et al. GAPDH controls extracellular vesicle biogenesis and enhances the therapeutic potential of EV mediated siRNA delivery to the brain. Nat. Commun. 2021, 12, 6666. [Google Scholar] [CrossRef]
- Muralidharan-Chari, V.; Clancy, J.; Plou, C.; Romao, M.; Chavrier, P.; Raposo, G.; D’Souza-Schorey, C. ARF6-regulated shedding of tumor cell-derived plasma membrane microvesicles. Curr. Biol. 2009, 19, 1875–1885. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Antonyak, M.A.; Zhang, J.; Cerione, R.A. RhoA triggers a specific signaling pathway that generates transforming microvesicles in cancer cells. Oncogene 2012, 31, 4740–4749. [Google Scholar] [CrossRef] [Green Version]
- Di Vizio, D.; Kim, J.; Hager, M.H.; Morello, M.; Yang, W.; Lafargue, C.J.; True, L.D.; Rubin, M.A.; Adam, R.M.; Beroukhim, R.; et al. Oncosome formation in prostate cancer: Association with a region of frequent chromosomal deletion in metastatic disease. Cancer Res. 2009, 69, 5601–5609. [Google Scholar] [CrossRef] [Green Version]
- Sedgwick, A.E.; Clancy, J.W.; Olivia Balmert, M.; D’Souza-Schorey, C. Extracellular microvesicles and invadopodia mediate non-overlapping modes of tumor cell invasion. Sci. Rep. 2015, 5, 14748. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Zhuang, X.; Greene, K.S.; Si, H.; Antonyak, M.A.; Druso, J.E.; Wilson, K.F.; Cerione, R.A.; Feng, Q.; Wang, H. Cdc42 functions as a regulatory node for tumour-derived microvesicle biogenesis. J. Extracell. Vesicles 2021, 10, e12051. [Google Scholar] [CrossRef]
- Mulcahy, L.A.; Pink, R.C.; Carter, D.R. Routes and mechanisms of extracellular vesicle uptake. J. Extracell Vesicles 2014, 3, 21829. [Google Scholar] [CrossRef] [Green Version]
- Hoshino, A.; Costa-Silva, B.; Shen, T.L.; Rodrigues, G.; Hashimoto, A.; Tesic Mark, M.; Molina, H.; Kohsaka, S.; Di Giannatale, A.; Ceder, S.; et al. Tumour exosome integrins determine organotropic metastasis. Nature 2015, 527, 329–335. [Google Scholar] [CrossRef] [Green Version]
- Christianson, H.C.; Svensson, K.J.; van Kuppevelt, T.H.; Li, J.P.; Belting, M. Cancer cell exosomes depend on cell-surface heparan sulfate proteoglycans for their internalization and functional activity. Proc. Natl. Acad. Sci. USA 2013, 110, 17380–17385. [Google Scholar] [CrossRef] [Green Version]
- Cardeñes, B.; Clares, I.; Bezos, T.; Toribio, V.; López-Martín, S.; Rocha, A.; Peinado, H.; Yáñez-Mó, M.; Cabañas, C. ALCAM/CD166 Is Involved in the Binding and Uptake of Cancer-Derived Extracellular Vesicles. Int. J. Mol. Sci. 2022, 23, 5753. [Google Scholar] [CrossRef]
- Wang, T.; Wang, X.; Wang, H.; Li, L.; Zhang, C.; Xiang, R.; Tan, X.; Li, Z.; Jiang, C.; Zheng, L.; et al. High TSPAN8 expression in epithelial cancer cell-derived small extracellular vesicles promote confined diffusion and pronounced uptake. J. Extracell. Vesicles 2021, 10, e12167. [Google Scholar] [CrossRef]
- Tkach, M.; Kowal, J.; Zucchetti, A.E.; Enserink, L.; Jouve, M.; Lankar, D.; Saitakis, M.; Martin-Jaular, L.; Théry, C. Qualitative differences in T-cell activation by dendritic cell-derived extracellular vesicle subtypes. Embo. J. 2017, 36, 3012–3028. [Google Scholar] [CrossRef]
- Joshi, B.S.; de Beer, M.A.; Giepmans, B.N.G.; Zuhorn, I.S. Endocytosis of Extracellular Vesicles and Release of Their Cargo from Endosomes. ACS Nano 2020, 14, 4444–4455. [Google Scholar] [CrossRef] [Green Version]
- Sung, B.H.; von Lersner, A.; Guerrero, J.; Krystofiak, E.S.; Inman, D.; Pelletier, R.; Zijlstra, A.; Ponik, S.M.; Weaver, A.M. A live cell reporter of exosome secretion and uptake reveals pathfinding behavior of migrating cells. Nat. Commun. 2020, 11, 2092. [Google Scholar] [CrossRef]
- O′Brien, K.; Ughetto, S.; Mahjoum, S.; Nair, A.V.; Breakefield, X.O. Uptake, functionality, and re-release of extracellular vesicle-encapsulated cargo. Cell Rep. 2022, 39, 110651. [Google Scholar] [CrossRef]
- Zhou, X.; Miao, Y.; Wang, Y.; He, S.; Guo, L.; Mao, J.; Chen, M.; Yang, Y.; Zhang, X.; Gan, Y. Tumour-derived extracellular vesicle membrane hybrid lipid nanovesicles enhance siRNA delivery by tumour-homing and intracellular freeway transportation. J. Extracell. Vesicles 2022, 11, e12198. [Google Scholar] [CrossRef]
- He, C.; Zheng, S.; Luo, Y.; Wang, B. Exosome Theranostics: Biology and Translational Medicine. Theranostics 2018, 8, 237–255. [Google Scholar] [CrossRef]
- Nakagawa, Y.; Arafiles, J.V.V.; Kawaguchi, Y.; Nakase, I.; Hirose, H.; Futaki, S. Stearylated Macropinocytosis-Inducing Peptides Facilitating the Cellular Uptake of Small Extracellular Vesicles. Bioconjug. Chem. 2022, 33, 869–880. [Google Scholar] [CrossRef]
- Shimoda, A.; Miura, R.; Tateno, H.; Seo, N.; Shiku, H.; Sawada, S.I.; Sasaki, Y.; Akiyoshi, K. Assessment of Surface Glycan Diversity on Extracellular Vesicles by Lectin Microarray and Glycoengineering Strategies for Drug Delivery Applications. Small Methods 2022, 6, e2100785. [Google Scholar] [CrossRef]
- Rumi, E.; Pietra, D.; Ferretti, V.; Klampfl, T.; Harutyunyan, A.S.; Milosevic, J.D.; Them, N.C.; Berg, T.; Elena, C.; Casetti, I.C.; et al. JAK2 or CALR mutation status defines subtypes of essential thrombocythemia with substantially different clinical course and outcomes. Blood 2014, 123, 1544–1551. [Google Scholar] [CrossRef]
- Karantanos, T.; Moliterno, A.R. The roles of JAK2 in DNA damage and repair in the myeloproliferative neoplasms: Opportunities for targeted therapy. Blood Rev. 2018, 32, 426–432. [Google Scholar] [CrossRef]
- Karantanos, T.; Chaturvedi, S.; Braunstein, E.M.; Spivak, J.; Resar, L.; Karanika, S.; Williams, D.M.; Rogers, O.; Gocke, C.D.; Moliterno, A.R. Sex determines the presentation and outcomes in MPN and is related to sex-specific differences in the mutational burden. Blood Adv. 2020, 4, 2567–2576. [Google Scholar] [CrossRef]
- Accurso, V.; Santoro, M.; Mancuso, S.; Napolitano, M.; Carlisi, M.; Mattana, M.; Russo, C.; Di Stefano, A.; Sirocchi, D.; Siragusa, S. The Essential Thrombocythemia in 2020: What We Know and Where We Still Have to Dig Deep. Clin. Med. Insights Blood Disord. 2020, 13, 2634853520978210. [Google Scholar] [CrossRef]
- Tefferi, A.; Vannucchi, A.M.; Barbui, T. Polycythemia vera: Historical oversights, diagnostic details, and therapeutic views. Leukemia 2021, 35, 3339–3351. [Google Scholar] [CrossRef]
- Tefferi, A. Primary myelofi.ibrosis: 2021 update on diagnosis, risk-stratification and management. Am. J. Hematol. 2021, 96, 145–162. [Google Scholar] [CrossRef]
- Gersuk, G.M.; Carmel, R.; Pattamakom, S.; Challita, P.M.; Rabinowitz, A.P.; Pattengale, P.K. Quantitative and functional studies of impaired natural killer (NK) cells in patients with myelofibrosis, essential thrombocythemia, and polycythemia vera. I. A potential role for platelet-derived growth factor in defective NK cytotoxicity. Nat. Immun. 1993, 12, 136–151. [Google Scholar]
- Fisher, D.A.C.; Fowles, J.S.; Zhou, A.; Oh, S.T. Inflammatory Pathophysiology as a Contributor to Myeloproliferative Neoplasms. Front. Immunol. 2021, 12, 683401. [Google Scholar] [CrossRef]
- Mascarenhas, J.; Li, T.; Sandy, L.; Newsom, C.; Petersen, B.; Godbold, J.; Hoffman, R. Anti-transforming growth factor-β therapy in patients with myelofibrosis. Leuk. Lymphoma 2014, 55, 450–452. [Google Scholar] [CrossRef]
- Gleitz, H.F.E.; Dugourd, A.J.F.; Leimkühler, N.B.; Snoeren, I.A.M.; Fuchs, S.N.R.; Menzel, S.; Ziegler, S.; Kröger, N.; Triviai, I.; Büsche, G.; et al. Increased CXCL4 expression in hematopoietic cells links inflammation and progression of bone marrow fibrosis in MPN. Blood 2020, 136, 2051–2064. [Google Scholar] [CrossRef]
- Janowska-Wieczorek, A.; Majka, M.; Kijowski, J.; Baj-Krzyworzeka, M.; Reca, R.; Turner, A.R.; Ratajczak, J.; Emerson, S.G.; Kowalska, M.A.; Ratajczak, M.Z. Platelet-derived microparticles bind to hematopoietic stem/progenitor cells and enhance their engraftment. Blood 2001, 98, 3143–3149. [Google Scholar] [CrossRef] [Green Version]
- Baj-Krzyworzeka, M.; Majka, M.; Pratico, D.; Ratajczak, J.; Vilaire, G.; Kijowski, J.; Reca, R.; Janowska-Wieczorek, A.; Ratajczak, M.Z. Platelet-derived microparticles stimulate proliferation, survival, adhesion, and chemotaxis of hematopoietic cells. Exp. Hematol. 2002, 30, 450–459. [Google Scholar] [CrossRef]
- Boyiadzis, M.; Whiteside, T.L. Information transfer by exosomes: A new frontier in hematologic malignancies. Blood Rev. 2015, 29, 281–290. [Google Scholar] [CrossRef]
- Catani, L.; Cavo, M.; Palandri, F. The Power of Extracellular Vesicles in Myeloproliferative Neoplasms: “Crafting” a Microenvironment That Matters. Cells 2021, 10, 2316. [Google Scholar] [CrossRef]
- Connor, D.E.; Ma, D.D.; Joseph, J.E. Flow cytometry demonstrates differences in platelet reactivity and microparticle formation in subjects with thrombocytopenia or thrombocytosis due to primary haematological disorders. Thromb. Res. 2013, 132, 572–577. [Google Scholar] [CrossRef]
- Trappenburg, M.C.; van Schilfgaarde, M.; Marchetti, M.; Spronk, H.M.; ten Cate, H.; Leyte, A.; Terpstra, W.E.; Falanga, A. Elevated procoagulant microparticles expressing endothelial and platelet markers in essential thrombocythemia. Haematologica 2009, 94, 911–918. [Google Scholar] [CrossRef] [Green Version]
- Marchetti, M.; Tartari, C.J.; Russo, L.; Panova-Noeva, M.; Leuzzi, A.; Rambaldi, A.; Finazzi, G.; Woodhams, B.; Falanga, A. Phospholipid-dependent procoagulant activity is highly expressed by circulating microparticles in patients with essential thrombocythemia. Am. J. Hematol. 2014, 89, 68–73. [Google Scholar] [CrossRef] [Green Version]
- Piccin, A.; Steurer, M.; Feistritzer, C.; Murphy, C.; Eakins, E.; Van Schilfgaarde, M.; Corvetta, D.; Di Pierro, A.M.; Pusceddu, I.; Marcheselli, L.; et al. Observational retrospective study of vascular modulator changes during treatment in essential thrombocythemia. Transl. Res. 2017, 184, 21–34. [Google Scholar] [CrossRef]
- French, K.C.; Antonyak, M.A.; Cerione, R.A. Extracellular vesicle docking at the cellular port: Extracellular vesicle binding and uptake. Semin Cell Dev. Biol. 2017, 67, 48–55. [Google Scholar] [CrossRef]
- Wang, Q.; Yu, G.; He, H.; Zheng, Z.; Li, X.; Lin, R.; Xu, D. Differential expression of circular RNAs in bone marrow-derived exosomes from essential thrombocythemia patients. Cell Biol. Int. 2021, 45, 869–881. [Google Scholar] [CrossRef]
- Ahadon, M.; Abdul Aziz, S.; Wong, C.L.; Leong, C.F. Plasma-derived microparticles in polycythaemia vera. Malays. J. Pathol. 2018, 40, 41–48. [Google Scholar]
- Fel, A.; Lewandowska, A.E.; Petrides, P.E.; Wiśniewski, J.R. Comparison of Proteome Composition of Serum Enriched in Extracellular Vesicles Isolated from Polycythemia Vera Patients and Healthy Controls. Proteomes 2019, 7, 20. [Google Scholar] [CrossRef] [Green Version]
- Barone, M.; Barone, M.; Ricci, F.; Auteri, G.; Corradi, G.; Fabbri, F.; Papa, V.; Bandini, E.; Cenacchi, G.; Tazzari, P.L.; et al. An Abnormal Host/Microbiomes Signature of Plasma-Derived Extracellular Vesicles Is Associated to Polycythemia Vera. Front. Oncol. 2021, 11, 715217. [Google Scholar] [CrossRef]
- Sinauridze, E.I.; Kireev, D.A.; Popenko, N.Y.; Pichugin, A.V.; Panteleev, M.A.; Krymskaya, O.V.; Ataullakhanov, F.I. Platelet microparticle membranes have 50- to 100-fold higher specific procoagulant activity than activated platelets. Thromb. Haemost. 2007, 97, 425–434. [Google Scholar]
- Moliterno, A.R.; Ginzburg, Y.Z.; Hoffman, R. Clinical insights into the origins of thrombosis in myeloproliferative neoplasms. Blood 2021, 137, 1145–1153. [Google Scholar] [CrossRef]
- Gangat, N.; Tefferi, A. Myelofibrosis biology and contemporary management. Br. J. Haematol. 2020, 191, 152–170. [Google Scholar] [CrossRef]
- Baraga, J.J.; Rava, R.P.; Taroni, P.; Kittrell, C.; Fitzmaurice, M.; Feld, M.S. Laser induced fluorescence spectroscopy of normal and atherosclerotic human aorta using 306-310 nm excitation. Lasers Surg. Med. 1990, 10, 245–261. [Google Scholar] [CrossRef]
- Caivano, A.; Laurenzana, I.; De Luca, L.; La Rocca, F.; Simeon, V.; Trino, S.; D’Auria, F.; Traficante, A.; Maietti, M.; Izzo, T.; et al. High serum levels of extracellular vesicles expressing malignancy-related markers are released in patients with various types of hematological neoplastic disorders. Tumour. Biol. 2015, 36, 9739–9752. [Google Scholar] [CrossRef]
- Zhang, W.; Qi, J.; Zhao, S.; Shen, W.; Dai, L.; Han, W.; Huang, M.; Wang, Z.; Ruan, C.; Wu, D.; et al. Clinical significance of circulating microparticles in Ph(-) myeloproliferative neoplasms. Oncol. Lett. 2017, 14, 2531–2536. [Google Scholar] [CrossRef] [Green Version]
- Barone, M.; Ricci, F.; Sollazzo, D.; Ottaviani, E.; Romano, M.; Auteri, G.; Bartoletti, D.; Reggiani, M.L.B.; Vianelli, N.; Tazzari, P.L.; et al. Circulating megakaryocyte and platelet microvesicles correlate with response to ruxolitinib and distinct disease severity in patients with myelofibrosis. Br. J. Haematol. 2019, 185, 987–991. [Google Scholar] [CrossRef]
- Forte, D.; Barone, M.; Morsiani, C.; Simonetti, G.; Fabbri, F.; Bruno, S.; Bandini, E.; Sollazzo, D.; Collura, S.; Deregibus, M.C.; et al. Distinct profile of CD34(+) cells and plasma-derived extracellular vesicles from triple-negative patients with Myelofibrosis reveals potential markers of aggressive disease. J. Exp. Clin. Cancer Res. 2021, 40, 49. [Google Scholar] [CrossRef]
- Su, C.; Zhang, J.; Yarden, Y.; Fu, L. The key roles of cancer stem cell-derived extracellular vesicles. Signal. Transduct. Target. Ther. 2021, 6, 109. [Google Scholar] [CrossRef]
- Polverelli, N.; Breccia, M.; Benevolo, G.; Martino, B.; Tieghi, A.; Latagliata, R.; Sabattini, E.; Riminucci, M.; Godio, L.; Catani, L.; et al. Risk factors for infections in myelofibrosis: Role of disease status and treatment. A multicenter study of 507 patients. Am. J. Hematol. 2017, 92, 37–41. [Google Scholar] [CrossRef]
- Jabbour, E.; Kantarjian, H. Chronic myeloid leukemia: 2020 update on diagnosis, therapy and monitoring. Am. J. Hematol. 2020, 95, 691–709. [Google Scholar] [CrossRef]
- Berman, E. How I treat chronic-phase chronic myelogenous leukemia. Blood 2022, 139, 3138–3147. [Google Scholar] [CrossRef]
- Elrick, L.J.; Jorgensen, H.G.; Mountford, J.C.; Holyoake, T.L. Punish the parent not the progeny. Blood 2005, 105, 1862–1866. [Google Scholar] [CrossRef] [Green Version]
- Cortes, J.; Jabbour, E.; Kantarjian, H.; Yin, C.C.; Shan, J.; O’Brien, S.; Garcia-Manero, G.; Giles, F.; Breeden, M.; Reeves, N.; et al. Dynamics of BCR-ABL kinase domain mutations in chronic myeloid leukemia after sequential treatment with multiple tyrosine kinase inhibitors. Blood 2007, 110, 4005–4011. [Google Scholar] [CrossRef] [Green Version]
- Kumar, R.; Pereira, R.S.; Zanetti, C.; Minciacchi, V.R.; Merten, M.; Meister, M.; Niemann, J.; Dietz, M.S.; Rüssel, N.; Schnütgen, F.; et al. Specific, targetable interactions with the microenvironment influence imatinib-resistant chronic myeloid leukemia. Leukemia 2020, 34, 2087–2101. [Google Scholar] [CrossRef]
- Papayannopoulou, T.; Craddock, C.; Nakamoto, B.; Priestley, G.V.; Wolf, N.S. The VLA4/VCAM-1 adhesion pathway defines contrasting mechanisms of lodgement of transplanted murine hemopoietic progenitors between bone marrow and spleen. Proc. Natl. Acad. Sci. USA 1995, 92, 9647–9651. [Google Scholar] [CrossRef] [Green Version]
- Jin, L.; Tabe, Y.; Konoplev, S.; Xu, Y.; Leysath, C.E.; Lu, H.; Kimura, S.; Ohsaka, A.; Rios, M.B.; Calvert, L.; et al. CXCR4 up-regulation by imatinib induces chronic myelogenous leukemia (CML) cell migration to bone marrow stroma and promotes survival of quiescent CML cells. Mol. Cancer Ther. 2008, 7, 48–58. [Google Scholar] [CrossRef] [Green Version]
- Tarafdar, A.; Hopcroft, L.E.; Gallipoli, P.; Pellicano, F.; Cassels, J.; Hair, A.; Korfi, K.; Jørgensen, H.G.; Vetrie, D.; Holyoake, T.L.; et al. CML cells actively evade host immune surveillance through cytokine-mediated downregulation of MHC-II expression. Blood 2017, 129, 199–208. [Google Scholar] [CrossRef]
- Zhang, H.; Li, H.; Xi, H.S.; Li, S. HIF1α is required for survival maintenance of chronic myeloid leukemia stem cells. Blood 2012, 119, 2595–2607. [Google Scholar] [CrossRef] [Green Version]
- Raimondo, S.; Saieva, L.; Corrado, C.; Fontana, S.; Flugy, A.; Rizzo, A.; De Leo, G.; Alessandro, R. Chronic myeloid leukemia-derived exosomes promote tumor growth through an autocrine mechanism. Cell Commun. Signal. 2015, 13, 8. [Google Scholar] [CrossRef] [Green Version]
- Corrado, C.; Saieva, L.; Raimondo, S.; Santoro, A.; De Leo, G.; Alessandro, R. Chronic myelogenous leukaemia exosomes modulate bone marrow microenvironment through activation of epidermal growth factor receptor. J. Cell Mol. Med. 2016, 20, 1829–1839. [Google Scholar] [CrossRef] [Green Version]
- Jafarzadeh, N.; Safari, Z.; Pornour, M.; Amirizadeh, N.; Forouzandeh Moghadam, M.; Sadeghizadeh, M. Alteration of cellular and immune-related properties of bone marrow mesenchymal stem cells and macrophages by K562 chronic myeloid leukemia cell derived exosomes. J. Cell Physiol. 2019, 234, 3697–3710. [Google Scholar] [CrossRef]
- Aguayo, A.; Kantarjian, H.; Manshouri, T.; Gidel, C.; Estey, E.; Thomas, D.; Koller, C.; Estrov, Z.; O’Brien, S.; Keating, M.; et al. Angiogenesis in acute and chronic leukemias and myelodysplastic syndromes. Blood 2000, 96, 2240–2245. [Google Scholar]
- Ramos, T.L.; Sánchez-Abarca, L.I.; López-Ruano, G.; Muntión, S.; Preciado, S.; Hernández-Ruano, M.; Rosado, B.; de las Heras, N.; Chillón, M.C.; Hernández-Hernández, Á.; et al. Do endothelial cells belong to the primitive stem leukemic clone in CML? Role of extracellular vesicles. Leuk. Res. 2015, 39, 921–924. [Google Scholar] [CrossRef]
- Mineo, M.; Garfield, S.H.; Taverna, S.; Flugy, A.; De Leo, G.; Alessandro, R.; Kohn, E.C. Exosomes released by K562 chronic myeloid leukemia cells promote angiogenesis in a Src-dependent fashion. Angiogenesis 2012, 15, 33–45. [Google Scholar] [CrossRef] [Green Version]
- Javidi-Sharifi, N.; Martinez, J.; English, I.; Joshi, S.K.; Scopim-Ribeiro, R.; Viola, S.K.; Edwards, D.K.t.; Agarwal, A.; Lopez, C.; Jorgens, D.; et al. FGF2-FGFR1 signaling regulates release of Leukemia-Protective exosomes from bone marrow stromal cells. Elife 2019, 8, e40033. [Google Scholar] [CrossRef]
- Karantanos, T.; DeZern, A.E. Biology and clinical management of hypoplastic MDS: MDS as a bone marrow failure syndrome. Best Pract Res. Clin. Haematol. 2021, 34, 101280. [Google Scholar] [CrossRef]
- Greenberg, P.L.; Tuechler, H.; Schanz, J.; Sanz, G.; Garcia-Manero, G.; Solé, F.; Bennett, J.M.; Bowen, D.; Fenaux, P.; Dreyfus, F.; et al. Revised international prognostic scoring system for myelodysplastic syndromes. Blood 2012, 120, 2454–2465. [Google Scholar] [CrossRef]
- Karantanos, T.; Gondek, L.P.; Varadhan, R.; Moliterno, A.R.; DeZern, A.E.; Jones, R.J.; Jain, T. Gender-related differences in the outcomes and genomic landscape of patients with myelodysplastic syndrome/myeloproliferative neoplasm overlap syndromes. Br. J. Haematol. 2021, 193, 1142–1150. [Google Scholar] [CrossRef]
- Karantanos, T.; Jain, T.; Moliterno, A.R.; Jones, R.J.; DeZern, A.E. Sex-Related Differences in Chronic Myeloid Neoplasms: From the Clinical Observation to the Underlying Biology. Int. J. Mol. Sci. 2021, 22, 2595. [Google Scholar] [CrossRef]
- Wang, F.; Ni, J.; Wu, L.; Wang, Y.; He, B.; Yu, D. Gender disparity in the survival of patients with primary myelodysplastic syndrome. J. Cancer 2019, 10, 1325–1332. [Google Scholar] [CrossRef]
- Cazzola, M. Myelodysplastic Syndromes. N. Engl. J. Med. 2020, 383, 1358–1374. [Google Scholar] [CrossRef]
- Medyouf, H.; Mossner, M.; Jann, J.C.; Nolte, F.; Raffel, S.; Herrmann, C.; Lier, A.; Eisen, C.; Nowak, V.; Zens, B.; et al. Myelodysplastic cells in patients reprogram mesenchymal stromal cells to establish a transplantable stem cell niche disease unit. Cell Stem. Cell 2014, 14, 824–837. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, Y.; Kawabata, K.C.; Tanaka, Y.; Uehara, Y.; Mabuchi, Y.; Murakami, K.; Nishiyama, A.; Kiryu, S.; Yoshioka, Y.; Ota, Y.; et al. MDS cells impair osteolineage differentiation of MSCs via extracellular vesicles to suppress normal hematopoiesis. Cell Rep. 2022, 39, 110805. [Google Scholar] [CrossRef]
- Muto, T.; Walker, C.S.; Choi, K.; Hueneman, K.; Smith, M.A.; Gul, Z.; Garcia-Manero, G.; Ma, A.; Zheng, Y.; Starczynowski, D.T. Adaptive response to inflammation contributes to sustained myelopoiesis and confers a competitive advantage in myelodysplastic syndrome HSCs. Nat. Immunol. 2020, 21, 535–545. [Google Scholar] [CrossRef]
- Weidner, H.; Baschant, U.; Lademann, F.; Ledesma Colunga, M.G.; Balaian, E.; Hofbauer, C.; Misof, B.M.; Roschger, P.; Blouin, S.; Richards, W.G.; et al. Increased FGF-23 levels are linked to ineffective erythropoiesis and impaired bone mineralization in myelodysplastic syndromes. JCI Insight 2020, 5, e137062. [Google Scholar] [CrossRef]
- Verma, D.; Kumar, R.; Pereira, R.S.; Karantanou, C.; Zanetti, C.; Minciacchi, V.R.; Fulzele, K.; Kunz, K.; Hoelper, S.; Zia-Chahabi, S.; et al. Vitamin K antagonism impairs the bone marrow microenvironment and hematopoiesis. Blood 2019, 134, 227–238. [Google Scholar] [CrossRef]
- Schinke, C.; Giricz, O.; Li, W.; Shastri, A.; Gordon, S.; Barreyro, L.; Bhagat, T.; Bhattacharyya, S.; Ramachandra, N.; Bartenstein, M.; et al. IL8-CXCR2 pathway inhibition as a therapeutic strategy against MDS and AML stem cells. Blood 2015, 125, 3144–3152. [Google Scholar] [CrossRef] [Green Version]
- Karantanos, T.; Teodorescu, P.; Perkins, B.; Christodoulou, I.; Esteb, C.; Varadhan, R.; Helmenstine, E.; Rajkhowa, T.; Paun, B.C.; Bonifant, C.; et al. The role of the atypical chemokine receptor CCRL2 in myelodysplastic syndrome and secondary acute myeloid leukemia. Sci. Adv. 2022, 8, eabl8952. [Google Scholar] [CrossRef]
- Hrustincova, A.; Krejcik, Z.; Kundrat, D.; Szikszai, K.; Belickova, M.; Pecherkova, P.; Klema, J.; Vesela, J.; Hruba, M.; Cermak, J.; et al. Circulating Small Noncoding RNAs Have Specific Expression Patterns in Plasma and Extracellular Vesicles in Myelodysplastic Syndromes and Are Predictive of Patient Outcome. Cells 2020, 9, 794. [Google Scholar] [CrossRef] [Green Version]
- Muntión, S.; Ramos, T.L.; Diez-Campelo, M.; Rosón, B.; Sánchez-Abarca, L.I.; Misiewicz-Krzeminska, I.; Preciado, S.; Sarasquete, M.E.; de Las Rivas, J.; González, M.; et al. Microvesicles from Mesenchymal Stromal Cells Are Involved in HPC-Microenvironment Crosstalk in Myelodysplastic Patients. PLoS ONE 2016, 11, e0146722. [Google Scholar] [CrossRef]
- Saitoh, Y.; Umezu, T.; Imanishi, S.; Asano, M.; Yoshizawa, S.; Katagiri, S.; Suguro, T.; Fujimoto, H.; Akahane, D.; Kobayashi-Kawana, C.; et al. Downregulation of extracellular vesicle microRNA-101 derived from bone marrow mesenchymal stromal cells in myelodysplastic syndrome with disease progression. Oncol. Lett. 2020, 19, 2053–2061. [Google Scholar] [CrossRef]
- Meunier, M.; Guttin, A.; Ancelet, S.; Laurin, D.; Zannoni, J.; Lefebvre, C.; Tondeur, S.; Persoons, V.; Pezet, M.; Pernet-Gallay, K.; et al. Extracellular vesicles from myelodysplastic mesenchymal stromal cells induce DNA damage and mutagenesis of hematopoietic stem cells through miRNA transfer. Leukemia 2020, 34, 2249–2253. [Google Scholar] [CrossRef]
- Shallis, R.M.; Wang, R.; Davidoff, A.; Ma, X.; Zeidan, A.M. Epidemiology of acute myeloid leukemia: Recent progress and enduring challenges. Blood Rev. 2019, 36, 70–87. [Google Scholar] [CrossRef]
- Kantarjian, H.; Kadia, T.; DiNardo, C.; Daver, N.; Borthakur, G.; Jabbour, E.; Garcia-Manero, G.; Konopleva, M.; Ravandi, F. Acute myeloid leukemia: Current progress and future directions. Blood Cancer J. 2021, 11, 41. [Google Scholar] [CrossRef]
- Kayser, S.; Levis, M.J. Updates on targeted therapies for acute myeloid leukaemia. Br. J. Haematol. 2022, 196, 316–328. [Google Scholar] [CrossRef]
- Karantanos, T.; Jones, R.J. Acute Myeloid Leukemia Stem Cell Heterogeneity and Its Clinical Relevance. Adv. Exp. Med. Biol. 2019, 1139, 153–169. [Google Scholar] [CrossRef]
- Viñado, A.C.; Calvo, I.A.; Cenzano, I.; Olaverri, D.; Cocera, M.; San Martin-Uriz, P.; Romero, J.P.; Vilas-Zornoza, A.; Vera, L.; Gomez-Cebrian, N.; et al. The bone marrow niche regulates redox and energy balance in MLL::AF9 leukemia stem cells. Leukemia 2022. [Google Scholar] [CrossRef]
- Zhou, H.S.; Carter, B.Z.; Andreeff, M. Bone marrow niche-mediated survival of leukemia stem cells in acute myeloid leukemia: Yin and Yang. Cancer Biol. Med. 2016, 13, 248–259. [Google Scholar] [CrossRef] [Green Version]
- Karantanou, C.; Godavarthy, P.S.; Krause, D.S. Targeting the bone marrow microenvironment in acute leukemia. Leuk. Lymphoma 2018, 59, 2535–2545. [Google Scholar] [CrossRef]
- Whiteside, T.L. Exosomes and tumor-mediated immune suppression. J. Clin. Invest. 2016, 126, 1216–1223. [Google Scholar] [CrossRef] [Green Version]
- Costello, R.T.; Sivori, S.; Marcenaro, E.; Lafage-Pochitaloff, M.; Mozziconacci, M.J.; Reviron, D.; Gastaut, J.A.; Pende, D.; Olive, D.; Moretta, A. Defective expression and function of natural killer cell-triggering receptors in patients with acute myeloid leukemia. Blood 2002, 99, 3661–3667. [Google Scholar] [CrossRef]
- Szczepanski, M.J.; Szajnik, M.; Welsh, A.; Whiteside, T.L.; Boyiadzis, M. Blast-derived microvesicles in sera from patients with acute myeloid leukemia suppress natural killer cell function via membrane-associated transforming growth factor-beta1. Haematologica 2011, 96, 1302–1309. [Google Scholar] [CrossRef]
- Huan, J.; Hornick, N.I.; Shurtleff, M.J.; Skinner, A.M.; Goloviznina, N.A.; Roberts, C.T., Jr.; Kurre, P. RNA trafficking by acute myelogenous leukemia exosomes. Cancer Res. 2013, 73, 918–929. [Google Scholar] [CrossRef] [Green Version]
- Doron, B.; Abdelhamed, S.; Butler, J.T.; Hashmi, S.K.; Horton, T.M.; Kurre, P. Transmissible ER stress reconfigures the AML bone marrow compartment. Leukemia 2019, 33, 918–930. [Google Scholar] [CrossRef]
- Corazzari, M.; Gagliardi, M.; Fimia, G.M.; Piacentini, M. Endoplasmic Reticulum Stress, Unfolded Protein Response, and Cancer Cell Fate. Front. Oncol. 2017, 7, 78. [Google Scholar] [CrossRef] [Green Version]
- Avril, T.; Vauléon, E.; Chevet, E. Endoplasmic reticulum stress signaling and chemotherapy resistance in solid cancers. Oncogenesis 2017, 6, e373. [Google Scholar] [CrossRef]
- Jiang, D.; Wu, X.; Sun, X.; Tan, W.; Dai, X.; Xie, Y.; Du, A.; Zhao, Q. Bone mesenchymal stem cell-derived exosomal microRNA-7-5p inhibits progression of acute myeloid leukemia by targeting OSBPL11. J. Nanobiotechnol. 2022, 20, 29. [Google Scholar] [CrossRef]
- Cui, P.; Zhang, Y.; Cui, M.; Li, Z.; Ma, G.; Wang, R.; Wang, N.; Huang, S.; Gao, J. Leukemia cells impair normal hematopoiesis and induce functionally loss of hematopoietic stem cells through immune cells and inflammation. Leuk. Res. 2018, 65, 49–54. [Google Scholar] [CrossRef]
- Hornick, N.I.; Doron, B.; Abdelhamed, S.; Huan, J.; Harrington, C.A.; Shen, R.; Cambronne, X.A.; Chakkaramakkil Verghese, S.; Kurre, P. AML suppresses hematopoiesis by releasing exosomes that contain microRNAs targeting c-MYB. Sci. Signal. 2016, 9, ra88. [Google Scholar] [CrossRef] [Green Version]
- Abdelhamed, S.; Butler, J.T.; Doron, B.; Halse, A.; Nemecek, E.; Wilmarth, P.A.; Marks, D.L.; Chang, B.H.; Horton, T.; Kurre, P. Extracellular vesicles impose quiescence on residual hematopoietic stem cells in the leukemic niche. EMBO Rep. 2019, 20, e47546. [Google Scholar] [CrossRef]
- Kumar, B.; Garcia, M.; Weng, L.; Jung, X.; Murakami, J.L.; Hu, X.; McDonald, T.; Lin, A.; Kumar, A.R.; DiGiusto, D.L.; et al. Acute myeloid leukemia transforms the bone marrow niche into a leukemia-permissive microenvironment through exosome secretion. Leukemia 2018, 32, 575–587. [Google Scholar] [CrossRef]
- Huan, J.; Hornick, N.I.; Goloviznina, N.A.; Kamimae-Lanning, A.N.; David, L.L.; Wilmarth, P.A.; Mori, T.; Chevillet, J.R.; Narla, A.; Roberts, C.T., Jr.; et al. Coordinate regulation of residual bone marrow function by paracrine trafficking of AML exosomes. Leukemia 2015, 29, 2285–2295. [Google Scholar] [CrossRef] [Green Version]
- Chalmin, F.; Ladoire, S.; Mignot, G.; Vincent, J.; Bruchard, M.; Remy-Martin, J.P.; Boireau, W.; Rouleau, A.; Simon, B.; Lanneau, D.; et al. Membrane-associated Hsp72 from tumor-derived exosomes mediates STAT3-dependent immunosuppressive function of mouse and human myeloid-derived suppressor cells. J. Clin. Invest. 2010, 120, 457–471. [Google Scholar] [CrossRef]
- Marleau, A.M.; Chen, C.S.; Joyce, J.A.; Tullis, R.H. Exosome removal as a therapeutic adjuvant in cancer. J. Transl. Med. 2012, 10, 134. [Google Scholar] [CrossRef] [Green Version]
- Vader, P.; Breakefield, X.O.; Wood, M.J. Extracellular vesicles: Emerging targets for cancer therapy. Trends Mol. Med. 2014, 20, 385–393. [Google Scholar] [CrossRef] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Karantanou, C.; Minciacchi, V.R.; Karantanos, T. Extracellular Vesicles in Myeloid Neoplasms. Int. J. Mol. Sci. 2022, 23, 8827. https://doi.org/10.3390/ijms23158827
Karantanou C, Minciacchi VR, Karantanos T. Extracellular Vesicles in Myeloid Neoplasms. International Journal of Molecular Sciences. 2022; 23(15):8827. https://doi.org/10.3390/ijms23158827
Chicago/Turabian StyleKarantanou, Christina, Valentina René Minciacchi, and Theodoros Karantanos. 2022. "Extracellular Vesicles in Myeloid Neoplasms" International Journal of Molecular Sciences 23, no. 15: 8827. https://doi.org/10.3390/ijms23158827
APA StyleKarantanou, C., Minciacchi, V. R., & Karantanos, T. (2022). Extracellular Vesicles in Myeloid Neoplasms. International Journal of Molecular Sciences, 23(15), 8827. https://doi.org/10.3390/ijms23158827