
Transcriptome Profile Identifies Actin as an Essential Regulator of Cardiac Myosin Binding Protein C3 Hypertrophic Cardiomyopathy in a Zebrafish Model

 , , , , , , , ,
, , , , , , , ,  and
and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
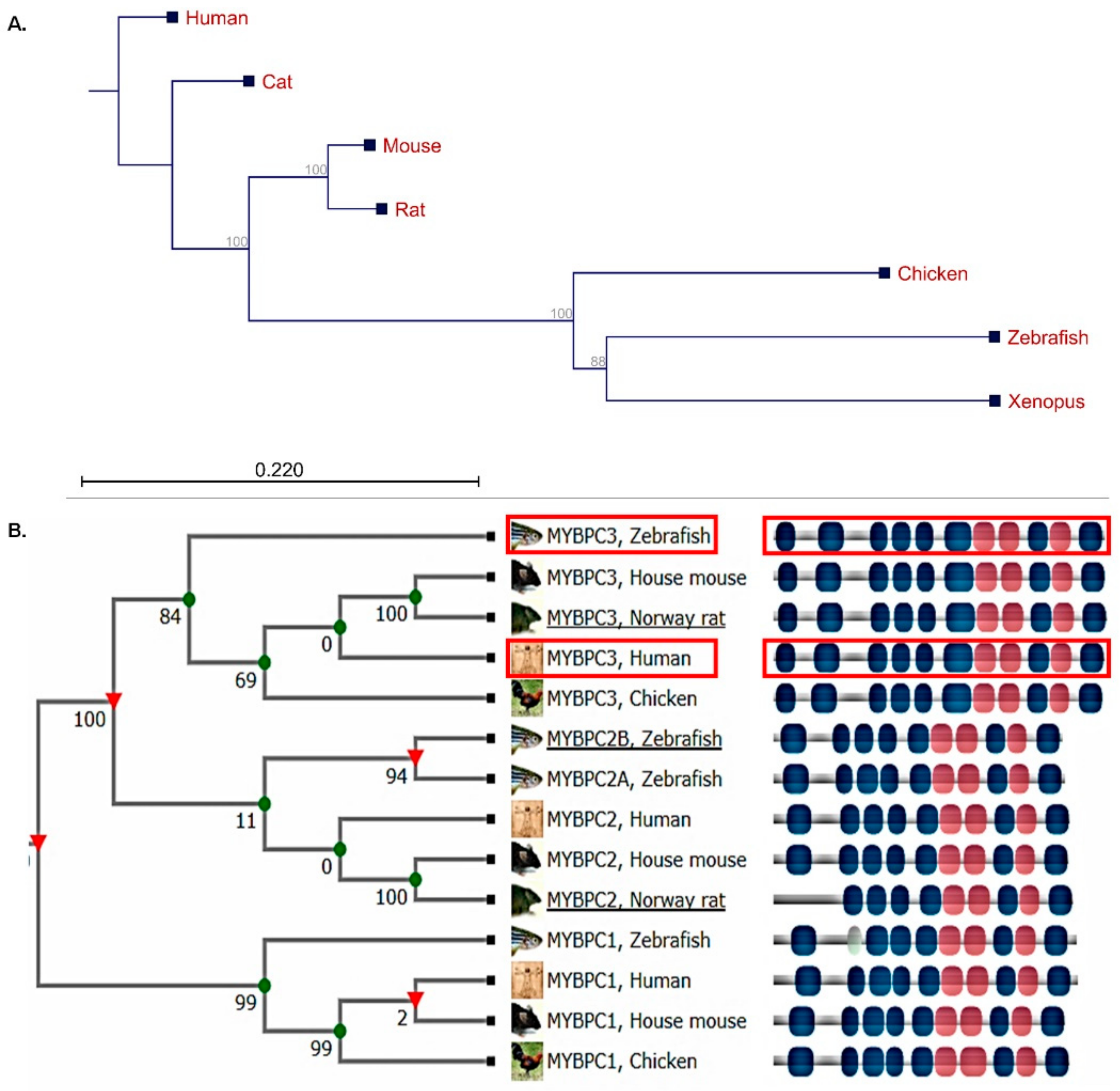
2.1. Human MyBP-C Is Highly Conserved in the Zebrafish Model
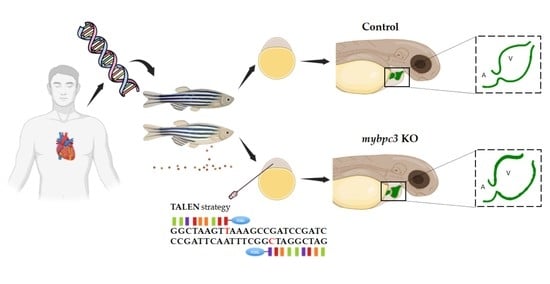
2.2. Zebrafish Mybpc3 Knockout (KO) Model Generation
2.3. Early Phenotype Analyses of mybpc3 Mutant Zebrafish
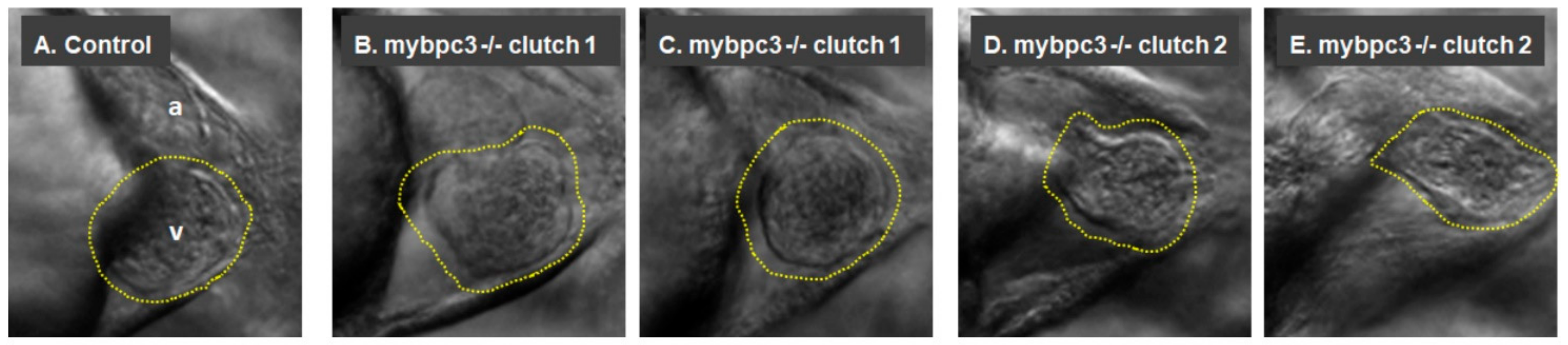
2.3.1. Cardiac Morphology
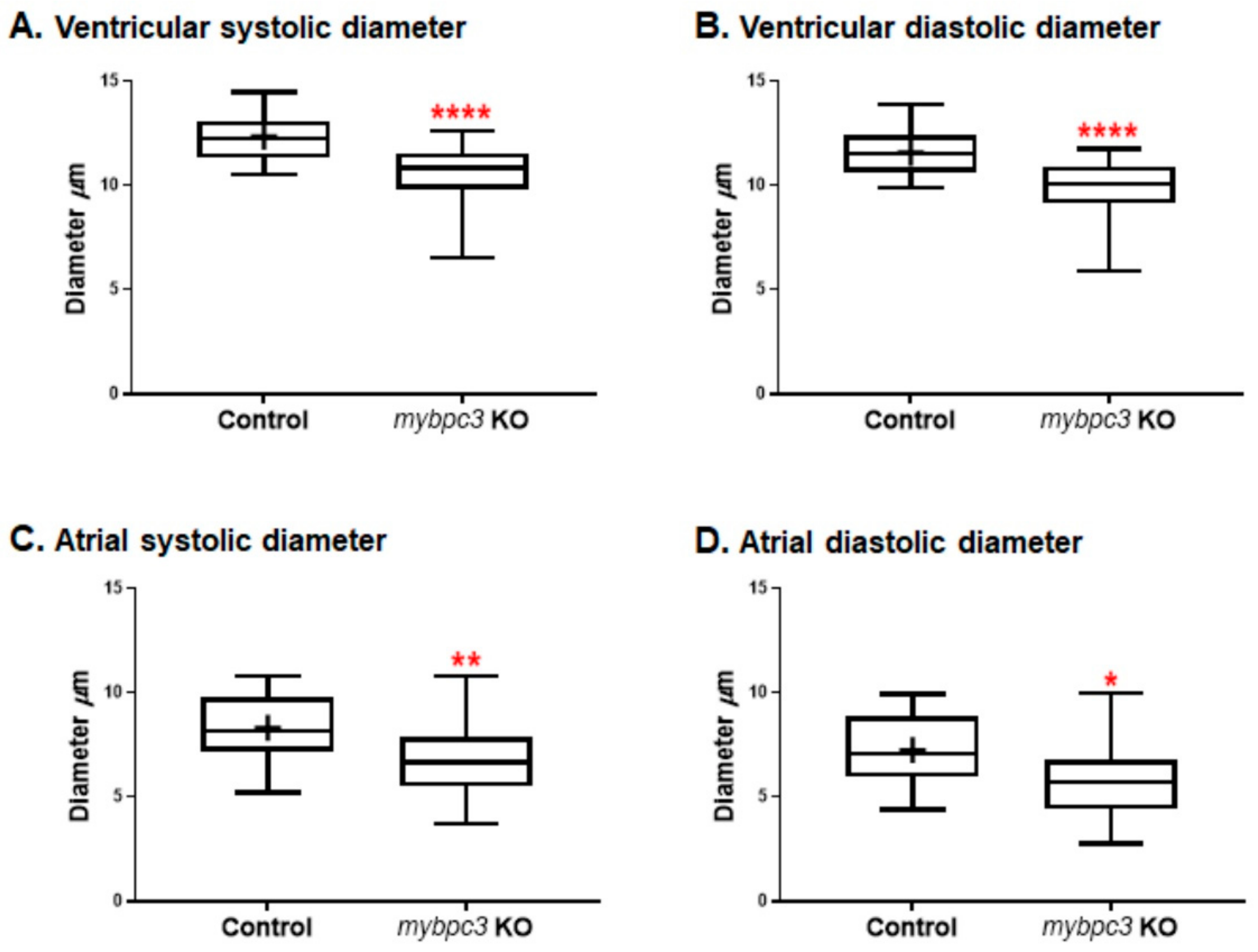
2.3.2. Cardiac Chambers
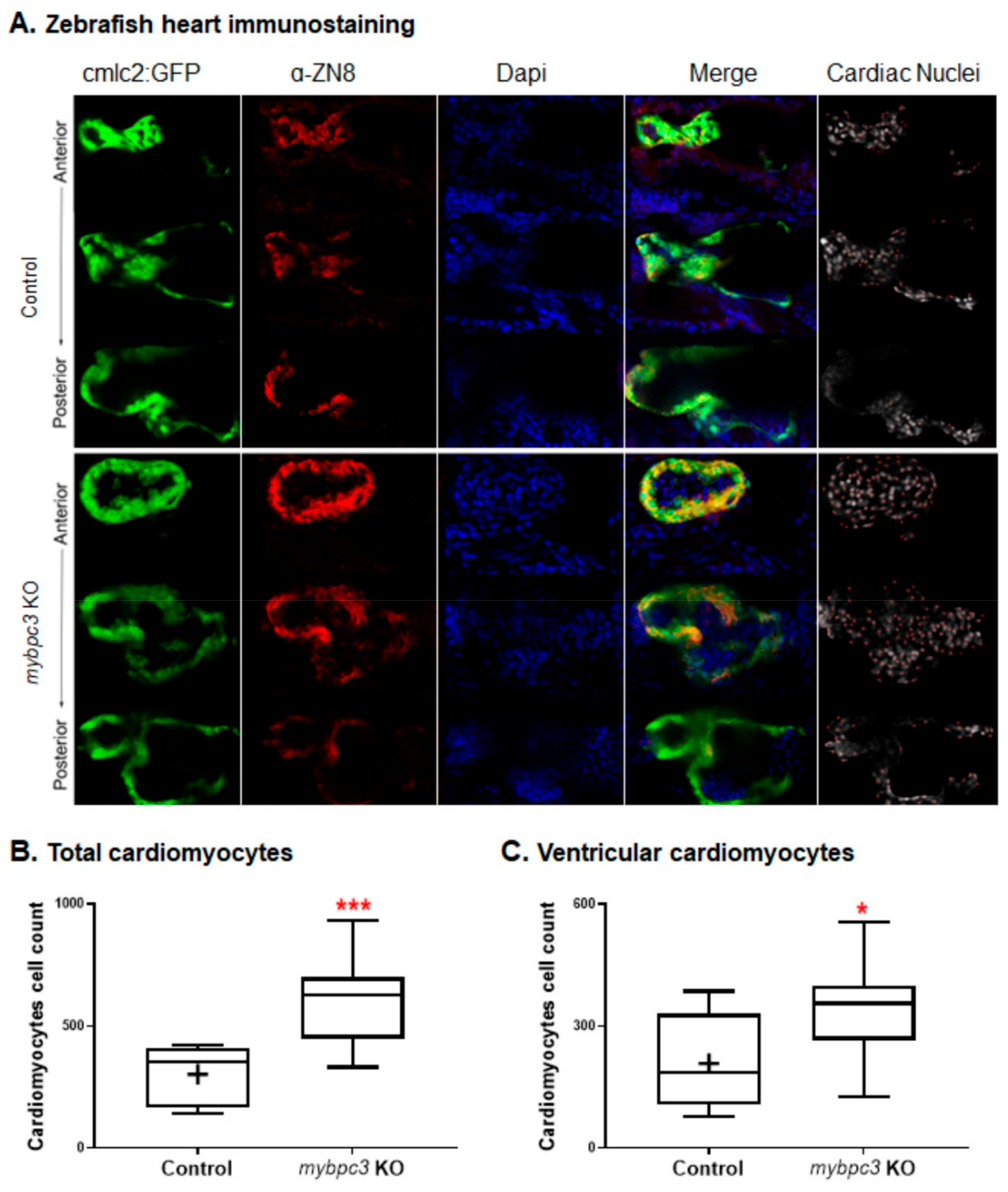
2.3.3. Cardiac Remodeling at the Cellular Level
2.3.4. Cardiac Contractility
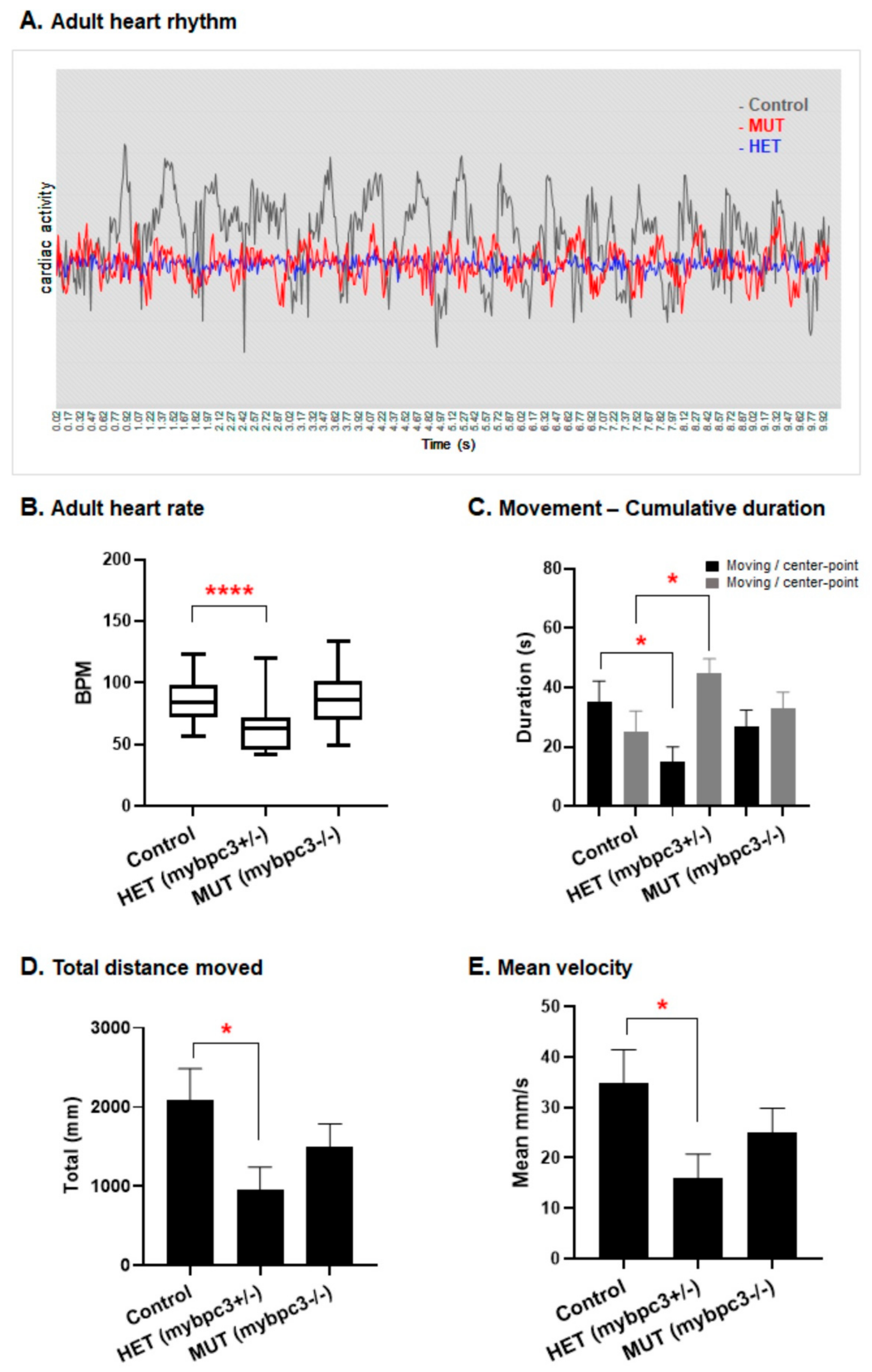
2.3.5. Cardiac Rhythm
2.3.6. Ejection Fraction
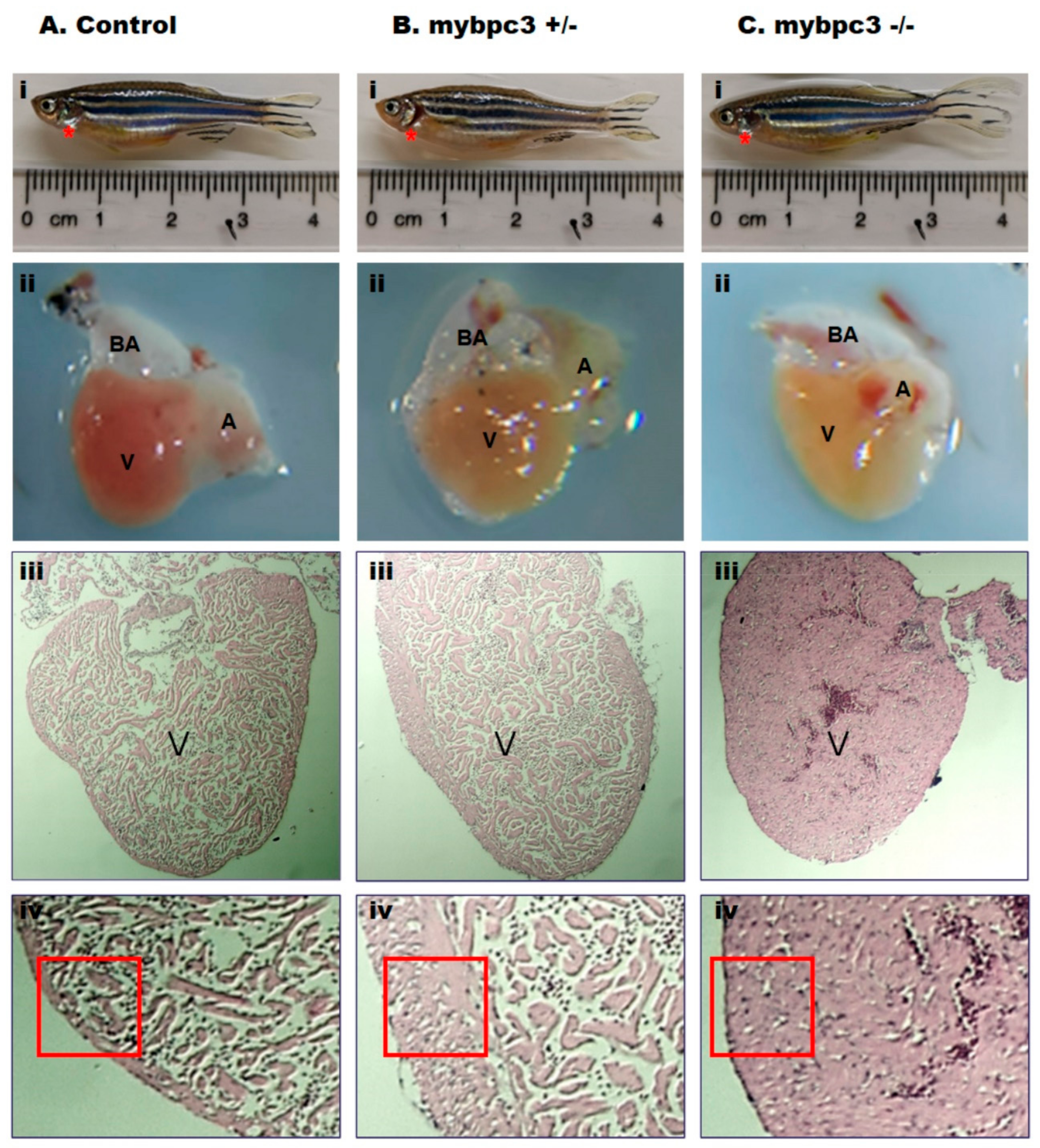
2.4. Ventricular Hypertrophy in Adult mybpc3 Mutant Zebrafish
2.5. Reduced Adult Swimming Performance in mybpc3 Mutant Zebrafish
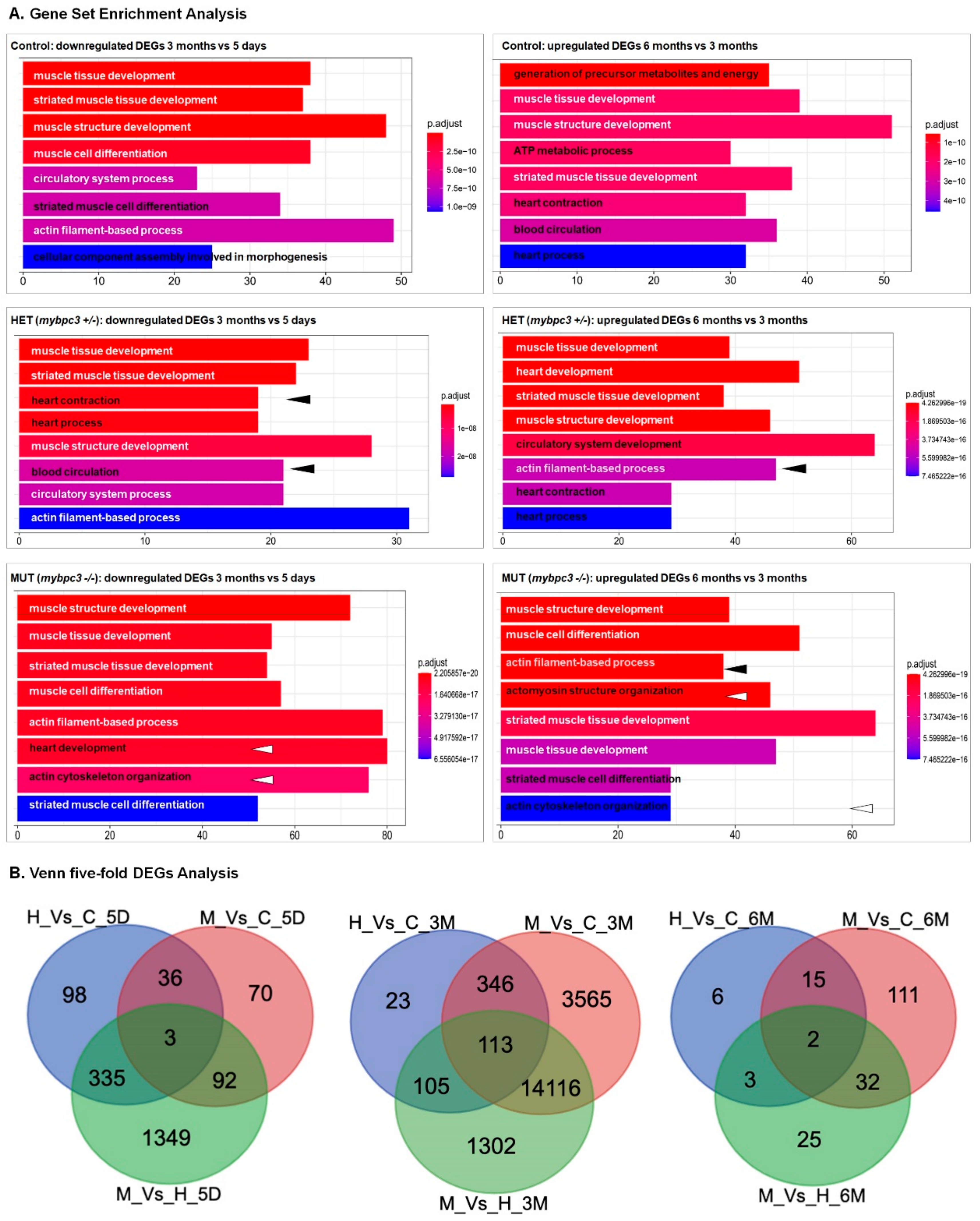
2.6. Differentially Expressed Genes through the Development of Hypertrophic Cardiomyopathy
3. Discussion
4. Materials and Methods
4.1. Zebrafish Lines and Maintenance
4.2. TALEN and Targeting-Vector Construction for mybpc3
4.3. TALEN Efficiency in Zebrafish Embryos
4.4. Genotyping of TALEN Edited mybpc3 Zebrafish Line
4.5. Generation of Zebrafish mybpc3 Mutant with Green-Fluorescent Hearts
4.6. Microscopic Assessment of Cardiac Phenotypes
4.7. Assessment of Cardiovascular Performance
4.8. Immunostaining of Zebrafish Mutants and Cardiomyocytes Count
4.9. The Heart Rate of Adult Zebrafish
4.10. Assessment of Swimming Performance and Endurance of Adult Zebrafish
4.11. Identification of Differentially Expressed Genes (DEGs) between HCM Zebrafish Mutants and Healthy Controls
4.12. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Semsarian, C.; Ingles, J.; Maron, M.S.; Maron, B.J. New perspectives on the prevalence of hypertrophic cardiomyopathy. J. Am. Coll. Cardiol. 2015, 65, 1249–1254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maron, B.J.; Gardin, J.M.; Flack, J.M.; Gidding, S.S.; Kurosaki, T.T.; Bild, D.E. Prevalence of hypertrophic cardiomyopathy in a general population of young adults. Echocardiographic analysis of 4111 subjects in the CARDIA study. Coronary artery risk development in (young) adults. Circulation 1995, 92, 785–789. [Google Scholar] [CrossRef]
- Basit, H.; Brito, D.; Sharma, S. Hypertrophic Cardiomyopathy. 2022. Available online: https://www.ncbi.nlm.nih.gov/books/NBK430788/ (accessed on 15 March 2022).
- Nagueh, S.F.; Bachinski, L.L.; Meyer, D.; Hill, R.; Zoghbi, W.A.; Tam, J.W.; Quiñones, M.A.; Roberts, R.; Marian, A. Tissue doppler imaging consistently detects myocardial abnormalities in patients with hypertrophic cardiomyopathy and provides a novel means for an early diagnosis before and independently of hypertrophy. Circulation 2001, 104, 128–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maron, B.J.; Ommen, S.R.; Semsarian, C.; Spirito, P.; Olivotto, I.; Maron, M.S. Hypertrophic cardiomyopathy: Present and future, with translation into contemporary cardiovascular medicine. J. Am. Coll. Cardiol. 2014, 64, 83–99. [Google Scholar] [PubMed] [Green Version]
- Marian, A.J.; Braunwald, E. Hypertrophic cardiomyopathy: Genetics, pathogenesis, clinical manifestations, diagnosis, and therapy. Circ. Res. 2017, 121, 749–770. [Google Scholar] [CrossRef]
- Schlossarek, S.; Mearini, G.; Carrier, L. Cardiac myosin-binding protein C in hypertrophic cardiomyopathy: Mechanisms and therapeutic opportunities. J. Mol. Cell. Cardiol. 2011, 50, 613–620. [Google Scholar] [CrossRef] [PubMed]
- Antunes, M.d.O.; Scudeler, T.L. Hypertrophic cardiomyopathy. Int. J. Cardiol. Heart Vasc. 2020, 27, 100503. [Google Scholar] [CrossRef] [PubMed]
- Sen-Chowdhry, S.; Jacoby, D.; Moon, J.; McKenna, W.J. Update on hypertrophic cardiomyopathy and a guide to the guidelines. Nat. Rev. Cardiol. 2016, 13, 651. [Google Scholar] [CrossRef]
- Sedaghat-Hamedani, F.; Kayvanpour, E.; Tugrul, O.F.; Lai, A.; Amr, A.; Haas, J.; Proctor, T.; Ehlermann, P.; Jensen, K.; Katus, H.A.; et al. Clinical outcomes associated with sarcomere mutations in hypertrophic cardiomyopathy: A meta-analysis on 7675 individuals. Clin. Res. Cardiol. 2018, 107, 30–41. [Google Scholar] [CrossRef]
- Tsang, D.C.; Link, M.S. Sudden cardiac death in athletes. Tex. Heart Inst. J. 2021, 48, e207513. [Google Scholar]
- Mohamed, I.A.; Krishnamoorthy, N.; Nasrallah, G.K.; Da’As, S.I. The role of cardiac myosin binding protein C3 in hypertrophic cardiomyopathy-progress and novel therapeutic opportunities. J. Cell. Physiol. 2017, 232, 1650–1659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maron, B.J. Clinical course and management of hypertrophic cardiomyopathy. N. Engl. J. Med. 2018, 379, 655–668. [Google Scholar] [CrossRef] [PubMed]
- Maron, B.J.; Maron, M.S.; Semsarian, C. Genetics of hypertrophic cardiomyopathy after 20 years: Clinical perspectives. J. Am. Coll. Cardiol. 2012, 60, 705–715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vikhorev, P.G.; Vikhoreva, N.N. Cardiomyopathies and related changes in contractility of human heart muscle. Int. J. Mol. Sci. 2018, 19, 2234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walsh, R.; Buchan, R.; Wilk, A.; John, S.; Felkin, L.E.; Thomson, K.; Chiaw, T.H.; Loong, C.C.W.; Pua, C.J.; Raphael, C.; et al. Defining the genetic architecture of hypertrophic cardiomyopathy: Re-evaluating the role of non-sarcomeric genes. Eur. Heart J. 2017, 38, 3461–3468. [Google Scholar] [CrossRef] [PubMed]
- Walsh, R.; Thomson, K.L.; Ware, J.S.; Funke, B.H.; Woodley, J.; McGuire, K.J.; Mazzarotto, F.; Blair, E.; Seller, A.; Taylor, J.C.; et al. Reassessment of Mendelian gene pathogenicity using 7855 cardiomyopathy cases and 60,706 reference samples. Genet. Med. 2017, 19, 192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harper, A.R.; HCMR Investigators; Goel, A.; Grace, C.; Thomson, K.L.; Petersen, S.E.; Xu, X.; Waring, A.; Ormondroyd, E.; Kramer, C.M.; et al. Common genetic variants and modifiable risk factors underpin hypertrophic cardiomyopathy susceptibility and expressivity. Nat. Genet. 2021, 53, 135–142. [Google Scholar] [CrossRef]
- Kassem, H.S.; Azer, R.S.; Ayad, M.S.; Moharem-Elgamal, S.; Magdy, G.; Elguindy, A.; Cecchi, F.; Olivotto, I.; Yacoub, M.H. Early results of sarcomeric gene screening from the egyptian national BA-HCM program. J. Cardiovasc. Transl. Res. 2012, 6, 65–80. [Google Scholar] [CrossRef] [Green Version]
- Heling, L.W.H.J.; Geeves, M.A.; Kad, N.M. MyBP-C: One protein to govern them all. J. Muscle Res. Cell Motil. 2020, 41, 91–101. [Google Scholar] [CrossRef] [Green Version]
- Hou, L.; Kumar, M.; Anand, P.; Chen, Y.; El-Bizri, N.; Pickens, C.J.; Seganish, W.M.; Sadayappan, S.; Swaminath, G. Modulation of myosin by cardiac myosin binding protein-C peptides improves cardiac contractility in ex-vivo experimental heart failure models. Sci. Rep. 2022, 12, 4337. [Google Scholar] [CrossRef]
- Luther, P.K.; Vydyanath, A. Myosin binding protein-C: An essential protein in skeletal and cardiac muscle. J. Muscle Res. Cell Motil. 2011, 31, 303–305. [Google Scholar] [CrossRef]
- Finley, N.L.; Cuperman, T.I. Cardiac myosin binding protein-C: A structurally dynamic regulator of myocardial contractility. Pflüg. Arch. Eur. J. Physiol. 2014, 466, 433–438. [Google Scholar] [CrossRef] [PubMed]
- Pfuhl, M.; Gautel, M. Structure, interactions and function of the N-terminus of cardiac myosin binding protein C (MyBP-C): Who does what, with what, and to whom? J. Muscle Res. Cell Motil. 2012, 33, 83–94. [Google Scholar] [CrossRef] [PubMed]
- Al-Khayat, H.A. Three-dimensional structure of the human myosin thick filament: Clinical implications. Glob. Cardiol. Sci. Pract. 2013, 2013, 280–302. [Google Scholar] [CrossRef]
- Lin, B.L.; Li, A.; Mun, J.Y.; Previs, M.J.; Previs, S.B.; Campbell, S.G.; Dos Remedios, C.G.; Tombe, P.D.P.; Craig, R.; Warshaw, D.M.; et al. Skeletal myosin binding protein-C isoforms regulate thin filament activity in a Ca2+-dependent manner. Sci. Rep. 2018, 8, 2604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaffer, J.F.; Kensler, R.W.; Harris, S.P. The myosin-binding protein C motif binds to F-actin in a phosphorylation-sensitive manner. J. Biol. Chem. 2009, 284, 12318–12327. [Google Scholar] [CrossRef] [Green Version]
- Belknap, B.; Harris, S.P.; White, H.D. Modulation of thin filament activation of myosin ATP hydrolysis by N-terminal domains of cardiac myosin binding protein-C. Biochemistry 2014, 53, 6717–6724. [Google Scholar] [CrossRef] [Green Version]
- Suay-Corredera, C.; Pricolo, M.R.; Velázquez-Carreras, D.; Pathak, D.; Nandwani, N.; Pimenta-Lopes, C.; Sánchez-Ortiz, D.; Urrutia-Irazabal, I.; Vilches, S.; Dominguez, F.; et al. Nanomechanical phenotypes in cardiac myosin-binding protein C mutants that cause hypertrophic cardiomyopathy. ACS Nano 2021, 15, 10203–10216. [Google Scholar] [CrossRef] [PubMed]
- Kampourakis, T.; Yan, Z.; Gautel, M.; Sun, Y.-B.; Irving, M. Myosin binding protein-C activates thin filaments and inhibits thick filaments in heart muscle cells. Proc. Natl. Acad. Sci. USA 2014, 111, 18763–18768. [Google Scholar] [CrossRef] [Green Version]
- Coulton, A.T.; Stelzer, J.E. Cardiac myosin binding protein C and its phosphorylation regulate multiple steps in the cross-bridge cycle of muscle contraction. Biochemistry 2012, 51, 3292–3301. [Google Scholar] [CrossRef] [Green Version]
- Sadayappan, S.; Gulick, J.; Osinska, H.; Barefield, D.; Cuello, F.; Avkiran, M.; Lasko, V.M.; Lorenz, J.N.; Maillet, M.; Martin, J.L.; et al. A critical function for Ser-282 in cardiac myosin binding protein-C phosphorylation and cardiac function novelty and significance. Circ. Res. 2011, 109, 141–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seeger, T.; Shrestha, R.; Lam, C.K.; Chen, C.; McKeithan, W.L.; Lau, E.; Wnorowski, A.; McMullen, G.; Greenhaw, M.; Lee, J.; et al. A premature termination codon mutation in MYBPC3 causes hypertrophic cardiomyopathy via chronic activation of nonsense-mediated decay. Circulation 2018, 139, 799–811. [Google Scholar] [CrossRef]
- Carrier, L.; Mearini, G.; Stathopoulou, K.; Cuello, F. Cardiac myosin-binding protein C (MYBPC3) in cardiac pathophysiology. Gene 2015, 573, 188–197. [Google Scholar] [CrossRef]
- Wijnker, P.J.M.; van der Velden, J. Mutation-specific pathology and treatment of hypertrophic cardiomyopathy in patients, mouse models and human engineered heart tissue. Biochim. Biophys. Acta BBA Mol. Basis Dis. 2020, 1866, 165774. [Google Scholar]
- Lu, Y.; Kwan, A.; Jeffries, C.M.; Guss, M.; Trewhella, J. The motif of human cardiac myosin-binding protein C is required for its Ca2+-dependent interaction with calmodulin. J. Biol. Chem. 2012, 287, 31596–31607. [Google Scholar] [CrossRef] [Green Version]
- Mun, J.Y.; Gulick, J.; Robbins, J.; Woodhead, J.; Lehman, W.; Craig, R. Electron microscopy and 3D reconstruction of F-actin decorated with cardiac myosin-binding protein C (cMyBP-C). J. Mol. Biol. 2011, 410, 214–225. [Google Scholar] [CrossRef] [Green Version]
- Poon, K.L.; Brand, T. The zebrafish model system in cardiovascular research: A tiny fish with mighty prospects. Glob. Cardiol. Sci. Pract. 2013, 2013, 4. [Google Scholar] [CrossRef] [Green Version]
- Amsterdam, A.; Hopkins, N. Mutagenesis strategies in zebrafish for identifying genes involved in development and disease. Trends Genet. 2006, 22, 473–478. [Google Scholar] [CrossRef]
- Da’as, S.I.; Yu, J.; Butcher, J.T.; Krishnamoorthy, N.; Al Suwaidi, J.A.S.; Kassem, H.; Al Shafai, K.N.; Al-Hashemi, M.A.; Shuayb, L.; Brand, T.; et al. Different human mutations in the myosin binding protein C3 (MYBPC3) produce specific cardiac phenotypes in the zebrafish. Circulation 2014, 130, A17545. [Google Scholar]
- Liu, X.; Jiang, T.; Piao, C.; Li, X.; Guo, J.; Zheng, S.; Zhang, X.; Cai, T.; Du, J. Screening mutations of MYBPC3 in 114 unrelated patients with hypertrophic cardiomyopathy by targeted capture and next-generation sequencing. Sci. Rep. 2015, 5, 11411. [Google Scholar] [CrossRef]
- Chi, N.C.; Shaw, R.M.; Jungblut, B.; Huisken, J.; Ferrer, T.; Arnaout, R.; Scott, I.; Beis, D.; Xiao, T.; Baier, H.; et al. Genetic and physiologic dissection of the vertebrate cardiac conduction system. PLoS Biol. 2008, 4, 262. [Google Scholar] [CrossRef] [Green Version]
- Sabater-Molina, M.; Pérez-Sánchez, I.; Hernandez Del Rincon, J.P.; Gimeno, J.R. Genetics of hypertrophic cardiomyopathy: A review of current state. Clin. Genet. 2018, 93, 3–14. [Google Scholar] [CrossRef]
- Frey, N.; Luedde, M.; Katus, H.A. Mechanisms of disease: Hypertrophic cardiomyopathy. Nat. Rev. Cardiol. 2012, 9, 91. [Google Scholar] [CrossRef]
- Von Gise, A.; Lin, Z.; Schlegelmilch, K.; Honor, L.B.; Pan, G.M.; Buck, J.N.; Ma, Q.; Ishiwata, T.; Zhou, B.; Camargo, F.D.; et al. YAP1, the nuclear target of Hippo signaling, stimulates heart growth through cardiomyocyte proliferation but not hypertrophy. Proc. Natl. Acad. Sci. USA 2012, 109, 2394–2399. [Google Scholar] [CrossRef] [Green Version]
- Bravo, P.E.; Pinheiro, A.; Higuchi, T.; Rischpler, C.; Merrill, J.; Santaularia-Tomas, M.; Abraham, M.R.; Wahl, R.L.; Abraham, T.P.; Bengel, F.M. PET/CT Assessment of symptomatic individuals with obstructive and nonobstructive hypertrophic cardiomyopathy. J. Nucl. Med. 2012, 53, 407–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farrell, E.T.; Grimes, A.C.; De Lange, W.J.; Armstrong, A.E.; Ralphe, J.C. Increased postnatal cardiac hyperplasia precedes cardiomyocyte hypertrophy in a model of hypertrophic cardiomyopathy. Front. Physiol. 2017, 8, 414. [Google Scholar] [CrossRef] [Green Version]
- McConnell, B.K.; Jones, K.A.; Fatkin, D.; Arroyo, L.H.; Lee, R.T.; Aristizabal, O.; Turnbull, D.H.; Georgakopoulos, D.; Kass, D.; Bond, M.; et al. Dilated cardiomyopathy in homozygous myosin-binding protein-C mutant mice. J. Clin. Investig. 1999, 104, 1235–1244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barefield, D.; Kumar, M.; Gorham, J.; Seidman, J.G.; Seidman, C.E.; de Tombe, P.P.; Sadayappan, S. Haploinsufficiency of MYBPC3 exacerbates the development of hypertrophic cardiomyopathy in heterozygous mice. J. Mol. Cell. Cardiol. 2015, 79, 234–243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, J.; Burgon, P.G.; Wakimoto, H.; Onoue, K.; Gorham, J.M.; O’Meara, C.C.; Fomovsky, G.; McConnell, B.K.; Lee, R.T.; Seidman, J.G.; et al. Cardiac myosin binding protein C regulates postnatal myocyte cytokinesis. Proc. Natl. Acad. Sci. USA 2015, 112, 9046–9051. [Google Scholar] [CrossRef] [Green Version]
- Farrell, E.; Armstrong, A.E.; Grimes, A.C.; Naya, F.J.; De Lange, W.J.; Ralphe, J.C. Transcriptome analysis of cardiac hypertrophic growth in MYBPC3-null mice suggests early responders in hypertrophic remodeling. Front. Physiol. 2018, 9, 1442. [Google Scholar] [CrossRef] [Green Version]
- Lyon, R.C.; Zanella, F.; Omens, J.H.; Sheikh, F. Mechanotransduction in cardiac hypertrophy and failure. Circ. Res. 2015, 116, 1462–1476. [Google Scholar] [CrossRef] [Green Version]
- Korte, F.S.; McDonald, K.S.; Harris, S.P.; Moss, R.L. Loaded shortening, power output, and rate of force redevelopment are increased with knockout of cardiac myosin binding protein-C. Circ. Res. 2003, 93, 752–758. [Google Scholar] [CrossRef] [Green Version]
- Kulikovskaya, I.; McClellan, G.; Flavigny, J.; Carrier, L.; Winegrad, S. Effect of MyBP-C binding to actin on contractility in heart muscle. J. Gen. Physiol. 2003, 122, 761–774. [Google Scholar] [CrossRef] [Green Version]
- Stelzer, J.E.; Dunning, S.B.; Moss, R.L. Ablation of cardiac myosin-binding protein-C accelerates stretch activation in murine skinned myocardium. Circ. Res. 2006, 98, 1212–1218. [Google Scholar] [CrossRef] [Green Version]
- Pohlmann, L.; Kroger, I.; Vignier, N.; Schlossarek, S.; Krämer, E.; Coirault, C.; Sultan, K.R.; El-Armouche, A.; Winegard, S.; Eschenhagen, T.; et al. Cardiac myosin-binding protein C is required for complete relaxation in intact myocytes. Circ. Res. 2007, 101, 928–938. [Google Scholar] [CrossRef] [Green Version]
- Van Dijk, S.J.; Paalberends, E.R.; Najafi, A.; Michels, M.; Sadayappan, S.; Carrier, L.; Boontje, N.M.; Kuster, D.W.; van Slegtenhorst, M.; Dooijes, D.; et al. Contractile dysfunction irrespective of the mutant protein in human hypertrophic cardiomyopathy with normal systolic function. Circ. Heart Fail. 2012, 5, 36–46. [Google Scholar] [CrossRef] [Green Version]
- Sadayappan, S.; de Tombe, P.P. Cardiac myosin binding protein-C: Redefining its structure and function. Biophys. Rev. 2012, 4, 93–106. [Google Scholar] [CrossRef] [PubMed]
- Westerfield, M. The Zebrafish Book. A Guide for the Laboratory Use of Zebrafish (Danio rerio); University of Oregon Press: Eugene, OR, USA, 2000. [Google Scholar]
- Kimmel, C.B.; Ballard, W.W.; Kimmel, S.R.; Ullmann, B.; Schilling, T.F. Stages of embryonic development of the zebrafish. Dev. Dyn. 1995, 203, 253–310. [Google Scholar] [CrossRef]
- Cornet, C.; Calzolari, S.; Miñana-Prieto, R.; Dyballa, S.; Van Doornmalen, E.; Rutjes, H.; Savy, T.; D’Amico, D.; Terriente, J. ZeGlobalTox: An innovative approach to address organ drug toxicity using zebrafish. Int. J. Mol. Sci. 2017, 18, 864. [Google Scholar] [CrossRef] [Green Version]
- Milan, D.J.; Jones, I.L.; Ellinor, P.; Macrae, C.A. In vivo recording of adult zebrafish electrocardiogram and assessment of drug-induced QT prolongation. Am. J. Physiol. Circ. Physiol. 2006, 291, H269–H273. [Google Scholar] [CrossRef]
- Dhillon, S.S.; Dóró, É.; Magyary, I.; Egginton, S.; Sík, A.; Müller, F. Optimisation of embryonic and larval ECG measurement in zebrafish for quantifying the effect of QT prolonging drugs. PLoS ONE 2013, 8, e60552. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Xu, X. Immunostaining of dissected zebrafish embryonic heart. J. Vis. Exp. 2012, 59, e3510. [Google Scholar] [CrossRef] [PubMed]
- Da’As, S.I.; Coombs, A.J.; Balci, T.B.; Grondin, C.A.; Ferrando, A.A.; Berman, J.N. The zebrafish reveals dependence of the mast cell lineage on Notch signaling in vivo. Blood 2012, 119, 3585–3594. [Google Scholar] [CrossRef]
- Huang, W.-C.; Hsieh, Y.-S.; Chen, I.-H.; Wang, C.-H.; Chang, H.-W.; Yang, C.-C.; Ku, T.-H.; Yeh, S.-R.; Chuang, Y.-J. Combined use of MS-222 (tricaine) and isoflurane extends anesthesia time and minimizes cardiac rhythm side effects in adult zebrafish. Zebrafish 2010, 7, 297–304. [Google Scholar] [CrossRef] [PubMed]
- Lee, L.; Genge, C.E.; Cua, M.; Sheng, X.; Rayani, K.; Beg, M.F.; Sarunic, M.V.; Tibbits, G.F. Functional assessment of cardiac responses of adult zebrafish (Danio rerio) to acute and chronic temperature change using high-resolution echocardiography. PLoS ONE 2016, 11, e0145163. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Da’as, S.I.; Hasan, W.; Salem, R.; Younes, N.; Abdelrahman, D.; Mohamed, I.A.; Aldaalis, A.; Temanni, R.; Mathew, L.S.; Lorenz, S.; et al. Transcriptome Profile Identifies Actin as an Essential Regulator of Cardiac Myosin Binding Protein C3 Hypertrophic Cardiomyopathy in a Zebrafish Model. Int. J. Mol. Sci. 2022, 23, 8840. https://doi.org/10.3390/ijms23168840
Da’as SI, Hasan W, Salem R, Younes N, Abdelrahman D, Mohamed IA, Aldaalis A, Temanni R, Mathew LS, Lorenz S, et al. Transcriptome Profile Identifies Actin as an Essential Regulator of Cardiac Myosin Binding Protein C3 Hypertrophic Cardiomyopathy in a Zebrafish Model. International Journal of Molecular Sciences. 2022; 23(16):8840. https://doi.org/10.3390/ijms23168840
Chicago/Turabian StyleDa’as, Sahar Isa, Waseem Hasan, Rola Salem, Nadine Younes, Doua Abdelrahman, Iman A. Mohamed, Arwa Aldaalis, Ramzi Temanni, Lisa Sara Mathew, Stephan Lorenz, and et al. 2022. "Transcriptome Profile Identifies Actin as an Essential Regulator of Cardiac Myosin Binding Protein C3 Hypertrophic Cardiomyopathy in a Zebrafish Model" International Journal of Molecular Sciences 23, no. 16: 8840. https://doi.org/10.3390/ijms23168840
APA StyleDa’as, S. I., Hasan, W., Salem, R., Younes, N., Abdelrahman, D., Mohamed, I. A., Aldaalis, A., Temanni, R., Mathew, L. S., Lorenz, S., Yacoub, M., Nomikos, M., Nasrallah, G. K., & Fakhro, K. A. (2022). Transcriptome Profile Identifies Actin as an Essential Regulator of Cardiac Myosin Binding Protein C3 Hypertrophic Cardiomyopathy in a Zebrafish Model. International Journal of Molecular Sciences, 23(16), 8840. https://doi.org/10.3390/ijms23168840