
Functional Expression of Choline Transporters in Microglia and Their Regulation of Microglial M1/M2 Polarization



Abstract
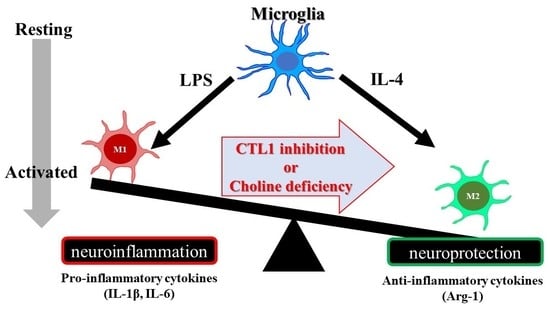
1. Introduction
2. Results
2.1. Expression of Choline Transporters in SIM-A9 Cells
2.2. Properties of [3H]Choline Uptake in SIM-A9 Cells
2.3. Effects of LPS and IL-4 on [3H]Choline Uptake in SIM-A9 Cells
2.4. Effects of Choline Deficiency on IL-1β, IL-6, and Arg-1 mRNA Expression in SIM-A9 Cells Stimulated with LPS and IL-4
2.5. Effect of HC-3 on IL-1β, IL-6, and Arg-1 mRNA Expression in SIM-A9 Cells Stimulated by LPS and IL-4
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cell Culture
4.3. RNA Extraction and Real-Time PCR Assay
4.4. Immunoblotting
4.5. Immunofluorescence Staining
4.6. Analysis of [3H]Choline Uptake into SIM-A9 Cells
4.7. Data Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Greter, M.; Merad, M. Regulation of microglia development and homeostasis. Glia 2013, 61, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Ginhoux, F.; Greter, M.; Leboeuf, M.; Nandi, S.; See, P.; Gokhan, S.; Mehler, M.F.; Conway, S.J.; Ng, L.G.; Stanley, E.R.; et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 2010, 330, 841–845. [Google Scholar] [CrossRef] [PubMed]
- Schulz, C.; Gomez Perdiguero, E.; Chorro, L.; Szabo-Rogers, H.; Cagnard, N.; Kierdorf, K.; Prinz, M.; Wu, B.; Jacobsen, S.E.; Pollard, J.W.; et al. A lineage of myeloid cells independent of Myb and hematopoietic stem cells. Science 2012, 336, 86–90. [Google Scholar] [CrossRef] [PubMed]
- Ginhoux, F.; Prinz, M. Origin of microglia: Current concepts and past controversies. Cold Spring Harb. Perspect. Biol. 2015, 7, a020537. [Google Scholar] [CrossRef]
- Eggen, B.J.L.; Boddeke, E.; Kooistra, S.M. Regulation of Microglia Identity from an Epigenetic and Transcriptomic Point of View. Neuroscience 2019, 405, 3–13. [Google Scholar] [CrossRef]
- Kettenmann, H.; Hanisch, U.K.; Noda, M.; Verkhratsky, A. Physiology of microglia. Physiol. Rev. 2011, 91, 461–553. [Google Scholar] [CrossRef]
- Tang, Y.; Le, W. Differential Roles of M1 and M2 Microglia in Neurodegenerative Diseases. Mol. Neurobiol. 2016, 53, 1181–1194. [Google Scholar] [CrossRef]
- Chu, A.J. Bacterial lipopolysaccharide stimulates phospholipid synthesis and phosphatidylcholine breakdown in cultured human leukemia monocytic THP-1 cells. Int. J. Biochem. 1992, 24, 317–323. [Google Scholar] [CrossRef]
- Grove, R.I.; Allegretto, N.J.; Kiener, P.A.; Warr, G.A. Lipopolysaccharide (LPS) alters phosphatidylcholine metabolism in elicited peritoneal macrophages. J. Leukoc. Biol. 1990, 48, 38–42. [Google Scholar] [CrossRef]
- Tian, Y.; Pate, C.; Andreolotti, A.; Wang, L.; Tuomanen, E.; Boyd, K.; Claro, E.; Jackowski, S. Cytokine secretion requires phosphatidylcholine synthesis. J. Cell Biol. 2008, 181, 945–957. [Google Scholar] [CrossRef]
- Inazu, M. Functional Expression of Choline Transporters in the Blood-Brain Barrier. Nutrients 2019, 11, 2265. [Google Scholar] [CrossRef]
- Yamamura, H.I.; Snyder, S.H. Choline: High-affinity uptake by rat brain synaptosomes. Science 1972, 178, 626–628. [Google Scholar] [CrossRef]
- Yamamura, H.I.; Snyder, S.H. High affinity transport of choline into synaptosomes of rat brain. J. Neurochem. 1973, 21, 1355–1374. [Google Scholar] [CrossRef]
- Michel, V.; Yuan, Z.; Ramsubir, S.; Bakovic, M. Choline transport for phospholipid synthesis. Exp. Biol. Med. 2006, 231, 490–504. [Google Scholar] [CrossRef]
- Orihuela, R.; McPherson, C.A.; Harry, G.J. Microglial M1/M2 polarization and metabolic states. Br. J. Pharmacol. 2016, 173, 649–665. [Google Scholar] [CrossRef]
- He, Y.; Gao, Y.; Zhang, Q.; Zhou, G.; Cao, F.; Yao, S. IL-4 Switches Microglia/macrophage M1/M2 Polarization and Alleviates Neurological Damage by Modulating the JAK1/STAT6 Pathway Following ICH. Neuroscience 2020, 437, 161–171. [Google Scholar] [CrossRef]
- Watanabe, S.; Nishijima, N.; Hirai, K.; Shibata, K.; Hase, A.; Yamanaka, T.; Inazu, M. Anticancer Activity of Amb4269951, a Choline Transporter-Like Protein 1 Inhibitor, in Human Glioma Cells. Pharmaceuticals 2020, 13, 104. [Google Scholar] [CrossRef]
- Hirai, K.; Watanabe, S.; Nishijima, N.; Shibata, K.; Hase, A.; Yamanaka, T.; Inazu, M. Molecular and Functional Analysis of Choline Transporters and Antitumor Effects of Choline Transporter-Like Protein 1 Inhibitors in Human Pancreatic Cancer Cells. Int. J. Mol. Sci. 2020, 21, 5190. [Google Scholar] [CrossRef]
- Shibata, K.; Nishijima, N.; Hirai, K.; Watanabe, S.; Yamanaka, T.; Chikazu, D.; Inazu, M. A Novel Plant-Derived Choline Transporter-like Protein 1 Inhibitor, Amb544925, Induces Apoptotic Cell Death via the Ceramide/Survivin Pathway in Tongue Squamous Cell Carcinoma. Cancers 2022, 14, 329. [Google Scholar] [CrossRef]
- Sanchez-Lopez, E.; Zhong, Z.; Stubelius, A.; Sweeney, S.R.; Booshehri, L.M.; Antonucci, L.; Liu-Bryan, R.; Lodi, A.; Terkeltaub, R.; Lacal, J.C.; et al. Choline Uptake and Metabolism Modulate Macrophage IL-1beta and IL-18 Production. Cell Metab. 2019, 29, 1350–1362.e7. [Google Scholar] [CrossRef]
- Hedtke, V.; Bakovic, M. Choline transport for phospholipid synthesis: An emerging role of choline transporter-like protein 1. Exp. Biol. Med. 2019, 244, 655–662. [Google Scholar] [CrossRef] [PubMed]
- Inazu, M. Choline transporter-like proteins CTLs/SLC44 family as a novel molecular target for cancer therapy. Biopharm. Drug Dispos. 2014, 35, 431–449. [Google Scholar] [CrossRef] [PubMed]
- Yara, M.; Iwao, B.; Hara, N.; Yamanaka, T.; Uchino, H.; Inazu, M. Molecular and functional characterization of choline transporter in the human trophoblastic cell line JEG-3 cells. Placenta 2015, 36, 631–637. [Google Scholar] [CrossRef] [PubMed]
- Welch, M.J.; Markham, C.H.; Jenden, D.J. Acetylcholine and choline in cerebrospinal fluid of patients with Parkinson’s disease and Huntington’s chorea. J. Neurol. Neurosurg. Psychiatry 1976, 39, 367–374. [Google Scholar] [CrossRef][Green Version]
- Tang, Y.; Le, W. “Good” and “Bad” Microglia in Parkinson’s Disease: An Understanding of Homeostatic Mechanisms in Immunomodulation. In Inflammation in Parkinson’s Disease; Springer: Berlin/Heidelberg, Germany, 2014; pp. 105–126. [Google Scholar]
- Zhang, L.; Zhang, J.; You, Z. Switching of the Microglial Activation Phenotype Is a Possible Treatment for Depression Disorder. Front. Cell. Neurosci. 2018, 12, 306. [Google Scholar] [CrossRef]
- Ohto, U.; Fukase, K.; Miyake, K.; Shimizu, T. Structural basis of species-specific endotoxin sensing by innate immune receptor TLR4/MD-2. Proc. Natl. Acad. Sci. USA 2012, 109, 7421–7426. [Google Scholar] [CrossRef]
- Gadani, S.P.; Cronk, J.C.; Norris, G.T.; Kipnis, J. IL-4 in the brain: A cytokine to remember. J. Immunol. 2012, 189, 4213–4219. [Google Scholar] [CrossRef]
- Kim, H.M.; Shin, H.Y.; Jeong, H.J.; An, H.J.; Kim, N.S.; Chae, H.J.; Kim, H.R.; Song, H.J.; Kim, K.Y.; Baek, S.H.; et al. Reduced IL-2 but elevated IL-4, IL-6, and IgE serum levels in patients with cerebral infarction during the acute stage. J. Mol. Neurosci. 2000, 14, 191–196. [Google Scholar] [CrossRef]
- Xiong, X.; Barreto, G.E.; Xu, L.; Ouyang, Y.B.; Xie, X.; Giffard, R.G. Increased brain injury and worsened neurological outcome in interleukin-4 knockout mice after transient focal cerebral ischemia. Stroke 2011, 42, 2026–2032. [Google Scholar] [CrossRef]
- Xiong, X.; Xu, L.; Wei, L.; White, R.E.; Ouyang, Y.B.; Giffard, R.G. IL-4 Is Required for Sex Differences in Vulnerability to Focal Ischemia in Mice. Stroke 2015, 46, 2271–2276. [Google Scholar] [CrossRef]
- Kabba, J.A.; Xu, Y.; Christian, H.; Ruan, W.; Chenai, K.; Xiang, Y.; Zhang, L.; Saavedra, J.M.; Pang, T. Microglia: Housekeeper of the Central Nervous System. Cell. Mol. Neurobiol. 2018, 38, 53–71. [Google Scholar] [CrossRef]
- Snider, S.A.; Margison, K.D.; Ghorbani, P.; LeBlond, N.D.; O’Dwyer, C.; Nunes, J.R.C.; Nguyen, T.; Xu, H.; Bennett, S.A.L.; Fullerton, M.D. Choline transport links macrophage phospholipid metabolism and inflammation. J. Biol. Chem. 2018, 293, 11600–11611. [Google Scholar] [CrossRef]
- Hu, X.; Li, P.; Guo, Y.; Wang, H.; Leak, R.K.; Chen, S.; Gao, Y.; Chen, J. Microglia/macrophage polarization dynamics reveal novel mechanism of injury expansion after focal cerebral ischemia. Stroke 2012, 43, 3063–3070. [Google Scholar] [CrossRef]
- Perego, C.; Fumagalli, S.; De Simoni, M.G. Temporal pattern of expression and colocalization of microglia/macrophage phenotype markers following brain ischemic injury in mice. J. Neuroinflammation 2011, 8, 174. [Google Scholar] [CrossRef]
- Kanazawa, M.; Ninomiya, I.; Hatakeyama, M.; Takahashi, T.; Shimohata, T. Microglia and Monocytes/Macrophages Polarization Reveal Novel Therapeutic Mechanism against Stroke. Int. J. Mol. Sci. 2017, 18, 2135. [Google Scholar] [CrossRef]
- Lan, X.; Han, X.; Li, Q.; Yang, Q.W.; Wang, J. Modulators of microglial activation and polarization after intracerebral haemorrhage. Nat. Rev. Neurol. 2017, 13, 420–433. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhang, Z.; Lu, H.; Yang, Q.; Wu, H.; Wang, J. Microglial Polarization and Inflammatory Mediators After Intracerebral Hemorrhage. Mol. Neurobiol. 2017, 54, 1874–1886. [Google Scholar] [CrossRef]
- Zhao, H.; Garton, T.; Keep, R.F.; Hua, Y.; Xi, G. Microglia/Macrophage Polarization After Experimental Intracerebral Hemorrhage. Transl. Stroke Res. 2015, 6, 407–409. [Google Scholar] [CrossRef]
- Kuzmich, N.N.; Sivak, K.V.; Chubarev, V.N.; Porozov, Y.B.; Savateeva-Lyubimova, T.N.; Peri, F. TLR4 Signaling Pathway Modulators as Potential Therapeutics in Inflammation and Sepsis. Vaccines 2017, 5, 34. [Google Scholar] [CrossRef]
- Nagamoto-Combs, K.; Kulas, J.; Combs, C.K. A novel cell line from spontaneously immortalized murine microglia. J. Neurosci. Methods 2014, 233, 187–198. [Google Scholar] [CrossRef]
- Fujita, Y.; Nagakura, T.; Uchino, H.; Inazu, M.; Yamanaka, T. Functional Expression of Choline Transporters in Human Neural Stem Cells and Its Link to Cell Proliferation, Cell Viability, and Neurite Outgrowth. Cells 2021, 10, 453. [Google Scholar] [CrossRef]
- Ishikawa, T.; Suwanai, H.; Shikuma, J.; Suzuki, R.; Yamanaka, T.; Odawara, M.; Inazu, M. Protein kinase C promotes choline transporterlike protein 1 function via improved cell surface expression in immortalized human hepatic cells. Mol. Med. Rep. 2020, 21, 777–785. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target Gene | Accession Number | Assay ID |
|---|---|---|
| CTL1 (slc44a1) | NM_001159633.1 | Mm01350815_m1 |
| CTL2 (slc44a2) | NM_001199186.1 | Mm00507664_m1 |
| CTL3 (slc44a3) | NM_145394.3 | Mm00520420_m1 |
| CTL4 (slc44a4) | NM_023557.3 | Mm00469893_m1 |
| CTL5 (slc44a5) | NM_001081263.1 | Mm01317372_m1 |
| CHT1 (slc5a7) | NM_022025.4 | Mm00452075_m1 |
| OCT1 (slc22a1) | NM_009202.5 | Mm00456303_m1 |
| OCT2 (slc22a2) | NM_013667.2 | Mm00457295_m1 |
| β-actin | AK078935.1 | Mm00607939_s1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Okada, T.; Muto, E.; Yamanaka, T.; Uchino, H.; Inazu, M. Functional Expression of Choline Transporters in Microglia and Their Regulation of Microglial M1/M2 Polarization. Int. J. Mol. Sci. 2022, 23, 8924. https://doi.org/10.3390/ijms23168924
Okada T, Muto E, Yamanaka T, Uchino H, Inazu M. Functional Expression of Choline Transporters in Microglia and Their Regulation of Microglial M1/M2 Polarization. International Journal of Molecular Sciences. 2022; 23(16):8924. https://doi.org/10.3390/ijms23168924
Chicago/Turabian StyleOkada, Toshio, Eisuke Muto, Tsuyoshi Yamanaka, Hiroyuki Uchino, and Masato Inazu. 2022. "Functional Expression of Choline Transporters in Microglia and Their Regulation of Microglial M1/M2 Polarization" International Journal of Molecular Sciences 23, no. 16: 8924. https://doi.org/10.3390/ijms23168924
APA StyleOkada, T., Muto, E., Yamanaka, T., Uchino, H., & Inazu, M. (2022). Functional Expression of Choline Transporters in Microglia and Their Regulation of Microglial M1/M2 Polarization. International Journal of Molecular Sciences, 23(16), 8924. https://doi.org/10.3390/ijms23168924