
Pathology and Pathogenesis of Brain Lesions Produced by Clostridium perfringens Type D Epsilon Toxin


Abstract
:1. Introduction
2. Epsilon Toxin
3. Intestinal Bacterial Proliferation and Toxin Production
4. The Enterotoxemic Phase
5. The Acute Neurologic Syndrome: Vasculocentric Brain Injury with Resulting Generalized Cerebral Edema
5.1. Neurologic Signs
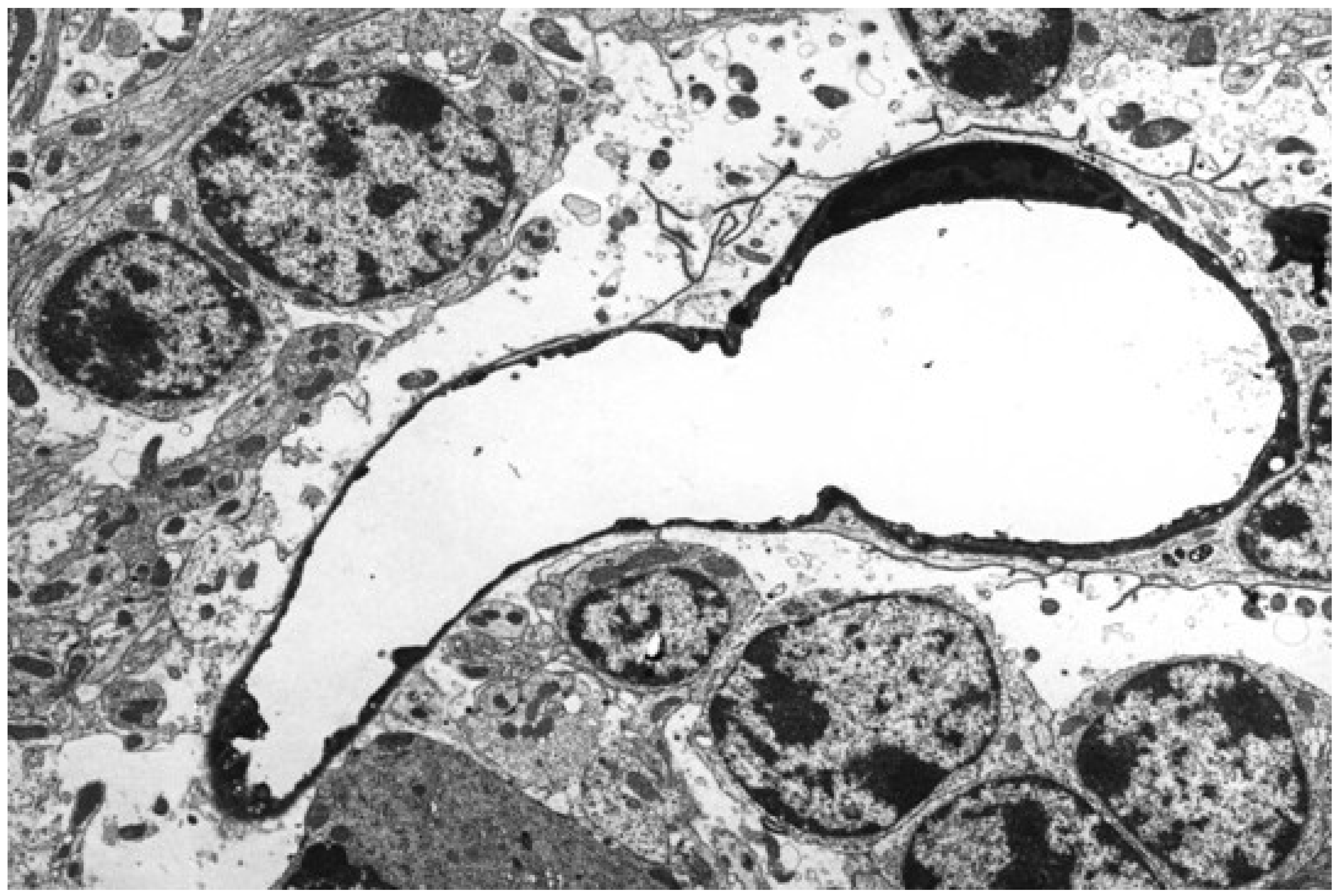
5.2. Neuropathology
5.3. Pathogenesis
6. The Protracted Neurologic Syndrome: Focal, Bilaterally Symmetrical, Regionally Selective, Neuroparenchymal Necrosis
6.1. Neurologic Signs
6.2. Neuropathology
6.3. Pathogenesis
7. Conclusions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rood, J.I.; Adams, V.; Lacey, J.; Lyras, D.; McClane, B.A.; Melville, S.B.; Moore, R.J.; Popoff, M.R.; Sarker, M.R.; Songer, J.G.; et al. Expansion of the Clostridium perfringens-based typing scheme. Anaerobe 2018, 53, 5–10. [Google Scholar] [CrossRef]
- Theoret, J.; McClane, B. Toxins of Clostridium perfringens. In Clostridial Diseases of Animals; Uzal, F.A., Songer, J.G., Prescott, J.F., Popoff, M.R., Eds.; Wiley-Blackwell: Ames, IA, USA, 2016; pp. 45–60. [Google Scholar]
- Garcia, J.P.; Adams, V.; Beingesser, J.; Hughes, M.L.; Poon, R.; Lyras, D.; Hill, A.; McClane, B.A.; Rood, J.I.; Uzal, F.A. Epsilon toxin is essential for the virulence of Clostridium perfringens type D infection in sheep, goats, and mice. Infect. Immun. 2013, 81, 2405–2414. [Google Scholar] [CrossRef]
- Songer, J.G. Clostridial enteric diseases of domestic animals. Clin. Microbiol. Rev. 1996, 9, 216–234. [Google Scholar] [CrossRef] [PubMed]
- Cantile, C.; Youssef, S. Nervous System. In Jubb, Kennedy and Palmer’s Pathology of Domestic Animals; Maxie, G., Ed.; Elsevier: St. Louis, MO, USA, 2016; pp. 307–308. [Google Scholar]
- Uzal, F.A.; Plattner, B.L.; Hostetter, J.M. Alimentary System. In Jubb, Kennedy and Palmer’s Pathology of Domestic Animals; Maxie, G., Ed.; Elsevier: St Louis, MO, USA, 2016; pp. 188–191. [Google Scholar]
- Uzal, F.A.; Kelly, W.R.; Morris, W.E.; Assis, R.A. Effects of intravenous injection of Clostridium perfringens type D epsilon toxin in calves. J. Comp. Pathol. 2002, 126, 71–75. [Google Scholar] [CrossRef] [PubMed]
- Filho, E.J.; Carvalho, A.V.; Assis, R.A.; Lobato, F.F.; Rachid, M.A.; Carvalho, A.A.; Ferreira, P.M.; Nascimento, R.A.; Fernandes, A.A.; Vidal, J.E.; et al. Clinicopathologic features of experimental Clostridium perfringens type D entertoxemia in cattle. Vet. Pathol. 2009, 46, 1213–1220. [Google Scholar] [CrossRef] [PubMed]
- Mete, A.; Garcia, J.; Ortega, J.; Lane, M.; Scholes, S.; Uzal, F.A. Brain lesions associated with Clostridium perfringens type D epsilon toxin in a Holstein heifer calf. Vet. Pathol. 2013, 50, 765–768. [Google Scholar] [CrossRef]
- Stubbings, D.P. Clostridium perfringens enterotoxemia in 2 young horses. Vet. Rec. 1990, 127, 431. [Google Scholar]
- Summers, B.A.; Cummings, J.F.; deLahunta, A. Veterinary Neuropathology; Mosby: St. Louis, MO, USA, 1995; pp. 269–270. [Google Scholar]
- Wagley, S.; Bokori-Brown, M.; Morcrette, H.; Malaspina, A.; D’Arcy, C.; Gnanapavan, S.; Lewis, N.; Popoff, M.R.; Raciborska, D.; Nicholas, R.; et al. Evidence of Clostridium perfringens epsilon toxin associated with multiple sclerosis. Mult. Scler. 2019, 25, 653–660. [Google Scholar] [CrossRef]
- Goldstein, J.; Morris, W.E.; Loidl, C.F.; Tironi-Farinati, C.; McClane, B.A.; Uzal, F.A.; Fernandez Miyakawa, M.E. Clostridium perfringens epsilon toxin increases the small intestinal permeability in mice and rats. PLoS ONE 2009, 4, e7065. [Google Scholar] [CrossRef]
- Finnie, J.W.; Navarro, M.A.; Uzal, F.A. Pathogenesis and diagnostic features of brain and ophthalmic damage produced by Clostridium perfringens type D epsilon toxin. J. Vet. Diagn. Investig. 2020, 32, 282–286. [Google Scholar] [CrossRef]
- Garcia, J.P.; Giannitti, F.; Finnie, J.W.; Manavis, J.; Beingesser, J.; Adams, V.; Rood, J.I.; Uzal, F.A. Comparative neuropathology of ovine enterotoxemia produced by Clostridium perfringens type D wild-type strain CN1020 and its genetically modified derivatives. Vet. Pathol. 2015, 52, 465–475. [Google Scholar] [CrossRef] [PubMed]
- Uzal, F.A.; Giannitti, F.; Finnie, J.W.; Garcia, J.P. Diseases Produced by Clostridium perfringens Type D. In Clostridial Diseases of Animals; Uzal, F.A., Songer, J.G., Prescott, J.F., Popoff, M.R., Eds.; Wiley-Blackwell: Ames, IA, USA, 2016; pp. 157–172. [Google Scholar]
- Uzal, F.A.; Navarro, M.A.; Li, J.; Freedman, J.C.; Shrestha, A.; McClane, B.A. Comparative pathogenesis of enteric clostridial infections in humans and animals. Anaerobe 2018, 53, 11–20. [Google Scholar] [CrossRef]
- Uzal, F.A.; Navarro, M.A.; Hostetter, J.M. Focus issue on clostridial disease. J. Vet. Diagn. Investig. 2020, 32, 173–174. [Google Scholar] [CrossRef]
- Munday, B.L.; Mason, R.W.; Hartley, W.J. Encephalopathies in cattle in Tasmania. Aust. Vet. J. 1976, 52, 92–96. [Google Scholar] [CrossRef]
- Hartley, W.J. A focal symmetrical encephalomalacia of lambs. N. Z. Vet. J. 1956, 4, 129–135. [Google Scholar] [CrossRef]
- Rood, J.I. General Physiological and Virulence Properties of the Pathogenic Clostridia. In Clostridial Diseases of Animals; Uzal, F.A., Songer, J.G., Prescott, J.F., Popoff, M.R., Eds.; Wiley-Blackwell: Ames, IA, USA, 2016; pp. 7–11. [Google Scholar]
- Finnie, J.W. Pathogenesis of brain damage produced in sheep by Clostridium perfringens type D epsilon toxin: A review. Aust. Vet. J. 2003, 81, 219–221. [Google Scholar] [CrossRef]
- Finnie, J.W. Neurological disorders produced by Clostridium perfringens type D epsilon toxin. Anaerobe 2004, 10, 145–150. [Google Scholar] [CrossRef] [PubMed]
- Minami, J.; Katayama, S.; Matsushita, O.; Matsushita, C.; Okabe, A. Lambda-toxin of Clostridium perfringens activates the precursor of epsilon-toxin by releasing its N- and C-terminal peptides. Microbiol. Immunol. 1997, 41, 527–535. [Google Scholar] [CrossRef] [PubMed]
- Bokori-Brown, M.; Savva, C.G.; Fernandes da Costa, S.P.; Naylor, C.E.; Basak, A.K.; Titball, R.W. Molecular basis of toxicity of Clostridium perfringens epsilon toxin. FEBS J. 2011, 278, 4589–4601. [Google Scholar] [CrossRef]
- Freedman, J.C.; McClane, B.A.; Uzal, F.A. New insights into Clostridium perfringens epsilon toxin activation and action on the brain during enterotoxemia. Anaerobe 2016, 41, 27–31. [Google Scholar] [CrossRef]
- Sayeed, S.; Fernandez-Miyakawa, M.E.; Fisher, D.J.; Adams, V.; Poon, R.; Rood, J.I.; Uzal, F.A.; McClane, B.A. Epsilon-toxin is required for most Clostridium perfringens type D vegetative culture supernatants to cause lethality in the mouse intravenous injection model. Infect. Immun. 2005, 73, 7413–7421. [Google Scholar] [CrossRef] [PubMed]
- Navarro, M.A.; McClane, B.A.; Uzal, F.A. Mechanisms of action and cell death associated with Clostridium perfringens toxins. Toxins 2018, 10, 212. [Google Scholar] [CrossRef] [PubMed]
- Buxton, D. Use of horseradish peroxidase to study the antagonism of Clostridium welchii (Cl. p.) type D epsilon toxin in mice by the formalinised epsilon prototoxin. J. Comp. Pathol. 1976, 86, 67–72. [Google Scholar] [CrossRef]
- Buxton, D. The use of an immunoperoxidase technique to investigate by light and electron microscopy the site of binding of Clostridium welchii type D epsilon toxin in mice. J. Med. Microbiol. 1978, 11, 289–292. [Google Scholar] [CrossRef]
- Buxton, D.; Morgan, K.T. Studies of lesions in the brains of colostrum deprived lambs by Clostridium welchii (Cl. p.) type D epsilon toxin. J. Comp. Pathol. 1976, 86, 435–447. [Google Scholar] [CrossRef]
- Tamai, E.; Ishida, T.; Miyata, S.; Matsushita, O.; Suda, H.; Kobayashi, S.; Sonobe, H.; Okabe, A. Accumulation of Clostridium perfringens epsilon-toxin in the mouse kidney and its possible biological significance. Infect. Immun. 2003, 71, 5371–5375. [Google Scholar] [CrossRef]
- Dorca-Arevalo, J.; Martin-Satue, M.; Blasi, J. Characterization of the high affinity binding of epsilon toxin from Clostridium perfringens to the renal system. Vet. Microbiol. 2012, 157, 179–189. [Google Scholar] [CrossRef]
- Soler-Jover, A.; Blasi, J.; Gomez de Aranda, I.; Navarro, P.; Gilbert, M.; Popoff, M.R.; Martin-Satue, M. Effect of epsilon toxin-GFP on MDCK cells and renal tubules in vivo. J. Histochem. Cytochem. 2004, 52, 931–942. [Google Scholar] [CrossRef]
- Dorca-Arevalo, J.; Soler-Jover, A.; Gilbert, M.; Popoff, M.R.; Martin-Satue, M.; Blasi, J. Binding of the epsilon-toxin from Clostridium perfringens in the nervous system. Vet. Microbiol. 2008, 131, 14–25. [Google Scholar] [CrossRef]
- Lonchamp, E.; Dupont, J.L.; Wioland, L.; Courjaret, R.; Mbebi-Liegeois, C.; Jover, E.; Doussau, F.; Popoff, M.R.; Bossu, J.L.; de Barry, J.; et al. Clostridium perfringens epsilon toxin targets granule cells in the mouse cerebellum and stimulates glutamate release. PLoS ONE 2010, 5, e13046. [Google Scholar] [CrossRef]
- Wioland, L.; Dupont, J.L.; Doussau, F.; Gaillard, S.; Heid, F.; Isope, P.; Pauillac, S.; Popoff, M.R.; Bossu, J.L.; Poulain, B. Epsilon toxin from Clostridium perfringens acts on oligodendrocytes without forming pores, and causes demyelination. Cell. Microbiol. 2015, 17, 369–388. [Google Scholar] [CrossRef] [PubMed]
- Linden, J.R.; Ma, Y.; Zhao, B.; Harris, J.M.; Rumah, K.R.; Schaeren-Wiemers, N.; Vartanian, T. Clostridium perfringens epsilon toxin causes selective death of mature oligodendrocytes and central nervous system demyelination. mBio 2015, 6, e02513. [Google Scholar] [CrossRef] [PubMed]
- Linden, J.R.; Flores, C.; Schmidt, E.F.; Uzal, F.A.; Michel, A.O.; Valenzuela, M.; Dobrow, S.; Vartanian, T. Clostridium perfringens epsilon toxin induces blood-brain barrier permeability via caveolae-dependent transcytosis and requires expression of MAL. PLoS Pathog. 2019, 15, e1008014. [Google Scholar] [CrossRef] [PubMed]
- Rumah, K.R.; Ma, Y.; Linden, J.R.; Oo, M.L.; Anrather, J.; Schaeren-Wiemers, N.; Alonso, M.A.; Fischetti, V.A.; McClain, M.S.; Vartanian, T. The myelin and lymphocyte protein MAL is required for binding and activity of Clostridium perfringens epsilon-toxin. PLoS Pathog. 2015, 11, e1004896. [Google Scholar] [CrossRef] [PubMed]
- Niilo, L. Clostridium perfringens in animal disease: A review of current knowledge. Can. Vet. J. 1980, 21, 141–148. [Google Scholar]
- Radostits, O.M.; Gay, C.C.; Hinchcliff, K.W.; Constable, P.D. Veterinary Medicine; Saunders Elsevier: Edinburgh, UK, 2007; pp. 841–844. [Google Scholar]
- Giannitti, F.; Garcia, J.P.; Rood, J.I.; Adams, V.; Armendano, J.I.; Beingesser, J.; Uzal, F.A. Cardiopulmonary lesions in sheep produced by experimental acute Clostridium perfringens type D enterotoxemia. Vet. Pathol. 2021, 58, 103–113. [Google Scholar] [CrossRef]
- Uzal, F.A.; Songer, J.G. Diagnosis of Clostridium perfringens intestinal infections in sheep and goats. J. Vet. Diagn. Investig. 2008, 20, 253–265. [Google Scholar] [CrossRef] [PubMed]
- Uzal, F.A.; Kelly, W.R.; Thomas, R.; Hornitsky, M.; Galea, F. Comparison of techniques for the detection of Clostridium perfringens type D epsilon toxin in intestinal contents and other body fluids in sheep and goats. J. Vet. Diagn. Investig. 2003, 15, 94–99. [Google Scholar] [CrossRef] [PubMed]
- Uzal, F.A. Diagnosis of Clostridium perfringens intestinal infections in sheep and goats. Anaerobe 2004, 10, 135–143. [Google Scholar] [CrossRef] [PubMed]
- Uzal, F.A.; Kelly, W.R.; Morris, W.E.; Bermudez, J.; Baison, M. The pathology of peracute experimental Clostridium perfringens type D enterotoxemia in sheep. J. Vet. Diagn. Investig. 2004, 16, 403–411. [Google Scholar] [CrossRef]
- Griner, L.A. Enterotoxemia of sheep. I. Effects of Clostridium perfringens type D toxin on the brains of sheep and mice. Am. J. Vet. Res. 1961, 22, 429–442. [Google Scholar] [PubMed]
- Griner, L.A.; Carlson, W.D. Enterotoxemia of sheep. II. Distribution of I131 radioiodinated serum albumin in brains of Clostridium perfringens type D intoxicated lambs. J. Am. Vet. Med. Assoc. 1961, 22, 443–446. [Google Scholar]
- Worthington, R.W.; Mulders, M.S.G. The effect of Clostridium perfringens epsilon toxin on the blood-brain barrier of mice. Onderstepoort J. Vet. Res. 1975, 42, 25–28. [Google Scholar] [PubMed]
- Morgan, K.T.; Kelly, B.G.; Buxton, D. Vascular leakage produced in the brains of mice by Clostridium welchii type D toxin. J. Comp. Pathol. 1973, 85, 461–466. [Google Scholar] [CrossRef]
- Morgan, K.T.; Kelly, B.G. Ultrastructural study of brain lesions produced in mice by the administration of Clostridium welchii type D toxin. J. Comp. Pathol. 1974, 84, 181–191. [Google Scholar] [CrossRef]
- Finnie, J.W. Histopathological changes in the brain of mice given Clostridium perfringens type D epsilon toxin. J. Comp. Pathol. 1984, 94, 363–370. [Google Scholar] [CrossRef]
- Finnie, J.W. Ultrastructural changes in the brain of mice given Clostridium perfringens type D epsilon toxin. J. Comp. Pathol. 1984, 94, 445–452. [Google Scholar] [CrossRef]
- Finnie, J.W.; Hajduk, P. An immunohistochemical study of plasma albumin extravasation in the brain of mice after the administration of Clostridium perfringens type D epsilon toxin. Aust. Vet. J. 1992, 69, 261–262. [Google Scholar] [CrossRef] [PubMed]
- Finnie, J.W.; Manavis, J.; Blumbergs, P.C. Aquaporin-4 in acute cerebral edema produced by Clostridium perfringens type D epsilon toxin. Vet. Pathol. 2008, 45, 307–309. [Google Scholar] [CrossRef] [PubMed]
- Gardner, D.E. Pathology of Clostridium welchii type D enterotoxemia. II. Structural and ultrastructural alterations in tissue of lambs and mice. J. Comp. Pathol. 1973, 83, 509–524. [Google Scholar] [CrossRef]
- Finnie, J.W.; Manavis, J.; Chidlow, G. Loss of endothelial barrier antigen (EBA) immunoreactivity as a marker of Clostridium perfringens type D epsilon toxin-induced microvascular damage in rat brain. J. Comp. Pathol. 2014, 151, 153–156. [Google Scholar] [CrossRef]
- Finnie, J.W.; Manavis, J.; Casson, R.J.; Chidlow, G. Retinal microvascular damage and vasogenic oedema produced by Clostridium perfringens type D epsilon toxin in rats. J. Vet. Diagn. Investig. 2014, 26, 470–472. [Google Scholar] [CrossRef]
- Mander, K.A.; Finnie, J.W. Loss of endothelial barrier antigen immunoreactivity in rat retinal microvessels is correlated with Clostridium perfringens type D epsilon toxin-induced damage to the blood-retinal barrier. J. Comp. Pathol. 2018, 158, 51–55. [Google Scholar] [CrossRef]
- Uzal, F.A.; Kelly, W.R. Effect of the intravenous administration of Clostridium perfringens type D epsilon toxin on young goats and lambs. J. Comp. Pathol. 1997, 116, 63–71. [Google Scholar] [CrossRef]
- Ghabriel, M.N.; Zhu, C.; Reilly, P.L.; Blumbergs, P.C.; Manavis, J.; Finnie, J.W. Toxin-induced vasogenic cerebral oedema in a rat model. Acta Neurochir. Suppl. 2000, 76, 231–236. [Google Scholar] [PubMed]
- Gould, D.H. The Development of the Early Brain Lesions Induced by the Toxin of Clostridium perfringens Type D in Mice and Lambs. Ph.D. Thesis, University of California Davis, Davis, CA, USA, 1972. [Google Scholar]
- Uzal, F.A.; Kelly, W.R. Experimental Clostridium perfringens type D enterotoxemia in goats. Vet. Pathol. 1998, 35, 132–140. [Google Scholar] [CrossRef]
- Ortega, J.; Verdes, J.M.; Morrell, E.L.; Finnie, J.W.; Manavis, J.; Uzal, F.A. Intramural vascular oedema in the brain of goats with Clostridium perfringens type D enterotoxaemia. Vet. Pathol. 2019, 56, 452–459. [Google Scholar] [CrossRef]
- Uzal, F.A.; Rolfe, B.E.; Smith, N.J.; Thomas, A.C.; Kelly, W.R. Resistance of ovine, caprine, and bovine endothelial cells to Clostridium perfringens type D epsilon toxin. Vet. Res. Commun. 1999, 23, 275–284. [Google Scholar] [CrossRef] [PubMed]
- Mander, K.A.; Uzal, F.A.; Williams, R.; Finnie, J.W. Clostridium perfringens type D epsilon toxin produces a rapid and dose-dependent cytotoxic effect on cerebral microvascular endothelial cells in vitro. J. Vet. Diagn. Investig. 2020, 32, 277–281. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Ghabriel, M.N.; Blumbergs, P.C.; Reilly, P.L.; Manavis, J.; Youssef, J.; Hatami, S.; Finnie, J.W. Clostridium perfringens prototoxin-induced alteration of endothelial barrier antigen (EBA) immunoreactivity at the blood-brain barrier (BBB). Exp. Neurol. 2001, 169, 72–82. [Google Scholar] [CrossRef]
- Wilhelm, I.; Nyul-Toth, A.; Sucui, M.; Hermenean, A.; Krizbai, I.A. Heterogeneity of the blood-brain barrier. Tissue Barriers 2016, 4, e1143544. [Google Scholar] [CrossRef] [PubMed]
- Kalaria, R.; Ferrer, I.; Love, S. Vascular Disease, Hypoxia and Related Conditions. In Greenfield’s Neuropathology; Love, S., Budka, H., Ironside, J.W., Perry, A., Eds.; CRC Press: Boca Raton, FL, USA, 2015; pp. 81, 150–152. [Google Scholar]
- Gay, C.C.; Blood, D.C.; Wilkinson, J.S. Clinical observations on sheep with focal symmetrical encephalomalacia. Aust. Vet. J. 1975, 51, 266–269. [Google Scholar] [CrossRef] [PubMed]
- Blackwell, T.E.; Butler, D.G. Clinical signs, treatment and post-mortem lesions in dairy goats with enterotoxemia in 13 cases (1979–1982). J. Am. Vet. Med. Assoc. 1992, 200, 214–217. [Google Scholar] [PubMed]
- Blackwell, T.E.; Butler, D.G.; Prescott, J.F.; Wilcock, B.P. Differences in signs and lesions in sheep and goats with enterotoxemia induced by infusion of Clostridium perfringens type D. Am. J. Vet. Res. 1991, 52, 1147–1152. [Google Scholar] [PubMed]
- Uzal, F.A.; Kelly, W.R. Enterotoxemia in goats. Vet. Res. Commun. 1996, 20, 481–492. [Google Scholar] [CrossRef]
- Uzal, F.A.; Glastonbury, J.R.; Kelly, W.R.; Thomas, R. Caprine enterotoxemia associated with cerebral microangiopathy. Vet. Rec. 1997, 141, 224–226. [Google Scholar] [CrossRef]
- Pienaar, J.G.; Thornton, D.J. Focal symmetrical encephalomalacia in sheep in South Africa. J. S. Afr. Vet. Med. Assoc. 1964, 35, 351–357. [Google Scholar]
- Buxton, D.; MacLeod, N.S.M.; Nicolson, T.B. Focal symmetrical encephalomalacia in young cattle. Vet. Rec. 1981, 108, 459. [Google Scholar] [CrossRef]
- Manavis, J.; Blumbergs, P.; Jerrett, I.; Hanshaw, D.; Uzal, F.; Finnie, J. Heterogeneous immunoreactivity of axonal spheroids in focal symmetrical encephalomalacia produced by Clostridium perfringens type D epsilon toxin in sheep. Vet. Pathol. 2022, 59, 328–332. [Google Scholar] [CrossRef]
- Vandevelde, M.; Higgins, R.J.; Oevermann, A. Veterinary Neuropathology. Essentials of Theory and Practice; Wiley-Blackwell: Ames, IA, USA, 2012; pp. 108, 187–189. [Google Scholar]
- Finnie, J.W.; Blumbergs, P.C.; Manavis, J. Neuronal damage produced in rat brains by Clostridium perfringens type D epsilon toxin. J. Comp. Pathol. 1999, 120, 415–420. [Google Scholar] [CrossRef]
- Morris, W.E.; Goldstein, J.; Redondo, L.M.; Cangelosi, A.; Geoghegan, P.; Brocco, M.; Loidl, F.C.; Fernandez-Miyakawa, M.E. Clostridium perfringens epsilon toxin induces permanent neuronal degeneration and behavioural changes. Toxicon 2017, 130, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Nagahama, M.; Sakurai, J. Effect of drugs acting on the central nervous system on the lethality in mice of Clostridium perfringens epsilon toxin. Toxicon 1993, 31, 427–435. [Google Scholar] [CrossRef]
- Miyamoto, O.; Minami, J.; Toyoshima, T.; Nakamura, T.; Masada, T.; Nagao, S.; Negi, T.; Itano, T.; Okabe, A. Neurotoxicity of Clostridium perfringens epsilon-toxin for the rat hippocampus via the glutamatergic system. Infect. Immune. 1998, 66, 2501–2508. [Google Scholar] [CrossRef]
- Miyamoto, O.; Sumitani, K.; Nakamura, T.; Yamagami, S.; Miyata, S.; Itano, T.; Negi, T.; Okabe, A. Clostridium perfringens epsilon toxin causes excessive release of glutamate in the mouse hippocampus. FEMS. Microbiol. Lett. 2000, 189, 109–113. [Google Scholar] [CrossRef]
- Auer, R.N.; Dunn, J.F.; Sutherland, G.R. Hypoxia and Related Conditions. In Greenfield’s Neuropathology; Love, S., Louis, D.N., Ellison, D.W., Eds.; Edward Arnold: London, UK, 2008; pp. 21–23, 103–105. [Google Scholar]
- Miller, J.D.; Ironside, J.W. Raised Intracranial Pressure, Oedema and Hydrocephalus. In Greenfield’s Neuropathology; Graham, D.I., Lantos, P.L., Eds.; Arnold: London, UK, 1997; pp. 166–173. [Google Scholar]
- Stiles, B.G.; Barth, G.; Barth, H.; Popoff, M.R. Clostridium perfringens epsilon toxin: A malevolent molecule for animals and man? Toxins 2013, 5, 2138–2160. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type | Typing Toxins | |||||
|---|---|---|---|---|---|---|
| Alpha (CPA) | Beta (CPB) | Epsilon (ETX) | Iota (ITX) | Enterotoxin (CPE) | Necrotic Enteritis B-like Net-B | |
| A | + | − | − | − | − | − |
| B | + | + | + | − | −/+ | − |
| C | + | + | − | − | −/+ | − |
| D | + | − | + | − | −/+ | − |
| E | + | − | − | + | −/+ | − |
| F | + | − | − | − | + | − |
| G | + | − | − | − | −/+ | + |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Finnie, J.W.; Uzal, F.A. Pathology and Pathogenesis of Brain Lesions Produced by Clostridium perfringens Type D Epsilon Toxin. Int. J. Mol. Sci. 2022, 23, 9050. https://doi.org/10.3390/ijms23169050
Finnie JW, Uzal FA. Pathology and Pathogenesis of Brain Lesions Produced by Clostridium perfringens Type D Epsilon Toxin. International Journal of Molecular Sciences. 2022; 23(16):9050. https://doi.org/10.3390/ijms23169050
Chicago/Turabian StyleFinnie, John W., and Francisco A. Uzal. 2022. "Pathology and Pathogenesis of Brain Lesions Produced by Clostridium perfringens Type D Epsilon Toxin" International Journal of Molecular Sciences 23, no. 16: 9050. https://doi.org/10.3390/ijms23169050
APA StyleFinnie, J. W., & Uzal, F. A. (2022). Pathology and Pathogenesis of Brain Lesions Produced by Clostridium perfringens Type D Epsilon Toxin. International Journal of Molecular Sciences, 23(16), 9050. https://doi.org/10.3390/ijms23169050