
Cilostazol Attenuates AngII-Induced Cardiac Fibrosis in apoE Deficient Mice



Abstract
:1. Introduction
2. Results
2.1. Cilostazol Had No Effect on Body Weight, Pulse Rate, Systolic Blood Pressure, and Lipid Profile
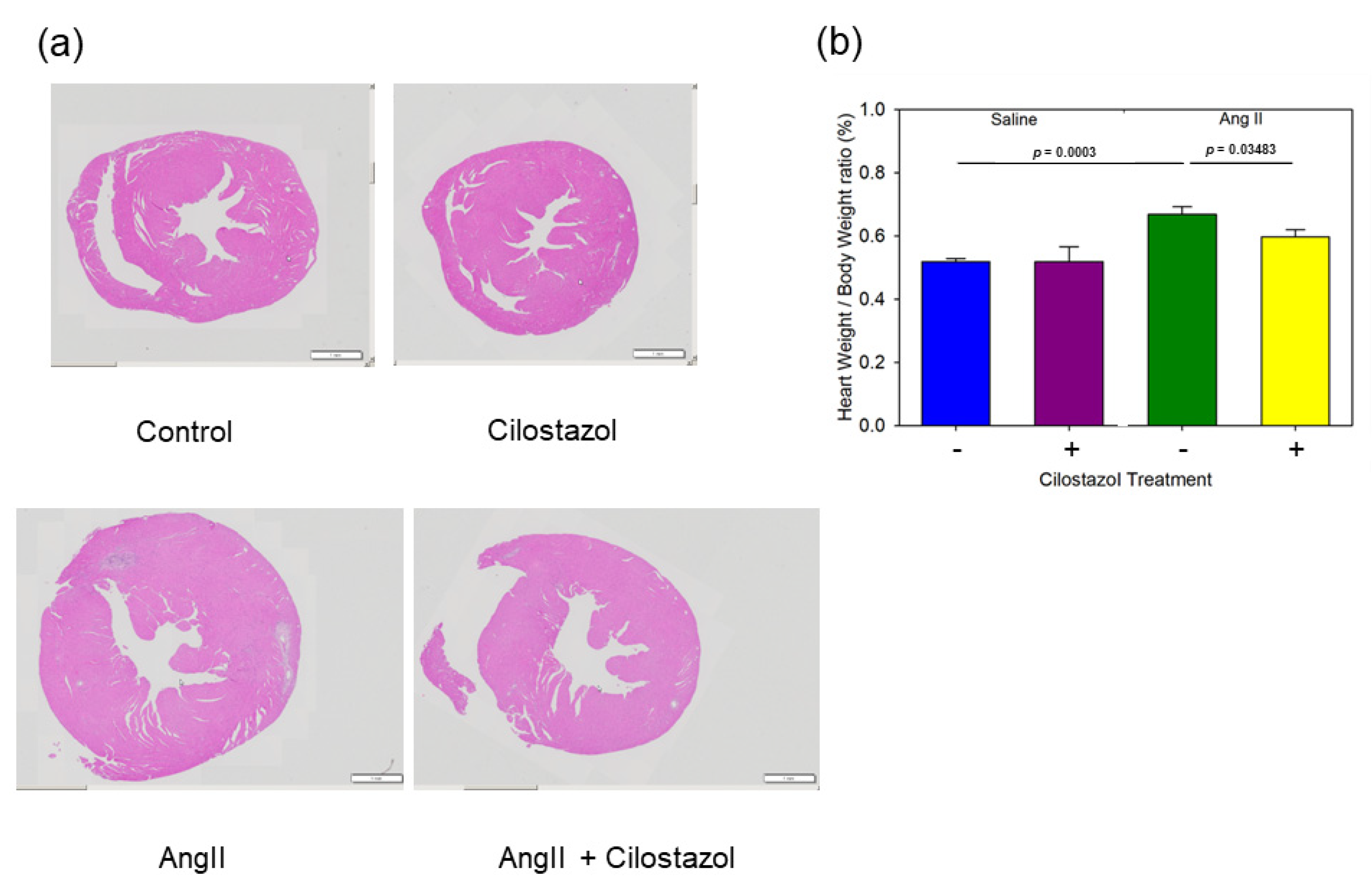
2.2. Cilostazol Attenuated AngII-Induced Cardiac Hypertrophy
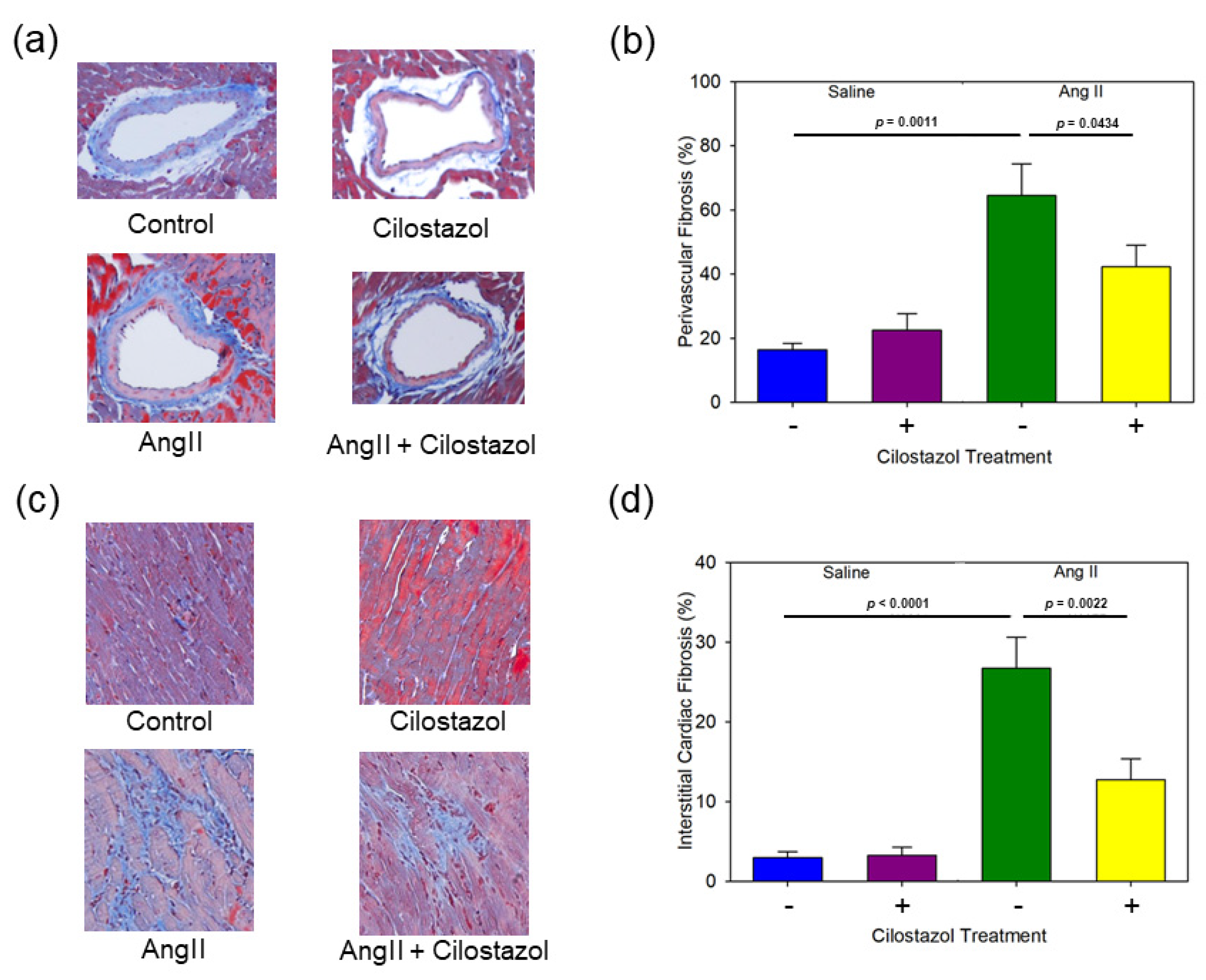
2.3. Cilostazol Suppressed AngII-Induced Cardiac Fibrosis
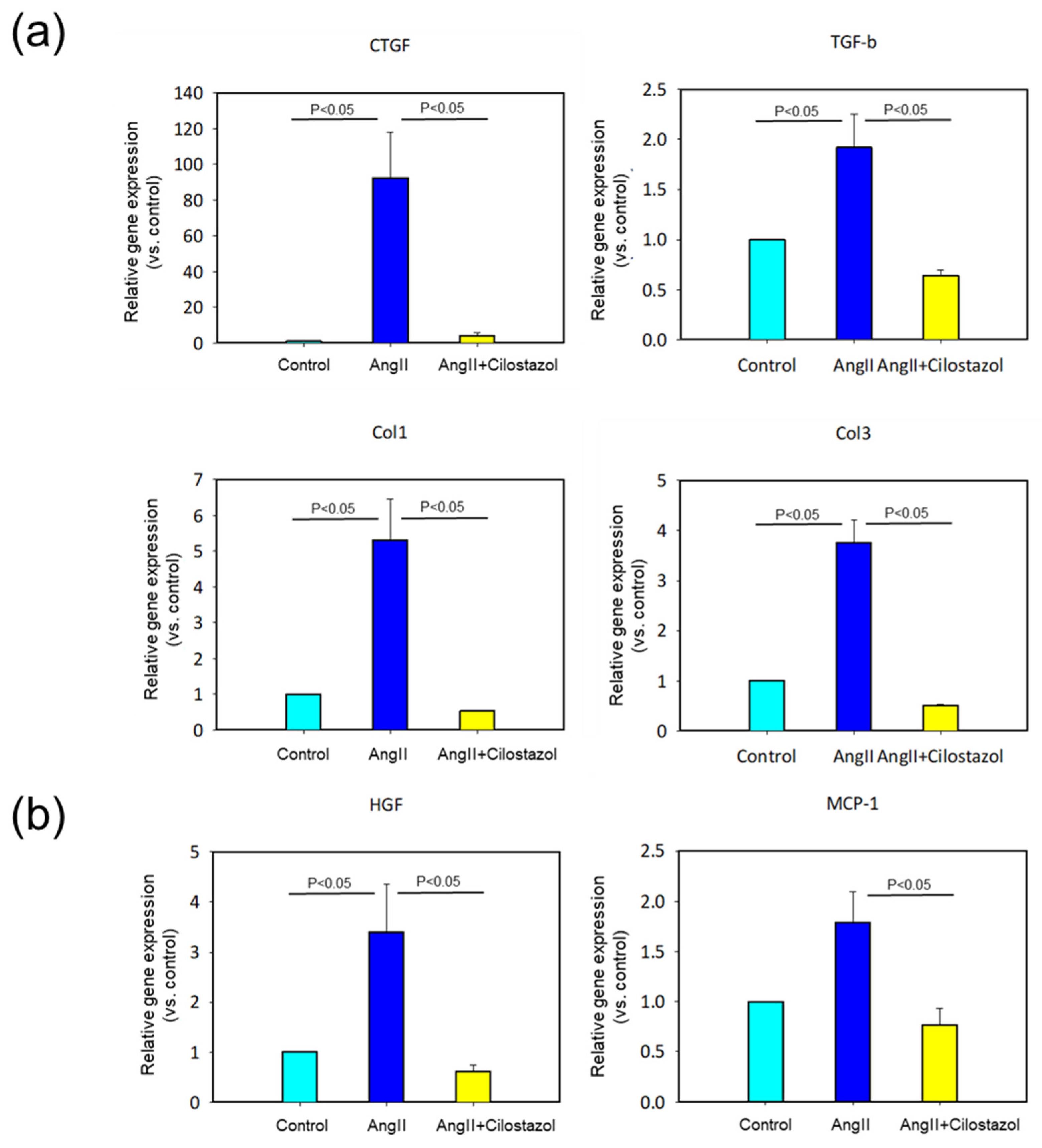
2.4. The Effect of Cilostazol on mRNA Expression in Heart Tissue
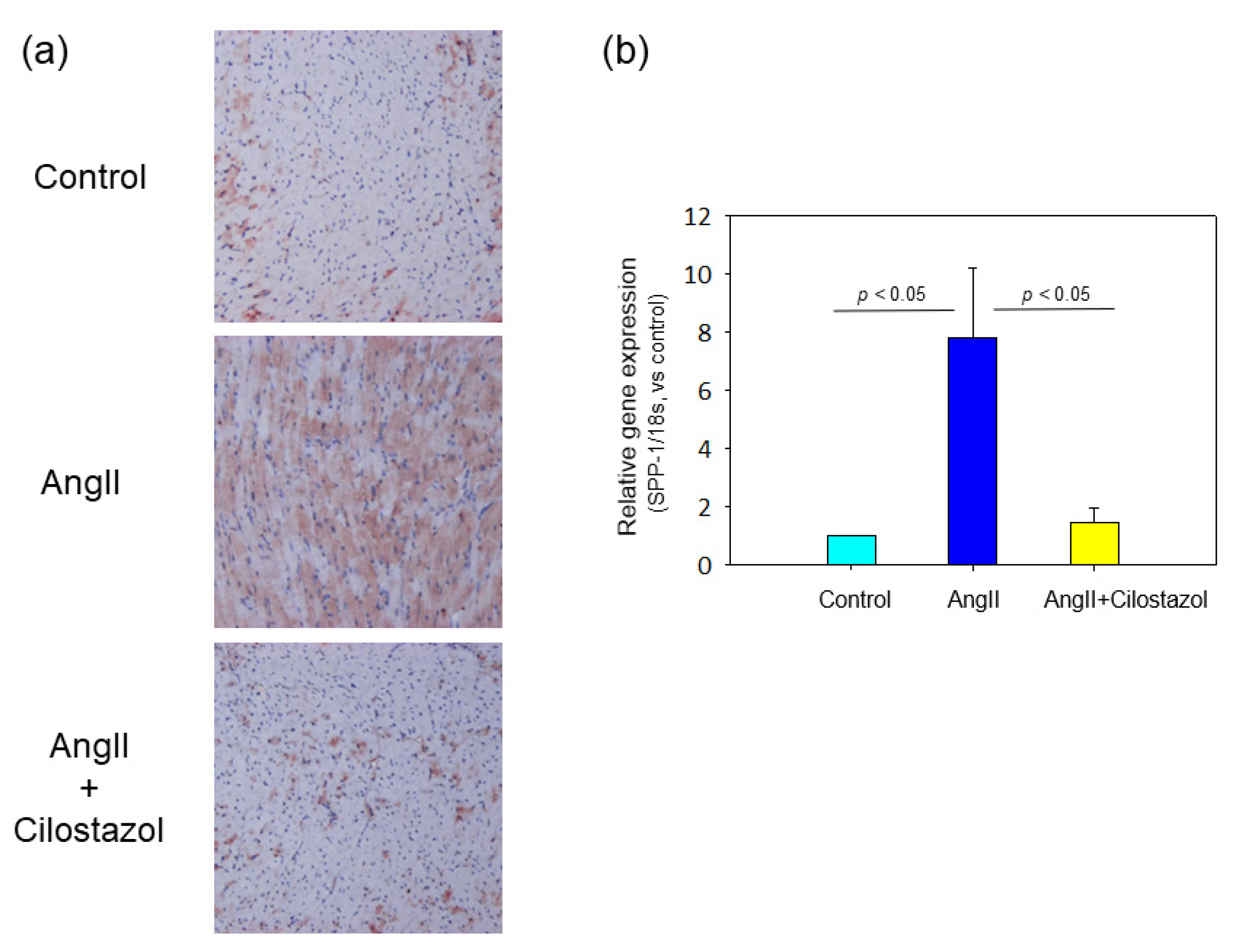
2.5. The Effect of Cilostazol on Osteopontin Expression in Heart Tissue
2.6. The Effect of Cilostazol on AngII-Induced OPN mRNA Expression in Human Cardiac Myocyte In Vitro Study
3. Discussion
4. Materials and Methods
4.1. Mice and Study Protocol
4.2. Blood Pressure Measurement
4.3. Lipid Measurements
4.4. Histological Analysis
4.5. Real-Time Polymerase Chain Reaction
4.6. Cell Culture and Treatment
4.7. Statistics
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Go, A.S.; Mozaffarian, D.; Roger, V.L.; Benjamin, E.J.; Berry, J.D.; Blaha, M.J.; Dai, S.; Ford, E.S.; Fox, C.S.; Franco, S.; et al. Heart disease and stroke statistics--2014 update: A report from the American Heart Association. Circulation 2014, 129, e28–e292. [Google Scholar] [CrossRef]
- Global, regional, and national age-sex specific all-cause and cause-specific mortality for 240 causes of death, 1990-2013: A systematic analysis for the Global Burden of Disease Study 2013. Lancet 2015, 385, 117–171. [CrossRef]
- Ponikowski, P.; Anker, S.D.; AlHabib, K.F.; Cowie, M.R.; Force, T.L.; Hu, S.; Jaarsma, T.; Krum, H.; Rastogi, V.; Rohde, L.E.; et al. Heart failure: Preventing disease and death worldwide. ESC Heart Fail. 2014, 1, 4–25. [Google Scholar] [CrossRef] [PubMed]
- Heymans, S.; González, A.; Pizard, A.; Papageorgiou, A.P.; López-Andrés, N.; Jaisser, F.; Thum, T.; Zannad, F.; Díez, J. Searching for new mechanisms of myocardial fibrosis with diagnostic and/or therapeutic potential. Eur. J. Heart Fail. 2015, 17, 764–771. [Google Scholar] [CrossRef] [PubMed]
- Kong, P.; Christia, P.; Frangogiannis, N.G. The pathogenesis of cardiac fibrosis. Cell. Mol. Life Sci. 2014, 71, 549–574. [Google Scholar] [CrossRef]
- Creemers, E.E.; Pinto, Y.M. Molecular mechanisms that control interstitial fibrosis in the pressure-overloaded heart. Cardiovasc. Res. 2011, 89, 265–272. [Google Scholar] [CrossRef] [PubMed]
- Wynn, T.A.; Ramalingam, T.R. Mechanisms of fibrosis: Therapeutic translation for fibrotic disease. Nat. Med. 2012, 18, 1028–1040. [Google Scholar] [CrossRef]
- Piek, A.; de Boer, R.A.; Silljé, H.H. The fibrosis-cell death axis in heart failure. Heart Fail. Rev. 2016, 21, 199–211. [Google Scholar] [CrossRef]
- Rienks, M.; Papageorgiou, A.P.; Frangogiannis, N.G.; Heymans, S. Myocardial extracellular matrix: An ever-changing and diverse entity. Circ. Res. 2014, 114, 872–888. [Google Scholar] [CrossRef] [PubMed]
- Bacmeister, L.; Schwarzl, M.; Warnke, S.; Stoffers, B.; Blankenberg, S.; Westermann, D.; Lindner, D. Inflammation and fibrosis in murine models of heart failure. Basic Res. Cardiol. 2019, 114, 19. [Google Scholar] [CrossRef]
- Schnee, J.M.; Hsueh, W.A. Angiotensin II, adhesion, and cardiac fibrosis. Cardiovasc. Res. 2000, 46, 264–268. [Google Scholar] [CrossRef]
- Yasuno, S.; Kuwahara, K.; Kinoshita, H.; Yamada, C.; Nakagawa, Y.; Usami, S.; Kuwabara, Y.; Ueshima, K.; Harada, M.; Nishikimi, T.; et al. Angiotensin II type 1a receptor signalling directly contributes to the increased arrhythmogenicity in cardiac hypertrophy. Br. J. Pharmacol. 2013, 170, 1384–1395. [Google Scholar] [CrossRef] [PubMed]
- Dawson, D.L.; Cutler, B.S.; Meissner, M.H.; Strandness, D.E., Jr. Cilostazol has beneficial effects in treatment of intermittent claudication: Results from a multicenter, randomized, prospective, double-blind trial. Circulation 1998, 98, 678–686. [Google Scholar] [CrossRef] [PubMed]
- Kambayashi, J.; Liu, Y.; Sun, B.; Shakur, Y.; Yoshitake, M.; Czerwiec, F. Cilostazol as a unique antithrombotic agent. Curr. Pharm. Des. 2003, 9, 2289–2302. [Google Scholar] [CrossRef] [PubMed]
- Da Motta, N.A.; de Brito, F.C. Cilostazol exerts antiplatelet and anti-inflammatory effects through AMPK activation and NF-kB inhibition on hypercholesterolemic rats. Fundam. Clin. Pharmacol. 2016, 30, 327–337. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, T.; Ohashi, W.; Tomita, K.; Hattori, K.; Matsuda, N.; Hattori, Y. Anti-inflammatory properties of cilostazol: Its interruption of DNA binding activity of NF-κB from the Toll-like receptor signaling pathways. Int. Immunopharmacol. 2018, 62, 120–131. [Google Scholar] [CrossRef] [PubMed]
- Kurtoglu, T.; Basoglu, H.; Ozkisacik, E.A.; Cetin, N.K.; Tataroglu, C.; Yenisey, C.; Discigil, B. Effects of cilostazol on oxidative stress, systemic cytokine release, and spinal cord injury in a rat model of transient aortic occlusion. Ann. Vasc. Surg. 2014, 28, 479–488. [Google Scholar] [CrossRef]
- Lee, Y.J.; Eun, J.R. Cilostazol Decreases Ethanol-Mediated TNFalpha Expression in RAW264.7 Murine Macrophage and in Liver from Binge Drinking Mice. Korean J. Physiol. Pharmacol. 2012, 16, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.M.; Shin, H.K.; Kim, K.Y.; Lee, J.H.; Hong, K.W. Neuroprotective effect of cilostazol against focal cerebral ischemia via antiapoptotic action in rats. J. Pharmacol. Exp. Ther. 2002, 300, 787–793. [Google Scholar] [CrossRef]
- Umebayashi, R.; Uchida, H.A.; Kakio, Y.; Subramanian, V.; Daugherty, A.; Wada, J. Cilostazol Attenuates Angiotensin II-Induced Abdominal Aortic Aneurysms but Not Atherosclerosis in Apolipoprotein E-Deficient Mice. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 903–912. [Google Scholar] [CrossRef]
- El Awdan, S.A.; Abdel Rahman, R.F.; Ibrahim, H.M.; Hegazy, R.R.; El Marasy, S.A.; Badawi, M.; Arbid, M.S. Regression of fibrosis by cilostazol in a rat model of thioacetamide-induced liver fibrosis: Up regulation of hepatic cAMP, and modulation of inflammatory, oxidative stress and apoptotic biomarkers. PLoS ONE 2019, 14, e0216301. [Google Scholar] [CrossRef] [PubMed]
- Han, K.; Zhang, Y.; Yang, Z. Cilostazol protects rats against alcohol-induced hepatic fibrosis via suppression of TGF-β1/CTGF activation and the cAMP/Epac1 pathway. Exp. Ther. Med. 2019, 17, 2381–2388. [Google Scholar] [CrossRef] [PubMed]
- Saito, S.; Hata, K.; Iwaisako, K.; Yanagida, A.; Takeiri, M.; Tanaka, H.; Kageyama, S.; Hirao, H.; Ikeda, K.; Asagiri, M.; et al. Cilostazol attenuates hepatic stellate cell activation and protects mice against carbon tetrachloride-induced liver fibrosis. Hepatol. Res. 2014, 44, 460–473. [Google Scholar] [CrossRef]
- Collins, A.R.; Schnee, J.; Wang, W.; Kim, S.; Fishbein, M.C.; Bruemmer, D.; Law, R.E.; Nicholas, S.; Ross, R.S.; Hsueh, W.A. Osteopontin modulates angiotensin II-induced fibrosis in the intact murine heart. J. Am. Coll. Cardiol. 2004, 43, 1698–1705. [Google Scholar] [CrossRef] [PubMed]
- Matsui, Y.; Jia, N.; Okamoto, H.; Kon, S.; Onozuka, H.; Akino, M.; Liu, L.; Morimoto, J.; Rittling, S.R.; Denhardt, D.; et al. Role of osteopontin in cardiac fibrosis and remodeling in angiotensin II-induced cardiac hypertrophy. Hypertension 2004, 43, 1195–1201. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Uchida, K.; Nakanishi, N.; Hattori, Y. Cilostazol activates AMP-activated protein kinase and restores endothelial function in diabetes. Am. J. Hypertens. 2008, 21, 451–457. [Google Scholar] [CrossRef] [PubMed]
- Weber, K.T.; Sun, Y.; Bhattacharya, S.K.; Ahokas, R.A.; Gerling, I.C. Myofibroblast-mediated mechanisms of pathological remodelling of the heart. Nat. Rev. Cardiol. 2013, 10, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Schelbert, E.B.; Fonarow, G.C.; Bonow, R.O.; Butler, J.; Gheorghiade, M. Therapeutic targets in heart failure: Refocusing on the myocardial interstitium. J. Am. Coll. Cardiol. 2014, 63, 2188–2198. [Google Scholar] [CrossRef]
- Butler, J.; Fonarow, G.C.; Zile, M.R.; Lam, C.S.; Roessig, L.; Schelbert, E.B.; Shah, S.J.; Ahmed, A.; Bonow, R.O.; Cleland, J.G.; et al. Developing therapies for heart failure with preserved ejection fraction: Current state and future directions. JACC Heart Fail. 2014, 2, 97–112. [Google Scholar] [CrossRef]
- Davis, J.; Molkentin, J.D. Myofibroblasts: Trust your heart and let fate decide. J. Mol. Cell. Cardiol. 2014, 70, 9–18. [Google Scholar] [CrossRef]
- Sopel, M.J.; Rosin, N.L.; Lee, T.D.; Légaré, J.F. Myocardial fibrosis in response to Angiotensin II is preceded by the recruitment of mesenchymal progenitor cells. Lab. Investig. 2011, 91, 565–578. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, A.; Tanaka, M.; Takeda, S.; Ito, H.; Nagano, K. Cilostazol Induces PGI2 Production via Activation of the Downstream Epac-1/Rap1 Signaling Cascade to Increase Intracellular Calcium by PLCε and to Activate p44/42 MAPK in Human Aortic Endothelial Cells. PLoS ONE 2015, 10, e0132835. [Google Scholar] [CrossRef]
- Lee, D.H.; Lee, H.R.; Shin, H.K.; Park, S.Y.; Hong, K.W.; Kim, E.K.; Bae, S.S.; Lee, W.S.; Rhim, B.Y.; Kim, C.D. Cilostazol enhances integrin-dependent homing of progenitor cells by activation of cAMP-dependent protein kinase in synergy with Epac1. J. Neurosci. Res. 2011, 89, 650–660. [Google Scholar] [CrossRef]
- Ke, K.; Safder, A.M.; Sul, O.J.; Suh, J.H.; Joe, Y.; Chung, H.T.; Choi, H.S. Cilostazol attenuates ovariectomy-induced bone loss by inhibiting osteoclastogenesis. PLoS ONE 2015, 10, e0124869. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.D.; Huang, H.F.; Yang, Q.; Chen, X.Q. Liraglutide improves myocardial fibrosis after myocardial infarction through inhibition of CTGF by activating cAMP in mice. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 4648–4656. [Google Scholar] [CrossRef] [PubMed]
- Weng, L.; Wang, W.; Su, X.; Huang, Y.; Su, L.; Liu, M.; Sun, Y.; Yang, B.; Zhou, H. The Effect of cAMP-PKA Activation on TGF-β1-Induced Profibrotic Signaling. Cell. Physiol. Biochem. 2015, 36, 1911–1927. [Google Scholar] [CrossRef] [PubMed]
- Satish, L.; Gallo, P.H.; Baratz, M.E.; Johnson, S.; Kathju, S. Reversal of TGF-β1 stimulation of α-smooth muscle actin and extracellular matrix components by cyclic AMP in Dupuytren’s-derived fibroblasts. BMC Musculoskelet. Disord. 2011, 12, 113. [Google Scholar] [CrossRef]
- Wolak, T.; Kim, H.; Ren, Y.; Kim, J.; Vaziri, N.D.; Nicholas, S.B. Osteopontin modulates angiotensin II-induced inflammation, oxidative stress, and fibrosis of the kidney. Kidney Int. 2009, 76, 32–43. [Google Scholar] [CrossRef]
- Rosenberg, M.; Zugck, C.; Nelles, M.; Juenger, C.; Frank, D.; Remppis, A.; Giannitsis, E.; Katus, H.A.; Frey, N. Osteopontin, a new prognostic biomarker in patients with chronic heart failure. Circ. Heart Fail. 2008, 1, 43–49. [Google Scholar] [CrossRef]
- Park, J.H.; Choi, B.H.; Ku, S.K.; Kim, D.H.; Jung, K.A.; Oh, E.; Kwak, M.K. Amelioration of high fat diet-induced nephropathy by cilostazol and rosuvastatin. Arch. Pharm. Res. 2017, 40, 391–402. [Google Scholar] [CrossRef]
- Pollard, C.M.; Desimine, V.L.; Wertz, S.L.; Perez, A.; Parker, B.M.; Maning, J.; McCrink, K.A.; Shehadeh, L.A.; Lymperopoulos, A. Deletion of Osteopontin Enhances β₂-Adrenergic Receptor-Dependent Anti-Fibrotic Signaling in Cardiomyocytes. Int. J. Mol. Sci. 2019, 20, 1396. [Google Scholar] [CrossRef] [PubMed]
- Brower, G.L.; Gardner, J.D.; Forman, M.F.; Murray, D.B.; Voloshenyuk, T.; Levick, S.P.; Janicki, J.S. The relationship between myocardial extracellular matrix remodeling and ventricular function. Eur. J. Cardiothorac. Surg. 2006, 30, 604–610. [Google Scholar] [CrossRef] [PubMed]
- Herum, K.M.; Romaine, A.; Wang, A.; Melleby, A.O.; Strand, M.E.; Pacheco, J.; Braathen, B.; Dunér, P.; Tønnessen, T.; Lunde, I.G.; et al. Syndecan-4 Protects the Heart From the Profibrotic Effects of Thrombin-Cleaved Osteopontin. J. Am. Heart Assoc. 2020, 9, e013518. [Google Scholar] [CrossRef] [PubMed]
- Lenga, Y.; Koh, A.; Perera, A.S.; McCulloch, C.A.; Sodek, J.; Zohar, R. Osteopontin expression is required for myofibroblast differentiation. Circ. Res. 2008, 102, 319–327. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, H.; Imanaka-Yoshida, K. Matricellular proteins: New molecular targets to prevent heart failure. Cardiovasc. Ther. 2012, 30, e198–e209. [Google Scholar] [CrossRef]
- López, B.; González, A.; Lindner, D.; Westermann, D.; Ravassa, S.; Beaumont, J.; Gallego, I.; Zudaire, A.; Brugnolaro, C.; Querejeta, R.; et al. Osteopontin-mediated myocardial fibrosis in heart failure: A role for lysyl oxidase? Cardiovasc. Res. 2013, 99, 111–120. [Google Scholar] [CrossRef]
- Rosenberg, M.; Meyer, F.J.; Gruenig, E.; Lutz, M.; Lossnitzer, D.; Wipplinger, R.; Katus, H.A.; Frey, N. Osteopontin predicts adverse right ventricular remodelling and dysfunction in pulmonary hypertension. Eur. J. Clin. Investig. 2012, 42, 933–942. [Google Scholar] [CrossRef]
- Das, S.; Aiba, T.; Rosenberg, M.; Hessler, K.; Xiao, C.; Quintero, P.A.; Ottaviano, F.G.; Knight, A.C.; Graham, E.L.; Boström, P.; et al. Pathological role of serum- and glucocorticoid-regulated kinase 1 in adverse ventricular remodeling. Circulation 2012, 126, 2208–2219. [Google Scholar] [CrossRef]
- Sawaki, D.; Czibik, G.; Pini, M.; Ternacle, J.; Suffee, N.; Mercedes, R.; Marcelin, G.; Surenaud, M.; Marcos, E.; Gual, P.; et al. Visceral Adipose Tissue Drives Cardiac Aging Through Modulation of Fibroblast Senescence by Osteopontin Production. Circulation 2018, 138, 809–822. [Google Scholar] [CrossRef]
- Okuyama, M.; Uchida, H.A.; Hada, Y.; Kakio, Y.; Otaka, N.; Umebayashi, R.; Tanabe, K.; Fujii, Y.; Kasahara, S.; Subramanian, V.; et al. Exogenous Vasohibin-2 Exacerbates Angiotensin II-Induced Ascending Aortic Dilation in Mice. Circ. Rep. 2019, 1, 155–161. [Google Scholar] [CrossRef]
- Hada, Y.; Uchida, H.A.; Mukai, T.; Kojima, F.; Yoshida, M.; Takeuchi, H.; Kakio, Y.; Otaka, N.; Morita, Y.; Wada, J. Inhibition of interleukin-6 signaling attenuates aortitis, left ventricular hypertrophy and arthritis in interleukin-1 receptor antagonist deficient mice. Clin. Sci. 2020, 134, 2771–2787. [Google Scholar] [CrossRef] [PubMed]
- Hada, Y.; Uchida, H.A.; Otaka, N.; Onishi, Y.; Okamoto, S.; Nishiwaki, M.; Takemoto, R.; Takeuchi, H.; Wada, J. The Protective Effect of Chlorogenic Acid on Vascular Senescence via the Nrf2/HO-1 Pathway. Int. J. Mol. Sci. 2020, 21, 4527. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Control | Cilostazol | AngII | AngII + Cilostazol | |
|---|---|---|---|---|
| n | 6 | 5 | 11 | 9 |
| BW (g) | 26.9 ± 1.0 | 26.1 ± 1.0 | 26.2 ± 0.6 | 26.8 ± 0.7 |
| SBP (mmHg) | 97 ± 3 | 97 ± 3 | 127 ± 8 * | 132 ± 7 ** |
| HR (bpm) | 621 ± 34 | 675 ± 23 | 645 ± 17 | 611 ± 15 |
| T-Cho (mg/dL) | 591 ± 48 | 584 ± 52 | 520 ± 109 | 524 ± 124 |
| TG (mg/dL) | 475 ± 190 | 391 ± 94 | 338 ± 78 | 379 ± 103 |
| HDL-C (mg/dL) | 22.0 ± 9.6 | 17.1 ± 6.5 | 17.4 ± 7.3 | 17.3 ± 4.9 |
| LDL-C (mg/dL) | 467 ± 31 | 484 ± 81 | 433 ± 97 | 426 ± 106 |
| LDL/HDL | 26.6 ± 17.3 | 32.7 ± 15.4 | 34.4 ± 28.0 | 25.6 ± 6.5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hada, Y.; Uchida, H.A.; Umebayashi, R.; Yoshida, M.; Wada, J. Cilostazol Attenuates AngII-Induced Cardiac Fibrosis in apoE Deficient Mice. Int. J. Mol. Sci. 2022, 23, 9065. https://doi.org/10.3390/ijms23169065
Hada Y, Uchida HA, Umebayashi R, Yoshida M, Wada J. Cilostazol Attenuates AngII-Induced Cardiac Fibrosis in apoE Deficient Mice. International Journal of Molecular Sciences. 2022; 23(16):9065. https://doi.org/10.3390/ijms23169065
Chicago/Turabian StyleHada, Yoshiko, Haruhito A. Uchida, Ryoko Umebayashi, Masashi Yoshida, and Jun Wada. 2022. "Cilostazol Attenuates AngII-Induced Cardiac Fibrosis in apoE Deficient Mice" International Journal of Molecular Sciences 23, no. 16: 9065. https://doi.org/10.3390/ijms23169065
APA StyleHada, Y., Uchida, H. A., Umebayashi, R., Yoshida, M., & Wada, J. (2022). Cilostazol Attenuates AngII-Induced Cardiac Fibrosis in apoE Deficient Mice. International Journal of Molecular Sciences, 23(16), 9065. https://doi.org/10.3390/ijms23169065