
High IGKC-Expressing Intratumoral Plasma Cells Predict Response to Immune Checkpoint Blockade

 , , , , , , , , , ,
, , , , , , , , , ,
Abstract
:1. Introduction
2. Results
2.1. Clinicopathological Variables Associated with Response and Prognosis
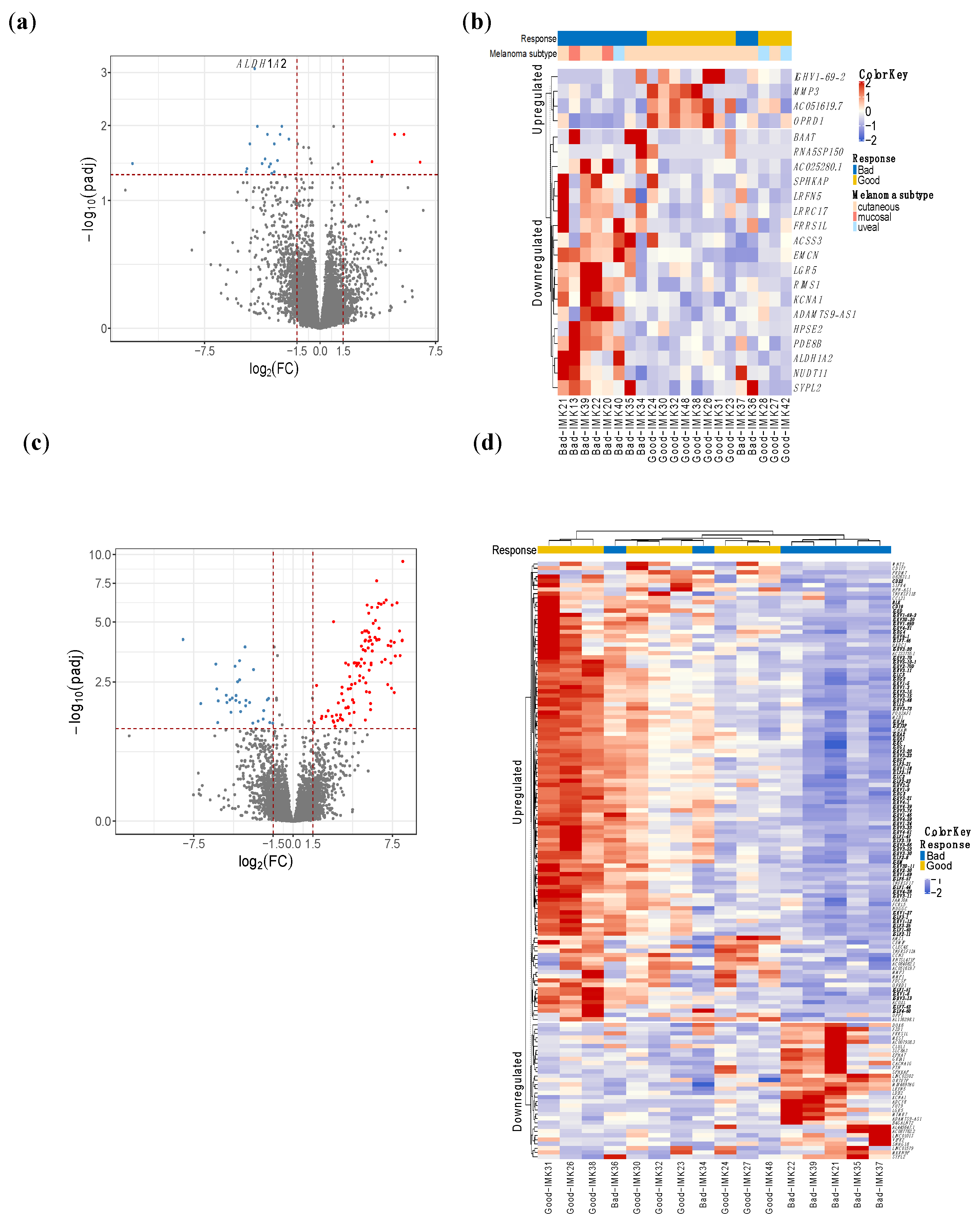
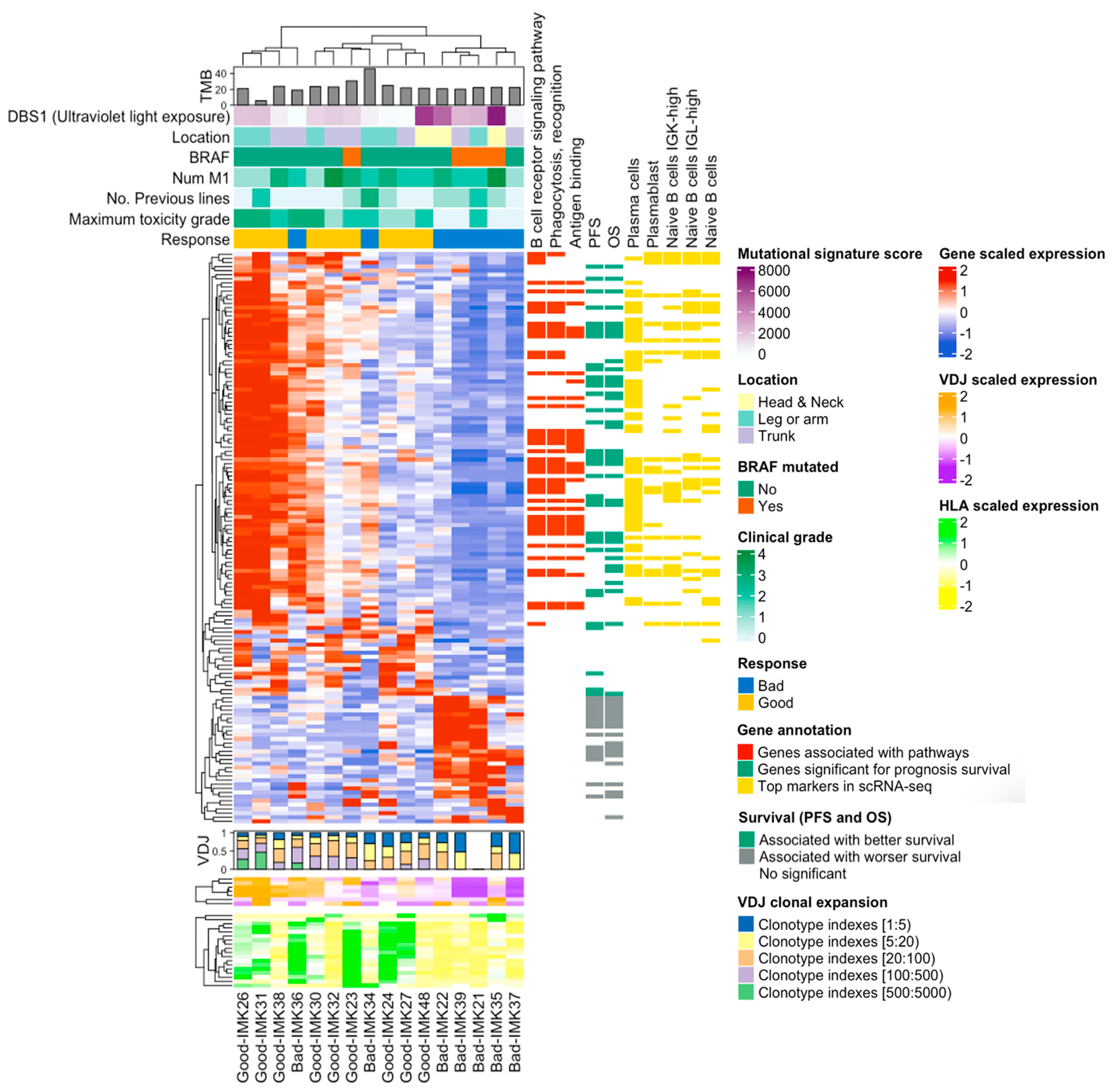
2.2. B-Cell Transcriptomic Signature of the Response to PD1 Blockade
2.3. TNFRSF11B, IGLV6-57, IGHA1 and GRIA1 Are Technically Validated as Promising Markers of Response to Nivolumab
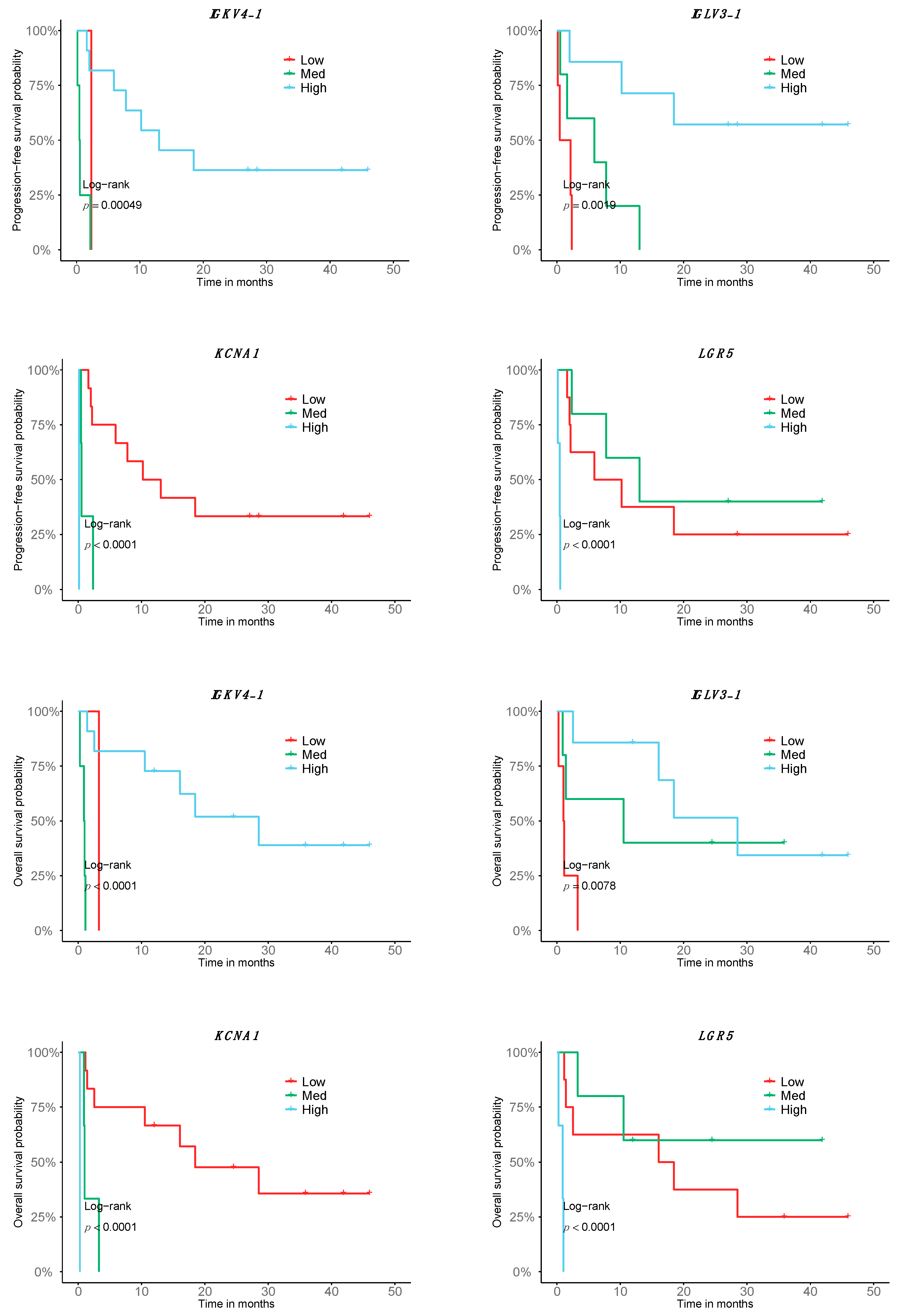
2.4. A Fraction of the Transcriptomic Signature of Response Is Prognostic
2.5. TMB Is Not Associated with Response to Nivolumab in Cutaneous Metastatic Melanoma Patients
2.6. Decoding the Response-Relevant Mutational Signatures in Patients with Cutaneous Metastatic Melanoma
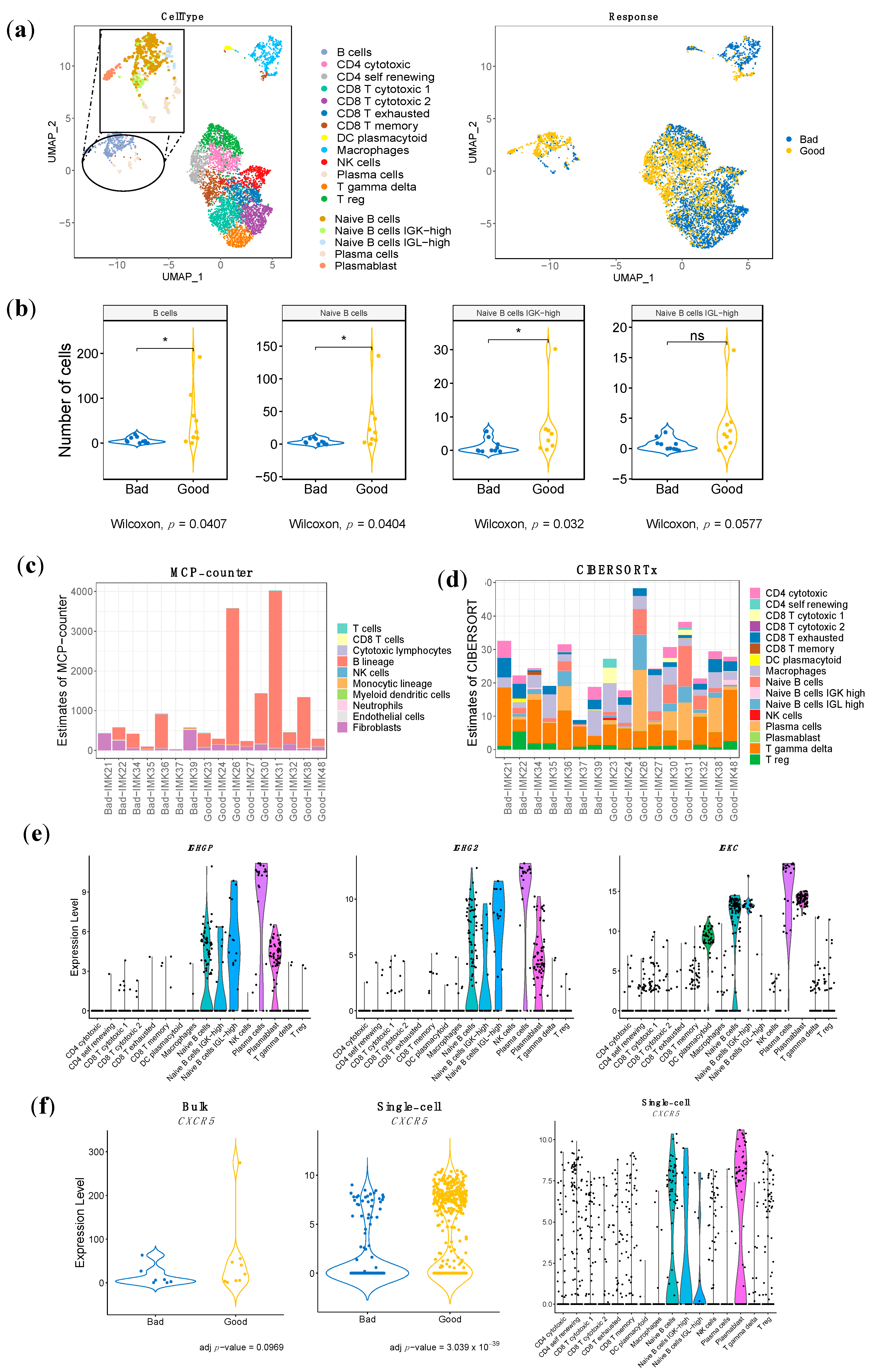
2.7. Identification of the Specific Response-Associated Stromal Cell Subtypes in Single Cell RNA-seq Data
2.8. Specific Stromal Cell Population Composition in the TME of Our Cohort of Metastatic Melanma Patents in Treament with Nivolumab
2.9. Validation of the Bulk RNA-seq Transcriptomic Gene Signature Using Single-Cell RNA-seq Data
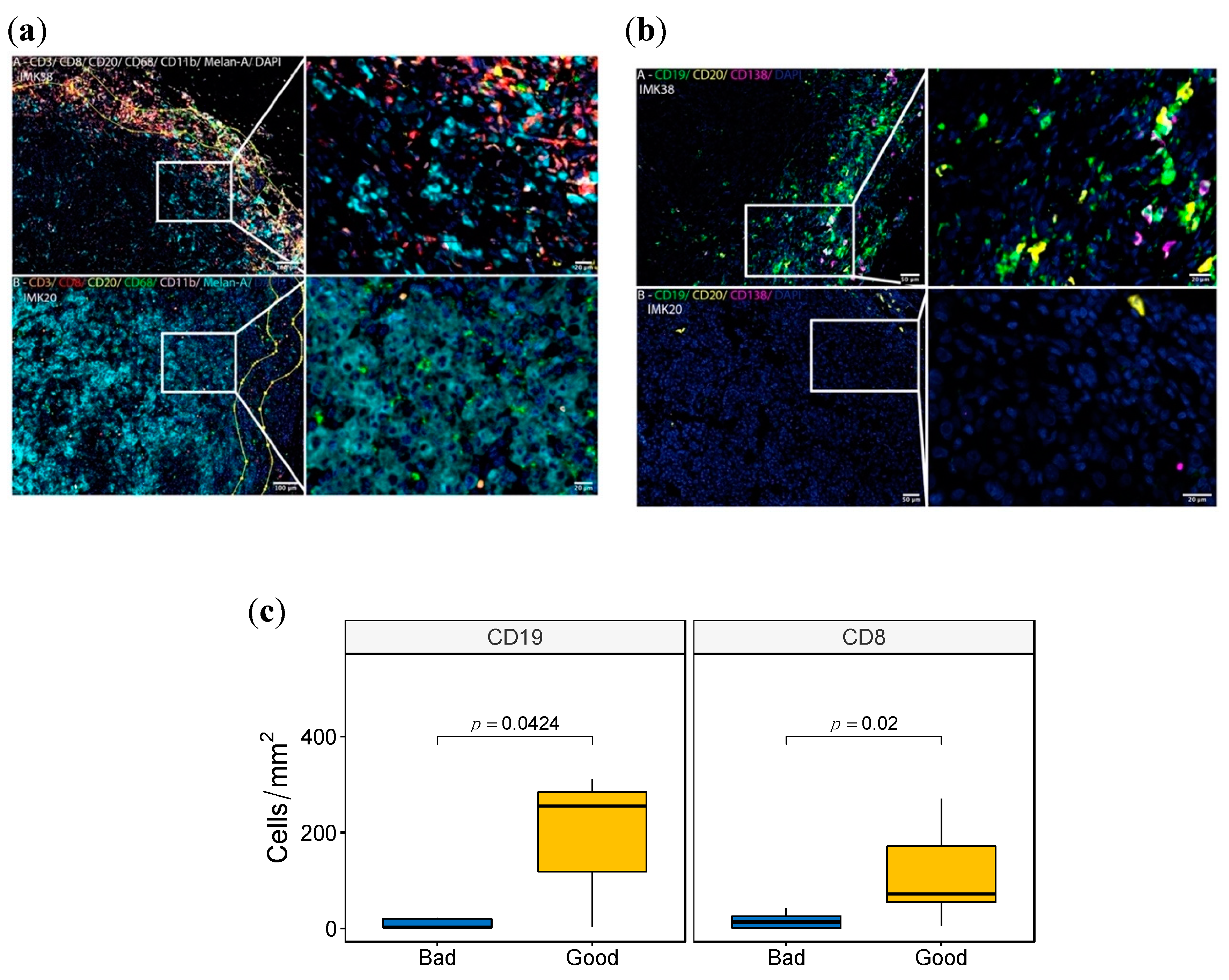
2.10. Validation of the B-Cell Signature by Multiplex Immunofluorescence
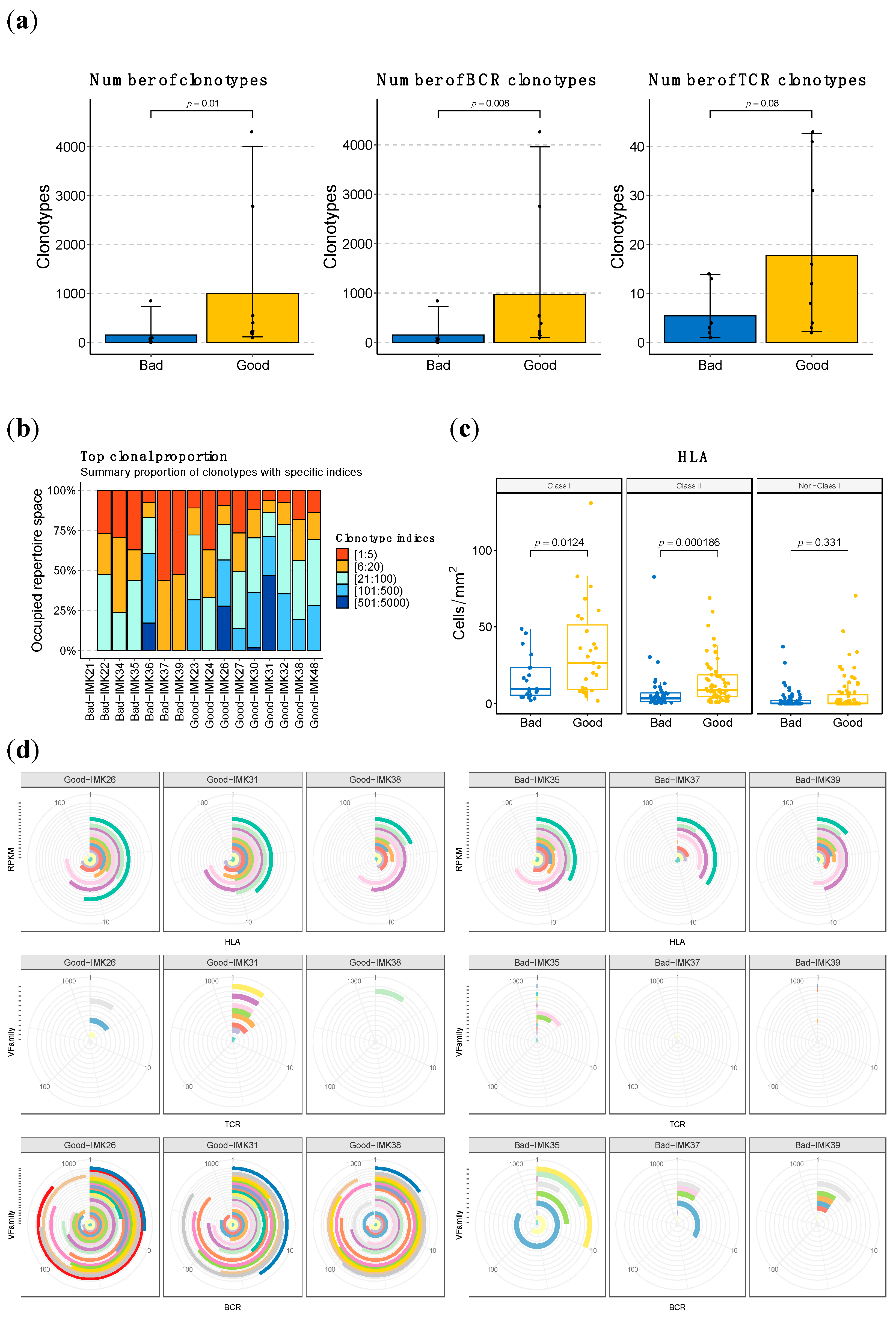
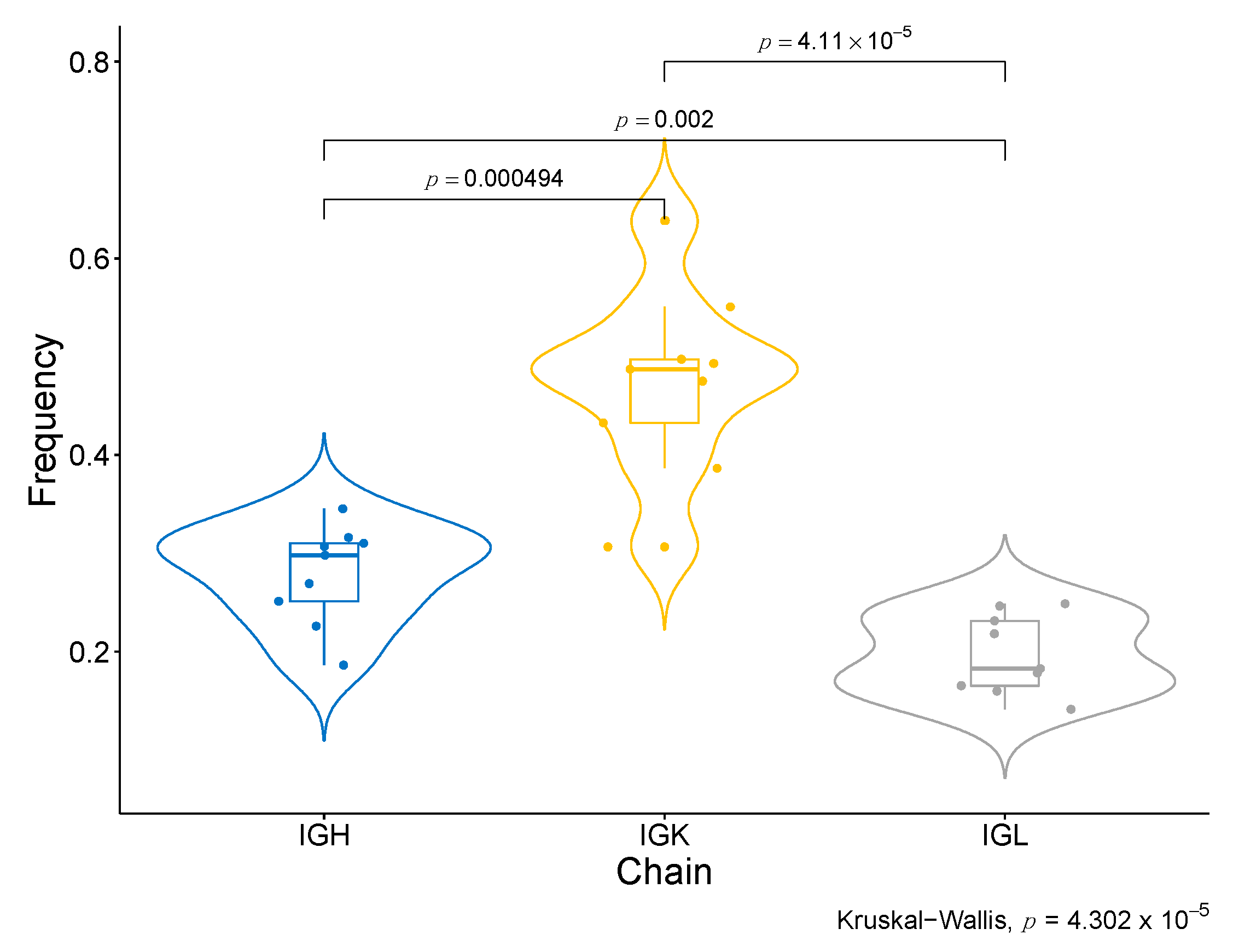
2.11. Higher Abundance and Diversity of BCR and HLA, but Not of TCR Clonotypes, Are Associated with Response to Nivolumab
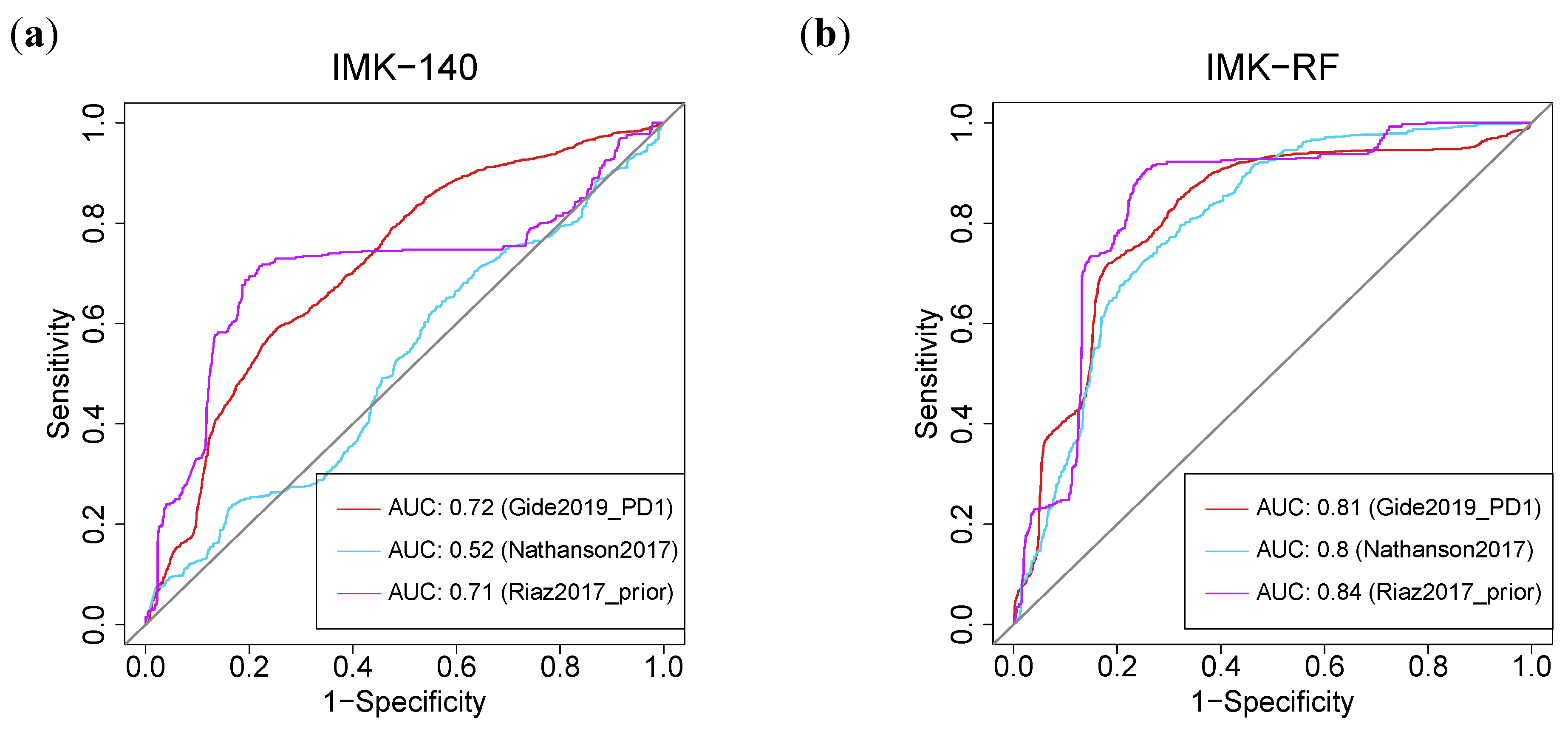
2.12. Machine-Learning-Based Models Based on Our Transcriptomics Gene Signature Predict Response in Eight External Melanoma Cohorts Treated with ICB
3. Discussion
4. Materials and Methods
4.1. Subjects
- Good responders: patients with maintained partial or complete response for a year or in treatment during at least one year.
- Bad responders: progression in less than 3 months from the start of IT. Of these, a subgroup of “severe” bad responders was defined as those who progressed in fewer than 60 days.
4.2. Nucleic Acid Extraction
4.3. Technical Validation
4.4. Next Generation Sequencing
4.5. Bioinformatic Analysis
4.6. Statistical Analysis
4.7. Multiplex Immunofluorescence
4.8. Tissue Imaging, Spectral Unmixing and Phenotyping
4.9. Special Case
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ribas, A.; Kefford, R.; Marshall, M.A.; Punt, C.J.A.; Haanen, J.B.; Marmol, M.; Garbe, C.; Gogas, H.; Schachter, J.; Linette, G.; et al. Phase III Randomized Clinical Trial Comparing Tremelimumab with Standard-of-Care Chemotherapy in Patients with Advanced Melanoma. J. Clin. Oncol. 2013, 31, 616–622. [Google Scholar] [CrossRef] [PubMed]
- Ribas, A.; Puzanov, I.; Dummer, R.; Schadendorf, D.; Hamid, O.; Robert, C.; Hodi, F.S.; Schachter, J.; Pavlick, A.C.; Lewis, K.D.; et al. Pembrolizumab versus Investigator-Choice Chemotherapy for Ipilimumab-Refractory Melanoma (KEYNOTE-002): A Randomised, Controlled, Phase 2 Trial. Lancet Oncol. 2015, 16, 908–918. [Google Scholar] [CrossRef]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.J.; Cowey, C.L.; Lao, C.D.; Schadendorf, D.; Dummer, R.; Smylie, M.; Rutkowski, P.; et al. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N. Engl. J. Med. 2015, 373, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Verma, V.; Sprave, T.; Haque, W.; Simone, C.B.; Chang, J.Y.; Welsh, J.W.; Thomas, C.R. A Systematic Review of the Cost and Cost-Effectiveness Studies of Immune Checkpoint Inhibitors. J. Immunother. Cancer 2018, 6, 128. [Google Scholar] [CrossRef]
- Mok, T.S.K.; Wu, Y.L.; Kudaba, I.; Kowalski, D.M.; Cho, B.C.; Turna, H.Z.; Castro, G.; Srimuninnimit, V.; Laktionov, K.K.; Bondarenko, I.; et al. Pembrolizumab versus Chemotherapy for Previously Untreated, PD-L1-Expressing, Locally Advanced or Metastatic Non-Small-Cell Lung Cancer (KEYNOTE-042): A Randomised, Open-Label, Controlled, Phase 3 Trial. Lancet 2019, 393, 1819–1830. [Google Scholar] [CrossRef]
- Burtness, B.; Harrington, K.J.; Greil, R.; Soulières, D.; Tahara, M.; de Castro, G.; Psyrri, A.; Basté, N.; Neupane, P.; Bratland, Å.; et al. Pembrolizumab Alone or with Chemotherapy versus Cetuximab with Chemotherapy for Recurrent or Metastatic Squamous Cell Carcinoma of the Head and Neck (KEYNOTE-048): A Randomised, Open-Label, Phase 3 Study. Lancet 2019, 394, 1915–1928. [Google Scholar] [CrossRef]
- Prelaj, A.; Tay, R.; Ferrara, R.; Chaput, N.; Besse, B.; Califano, R. Predictive Biomarkers of Response for Immune Checkpoint Inhibitors in Non–Small-Cell Lung Cancer. Eur. J. Cancer 2019, 106, 144–159. [Google Scholar] [CrossRef] [PubMed]
- Sacher, A.G.; Gandhi, L. Biomarkers for the Clinical Use of PD-1/PD-L1 Inhibitors in Non-Small-Cell Lung Cancer: A Review. JAMA Oncol. 2016, 2, 1217–1222. [Google Scholar] [CrossRef] [PubMed]
- Friedrich, M.J. Immunotherapy 2.0: Improving the Response to Checkpoint Inhibitors. J. Am. Med. Assoc. 2019, 321, 131–133. [Google Scholar] [CrossRef] [PubMed]
- Munari, E.; Zamboni, G.; Marconi, M.; Sommaggio, M.; Brunelli, M.; Martignoni, G.; Netto, G.J.; Moretta, F.; Mingari, M.C.; Salgarello, M.; et al. PD-L1 Expression Heterogeneity in Non-Small Cell Lung Cancer: Evaluation of Small Biopsies Reliability. Oncotarget 2017, 8, 90123–90131. [Google Scholar] [CrossRef]
- Borghaei, H.; Paz-Ares, L.; Horn, L.; Spigel, D.R.; Steins, M.; Ready, N.E.; Chow, L.Q.; Vokes, E.E.; Felip, E.; Holgado, E.; et al. Nivolumab versus Docetaxel in Advanced Nonsquamous Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2015, 373, 1627–1639. [Google Scholar] [CrossRef] [PubMed]
- Diem, S.; Kasenda, B.; Spain, L.; Martin-Liberal, J.; Marconcini, R.; Gore, M.; Larkin, J. Serum Lactate Dehydrogenase as an Early Marker for Outcome in Patients Treated with Anti-PD-1 Therapy in Metastatic Melanoma. Br. J. Cancer 2016, 114, 256–261. [Google Scholar] [CrossRef] [PubMed]
- Shields, B.D.; Mahmoud, F.; Taylor, E.M.; Byrum, S.D.; Sengupta, D.; Koss, B.; Baldini, G.; Ransom, S.; Cline, K.; Mackintosh, S.G.; et al. Indicators of Responsiveness to Immune Checkpoint Inhibitors. Sci. Rep. 2017, 7, 807. [Google Scholar] [CrossRef] [PubMed]
- Rizvi, H.; Sanchez-Vega, F.; La, K.; Chatila, W.; Jonsson, P.; Halpenny, D.; Plodkowski, A.; Long, N.; Sauter, J.L.; Rekhtman, N.; et al. Molecular Determinants of Response to Anti-Programmed Cell Death (PD)-1 and Anti-Programmed Death-Ligand 1 (PD-L1) Blockade in Patients with Non-Small-Cell Lung Cancer Profiled with Targeted next-Generation Sequencing. J. Clin. Oncol. 2018, 36, 633–641. [Google Scholar] [CrossRef]
- Gupta, S.; Vanderbilt, C.M.; Cotzia, P.; Arias Stella, J.A.; Chang, J.C.; Chen, Y.; Tang, L.H.; DeLair, D.F.; Yao, J.; Ladanyi, M.; et al. JAK2, PD-L1, and PD-L2 (9p24.1) Amplification in Metastatic Mucosal and Cutaneous Melanomas with Durable Response to Immunotherapy. Hum. Pathol. 2019, 88, 87–91. [Google Scholar] [CrossRef] [PubMed]
- Ascierto, M.L.; Makohon-Moore, A.; Lipson, E.J.; Taube, J.M.; McMiller, T.L.; Berger, A.E.; Fan, J.; Kaunitz, G.J.; Cottrell, T.R.; Kohutek, Z.A.; et al. Transcriptional Mechanisms of Resistance to Anti-PD-1 Therapy. Clin. Cancer Res. 2017, 23, 3168–3180. [Google Scholar] [CrossRef] [PubMed]
- Pabla, S.; Conroy, J.M.; Nesline, M.K.; Glenn, S.T.; Papanicolau-Sengos, A.; Burgher, B.; Hagen, J.; Giamo, V.; Andreas, J.; Lenzo, F.L.; et al. Proliferative Potential and Resistance to Immune Checkpoint Blockade in Lung Cancer Patients. J. Immunother. Cancer 2019, 7(1), 27. [Google Scholar] [CrossRef]
- Zaretsky, J.M.; Garcia-Diaz, A.; Shin, D.S.; Escuin-Ordinas, H.; Hugo, W.; Hu-Lieskovan, S.; Torrejon, D.Y.; Abril-Rodriguez, G.; Sandoval, S.; Barthly, L.; et al. Mutations Associated with Acquired Resistance to PD-1 Blockade in Melanoma. N. Engl. J. Med. 2016, 375, 819–829. [Google Scholar] [CrossRef] [PubMed]
- Snyder, A.; Makarov, V.; Merghoub, T.; Yuan, J.; Zaretsky, J.M.; Desrichard, A.; Walsh, L.A.; Postow, M.A.; Wong, P.; Ho, T.S.; et al. Genetic Basis for Clinical Response to CTLA-4 Blockade in Melanoma. N. Engl. J. Med. 2014, 371, 2189–2199. [Google Scholar] [CrossRef] [PubMed]
- Rizvi, N.A.; Hellmann, M.D.; Snyder, A.; Kvistborg, P.; Makarov, V.; Havel, J.J.; Lee, W.; Yuan, J.; Wong, P.; Ho, T.S.; et al. Mutational Landscape Determines Sensitivity to PD-1 Blockade in Non-Small Cell Lung Cancer. Science 2015, 348, 124–128. [Google Scholar] [CrossRef] [PubMed]
- De Coaña, Y.P.; Wolodarski, M.; Poschke, I.; Yoshimoto, Y.; Yang, Y.; Nyström, M.; Edbäck, U.; Brage, S.E.; Lundqvist, A.; Masucci, G.V.; et al. Ipilimumab Treatment Decreases Monocytic MDSCs and Increases CD8 Effector Memory T Cells in Long-Term Survivors with Advanced Melanoma. Oncotarget 2017, 8, 21539–21553. [Google Scholar] [CrossRef]
- Duruisseaux, M.; Martínez-Cardús, A.; Calleja-Cervantes, M.E.; Moran, S.; Castro de Moura, M.; Davalos, V.; Piñeyro, D.; Sanchez-Cespedes, M.; Girard, N.; Brevet, M.; et al. Epigenetic Prediction of Response to Anti-PD-1 Treatment in Non-Small-Cell Lung Cancer: A Multicentre, Retrospective Analysis. Lancet Respir. Med. 2018, 6, 771–781. [Google Scholar] [CrossRef]
- Hoffmann, F.; Zarbl, R.; Niebel, D.; Sirokay, J.; Fröhlich, A.; Posch, C.; Holderried, T.A.W.; Brossart, P.; Saavedra, G.; Kuster, P.; et al. Prognostic and Predictive Value of PD-L2 DNA Methylation and MRNA Expression in Melanoma. Clin. Epigenetics 2020, 12, 94. [Google Scholar] [CrossRef]
- Holderried, T.A.W.; De Vos, L.; Bawden, E.G.; Vogt, T.J.; Dietrich, J.; Zarbl, R.; Bootz, F.; Kristiansen, G.; Brossart, P.; Landsberg, J.; et al. Molecular and Immune Correlates of TIM-3 (HAVCR2) and Galectin 9 (LGALS9) MRNA Expression and DNA Methylation in Melanoma. Clin. Epigenetics 2019, 11, 161. [Google Scholar] [CrossRef]
- Marwitz, S.; Scheufele, S.; Perner, S.; Reck, M.; Ammerpohl, O.; Goldmann, T. Epigenetic Modifications of the Immune-Checkpoint Genes CTLA4 and PDCD1 in Non-Small Cell Lung Cancer Results in Increased Expression. Clin. Epigenetics 2017, 9, 51. [Google Scholar] [CrossRef] [PubMed]
- Anagnostou, V.; Smith, K.N.; Forde, P.M.; Niknafs, N.; Bhattacharya, R.; White, J.; Zhang, T.; Adleff, V.; Phallen, J.; Wali, N.; et al. Evolution of Neoantigen Landscape during Immune Checkpoint Blockade in Non-Small Cell Lung Cancer. Cancer Discov. 2016, 7, 264–276. [Google Scholar] [CrossRef]
- Simeone, E.; Gentilcore, G.; Giannarelli, D.; Grimaldi, A.M.; Caracò, C.; Curvietto, M.; Esposito, A.; Paone, M.; Palla, M.; Cavalcanti, E.; et al. Immunological and Biological Changes during Ipilimumab Treatment and Their Potential Correlation with Clinical Response and Survival in Patients with Advanced Melanoma. Cancer Immunol. Immunother. 2014, 63, 675–683. [Google Scholar] [CrossRef]
- Bratman, S.V.; Yang, S.Y.C.; Iafolla, M.A.J.; Liu, Z.; Hansen, A.R.; Bedard, P.L.; Lheureux, S.; Spreafico, A.; Razak, A.A.; Shchegrova, S.; et al. Personalized Circulating Tumor DNA Analysis as a Predictive Biomarker in Solid Tumor Patients Treated with Pembrolizumab. Nat. Cancer 2020, 1, 873–881. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Q.; Nobre, A.; Piñeiro, P.; Berciano-Guerrero, M.-Á.; Alba, E.; Cobo, M.; Lauschke, V.M.; Barragán, I. Genetic and Epigenetic Biomarkers of Immune Checkpoint Blockade Response. J. Clin. Med. 2020, 9, 286. [Google Scholar] [CrossRef]
- Sade-Feldman, M.; Yizhak, K.; Bjorgaard, S.L.; Ray, J.P.; de Boer, C.G.; Jenkins, R.W.; Lieb, D.J.; Chen, J.H.; Frederick, D.T.; Barzily-Rokni, M.; et al. Defining T Cell States Associated with Response to Checkpoint Immunotherapy in Melanoma. Cell 2018, 175, 998–1013. [Google Scholar] [CrossRef]
- Jerby-Arnon, L.; Shah, P.; Cuoco, M.S.; Rodman, C.; Su, M.J.; Melms, J.C.; Leeson, R.; Kanodia, A.; Mei, S.; Lin, J.R.; et al. A Cancer Cell Program Promotes T Cell Exclusion and Resistance to Checkpoint Blockade. Cell 2018, 175, 984–997. [Google Scholar] [CrossRef]
- Coelho, F.M.; Natale, D.; Soriano, S.F.; Hons, M.; Swoger, J.; Mayer, J.; Danuser, R.; Scandella, E.; Pieczyk, M.; Zerwes, H.G.; et al. Naive B-Cell Trafficking Is Shaped by Local Chemokine Availability and LFA-1-Independent Stromal Interactions. Blood 2013, 121, 4101–4109. [Google Scholar] [CrossRef]
- Griss, J.; Bauer, W.; Wagner, C.; Simon, M.; Chen, M.; Grabmeier-Pfistershammer, K.; Maurer-Granofszky, M.; Roka, F.; Penz, T.; Bock, C.; et al. B Cells Sustain Inflammation and Predict Response to Immune Checkpoint Blockade in Human Melanoma. Nat. Commun. 2019, 10, 4186. [Google Scholar] [CrossRef]
- Gide, T.N.; Quek, C.; Menzies, A.M.; Tasker, A.T.; Shang, P.; Holst, J.; Wilmott, J.S. Distinct Immune Cell Populations Define Response to Anti-PD-1 Monotherapy and Anti-PD-1/Anti-CTLA-4 Combined Therapy. Cancer Cell 2019, 35, 238–255. [Google Scholar] [CrossRef]
- Liu, D.; Schilling, B.; Liu, D.; Sucker, A.; Livingstone, E.; Jerby-Amon, L.; Schadendorf, D. Integrative Molecular and Clinical Modeling of Clinical Outcomes to PD1 Blockade in Patients with Metastatic Melanoma. Nat. Med. 2019, 25, 1916–1927. [Google Scholar] [CrossRef]
- Nathanson, T.; Ahuja, A.; Rubinsteyn, A.; Aksoy, B.A.; Hellmann, M.D.; Miao, D.; Hammerbacher, J. Somatic Mutations and Neoepitope Homology in Melanomas Treated with CTLA-4 Blockade. Cancer Immunol. Res. 2017, 5, 84–91. [Google Scholar] [CrossRef]
- Riaz, N.; Havel, J.J.; Makarov, V.; Desrichard, A.; Urba, W.J.; Sims, J.S.; Chan, T.A. Tumor and Microenvironment Evolution during Immunotherapy with Nivolumab. Cell 2017, 171, 934–949. [Google Scholar] [CrossRef]
- Hugo, W.; Zaretsky, J.M.; Sun, L.; Song, C.; Moreno, B.H.; Hu-Lieskovan, S.; Lo, R.S. Genomic and Transcriptomic Features of Response to Anti-PD-1 Therapy in Metastatic Melanoma. Cell 2016, 165, 35–44. [Google Scholar] [CrossRef]
- Jiang, P.; Gu, S.; Pan, D.; Fu, J.; Sahu, A.; Hu, X.; Li, Z.; Traugh, N.; Bu, X.; Li, B.; et al. Signatures of T Cell Dysfunction and Exclusion Predict Cancer Immunotherapy Response. Nat. Med. 2018, 24, 1550–1558. [Google Scholar] [CrossRef]
- Fu, J.; Li, K.; Zhang, W.; Wan, C.; Zhang, J.; Jiang, P.; Liu, X.S. Large-Scale Public Data Reuse to Model Immunotherapy Response and Resistance. Genome Med. 2020, 12, 21. [Google Scholar] [CrossRef]
- Choi, J.A.; Kwon, H.; Cho, H.; Chung, J.Y.; Hewitt, S.M.; Kim, J.H. ALDH1A2 Is a Candidate Tumor Suppressor Gene in Ovarian Cancer. Cancers 2019, 11, 1553. [Google Scholar] [CrossRef] [PubMed]
- Mowat, A.M.I. To Respond or Not to Respond—A Personal Perspective of Intestinal Tolerance. Nat. Rev. Immunol. 2018, 18, 405–415. [Google Scholar] [CrossRef] [PubMed]
- Gorantla, V.C.; Kirkwood, J.M. State of Melanoma. An Historic Overview of a Field in Transition. Hematol. Oncol. Clin. N. Am. 2014, 28, 415–435. [Google Scholar] [CrossRef] [PubMed]
- Shtivelman, E.; Davies, M.A.; Hwu, P.; Yang, J.; Lotem, M.; Oren, M.; Flaherty, K.T.; Fisher, D.E. Pathways and Therapeutic Targets in Melanoma. Oncotarget 2014, 5, 1701–1752. [Google Scholar] [CrossRef]
- Liu, Z.; Dai, W.; Jiang, L.; Cheng, Y. Over-Expression of LGR5 Correlates with Poor Survival of Colon Cancer in Mice as Well as in Patients. Neoplasma 2014, 61, 177–185. [Google Scholar] [CrossRef]
- Xi, H.Q.; Cui, J.X.; Shen, W.S.; Wu, X.S.; Bian, S.B.O.; Li, J.I.Y.; Song, Z.; Wei, B.O.; Chen, L. Increased Expression of Lgr5 Is Associated with Chemotherapy Resistance in Human Gastric Cancer. Oncol. Rep. 2014, 32, 181–188. [Google Scholar] [CrossRef] [PubMed]
- Schadendorf, D.; van Akkooi, A.C.J.; Berking, C.; Griewank, K.G.; Gutzmer, R.; Hauschild, A.; Stang, A.; Roesch, A.; Ugurel, S. Melanoma. Lancet 2018, 392, 971–984. [Google Scholar] [CrossRef]
- García-Mulero, S.; Alonso, M.H.; Pardo, J.; Santos, C.; Sanjuan, X.; Salazar, R.; Moreno, V.; Piulats, J.M.; Sanz-Pamplona, R. Lung Metastases Share Common Immune Features Regardless of Primary Tumor Origin. J. Immunother. Cancer 2020, 8, e000491. [Google Scholar] [CrossRef] [PubMed]
- Keenan, T.E.; Burke, K.P.; Van Allen, E.M. Genomic Correlates of Response to Immune Checkpoint Blockade. Nat. Med. 2019, 25, 389–402. [Google Scholar] [CrossRef]
- Anagnostou, V.; Bruhm, D.C.; Niknafs, N.; White, J.R.; Sidhom, J.-W.; Stein, J.E.; Tsai, H.-L.; Wang, H.; Belcaid, Z.; Murray, J.C.; et al. Integrative Tumor and Immune Cell Multi-Omic Analyses to Predict Melanoma Response to Immune Checkpoint Blockade. J. Clin. Oncol. 2020, 38, 10009. [Google Scholar] [CrossRef]
- Schmidt, M.; Hellwig, B.; Hammad, S.; Othman, A.; Lohr, M.; Chen, Z.; Boehm, D.; Gebhard, S.; Petry, I.; Lebrecht, A.; et al. A Comprehensive Analysis of Human Gene Expression Profiles Identifies Stromal Immunoglobulin κ C as a Compatible Prognostic Marker in Human Solid Tumors. Clin. Cancer Res. 2012, 18, 2695–2703. [Google Scholar] [CrossRef] [PubMed]
- Whiteside, T.L. Immune Responses to Cancer: Are They Potential Biomarkers of Prognosis? Front. Oncol. 2013, 3, 107. [Google Scholar] [CrossRef] [PubMed]
- Fristedt, R.; Borg, D.; Hedner, C.; Berntsson, J.; Nodin, B.; Eberhard, J.; Micke, P.; Jirström, K. Prognostic Impact of Tumour-Associated B Cells and Plasma Cells in Oesophageal and Gastric Adenocarcinoma. J. Gastrointest. Oncol. 2016, 7, 848. [Google Scholar] [CrossRef]
- Lohr, M.; Edlund, K.; Botling, J.; Hammad, S.; Hellwig, B.; Othman, A.; Berglund, A.; Lambe, M.; Holmberg, L.; Ekman, S.; et al. The Prognostic Relevance of Tumour-Infiltrating Plasma Cells and Immunoglobulin Kappa C Indicates an Important Role of the Humoral Immune Response in Non-Small Cell Lung Cancer. Cancer Lett. 2013, 333, 222–228. [Google Scholar] [CrossRef]
- Kinker, G.S.; Vitiello, G.A.F.; Ferreira, W.A.S.; Chaves, A.S.; Cordeiro de Lima, V.C.; Medina, T.D.S. B Cell Orchestration of Anti-Tumor Immune Responses: A Matter of Cell Localization and Communication. Front. Cell Dev. Biol. 2021, 9, 1282. [Google Scholar] [CrossRef]
- Cabrita, R.; Lauss, M.; Sanna, A.; Donia, M.; Skaarup Larsen, M.; Mitra, S.; Johansson, I.; Phung, B.; Harbst, K.; Vallon-Christersson, J.; et al. Tertiary Lymphoid Structures Improve Immunotherapy and Survival in Melanoma. Nature 2020, 577, 561–565. [Google Scholar] [CrossRef]
- Schalper, K.A.; Rodriguez-Ruiz, M.E.; Diez-Valle, R.; López-Janeiro, A.; Porciuncula, A.; Idoate, M.A.; Inogés, S.; de Andrea, C.; López-Diaz de Cerio, A.; Tejada, S.; et al. Neoadjuvant Nivolumab Modifies the Tumor Immune Microenvironment in Resectable Glioblastoma. Nat. Med. 2019, 25, 470–476. [Google Scholar] [CrossRef] [PubMed]
- Abengozar-Muela, M.; Esparza, M.V.; Garcia-Ros, D.; Vásquez, C.E.; Echeveste, J.I.; Idoate, M.A.; Lozano, M.D.; Melero, I.; de Andrea, C.E. Diverse Immune Environments in Human Lung Tuberculosis Granulomas Assessed by Quantitative Multiplexed Immunofluorescence. Mod. Pathol. 2020, 33, 2507–2519. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Good Responders Count | Bad Responders Count | Clonotype Composition |
|---|---|---|
| 1555 | 87 | IGKV3-20, IGKJ1, IGKC |
| 1364 | 8 | IGKV1-33, IGKJ4, IGKC |
| 917 | 129 | IGKV1-39, IGKJ2, IGKC |
| 818 | 49 | IGKV1-5, IGKJ1, IGKC |
| 816 | 47 | IGKV3-15, IGKJ2, IGKC |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Onieva, J.L.; Xiao, Q.; Berciano-Guerrero, M.-Á.; Laborda-Illanes, A.; de Andrea, C.; Chaves, P.; Piñeiro, P.; Garrido-Aranda, A.; Gallego, E.; Sojo, B.; et al. High IGKC-Expressing Intratumoral Plasma Cells Predict Response to Immune Checkpoint Blockade. Int. J. Mol. Sci. 2022, 23, 9124. https://doi.org/10.3390/ijms23169124
Onieva JL, Xiao Q, Berciano-Guerrero M-Á, Laborda-Illanes A, de Andrea C, Chaves P, Piñeiro P, Garrido-Aranda A, Gallego E, Sojo B, et al. High IGKC-Expressing Intratumoral Plasma Cells Predict Response to Immune Checkpoint Blockade. International Journal of Molecular Sciences. 2022; 23(16):9124. https://doi.org/10.3390/ijms23169124
Chicago/Turabian StyleOnieva, Juan Luis, Qingyang Xiao, Miguel-Ángel Berciano-Guerrero, Aurora Laborda-Illanes, Carlos de Andrea, Patricia Chaves, Pilar Piñeiro, Alicia Garrido-Aranda, Elena Gallego, Belén Sojo, and et al. 2022. "High IGKC-Expressing Intratumoral Plasma Cells Predict Response to Immune Checkpoint Blockade" International Journal of Molecular Sciences 23, no. 16: 9124. https://doi.org/10.3390/ijms23169124