
Vascular Smooth Muscle Cell Neutral Sphingomyelinase 2 in the Release of Exosomes and Vascular Calcification


 and
and
Abstract
:1. Introduction
2. Vascular Smooth Muscle Cells in Vascular Calcification
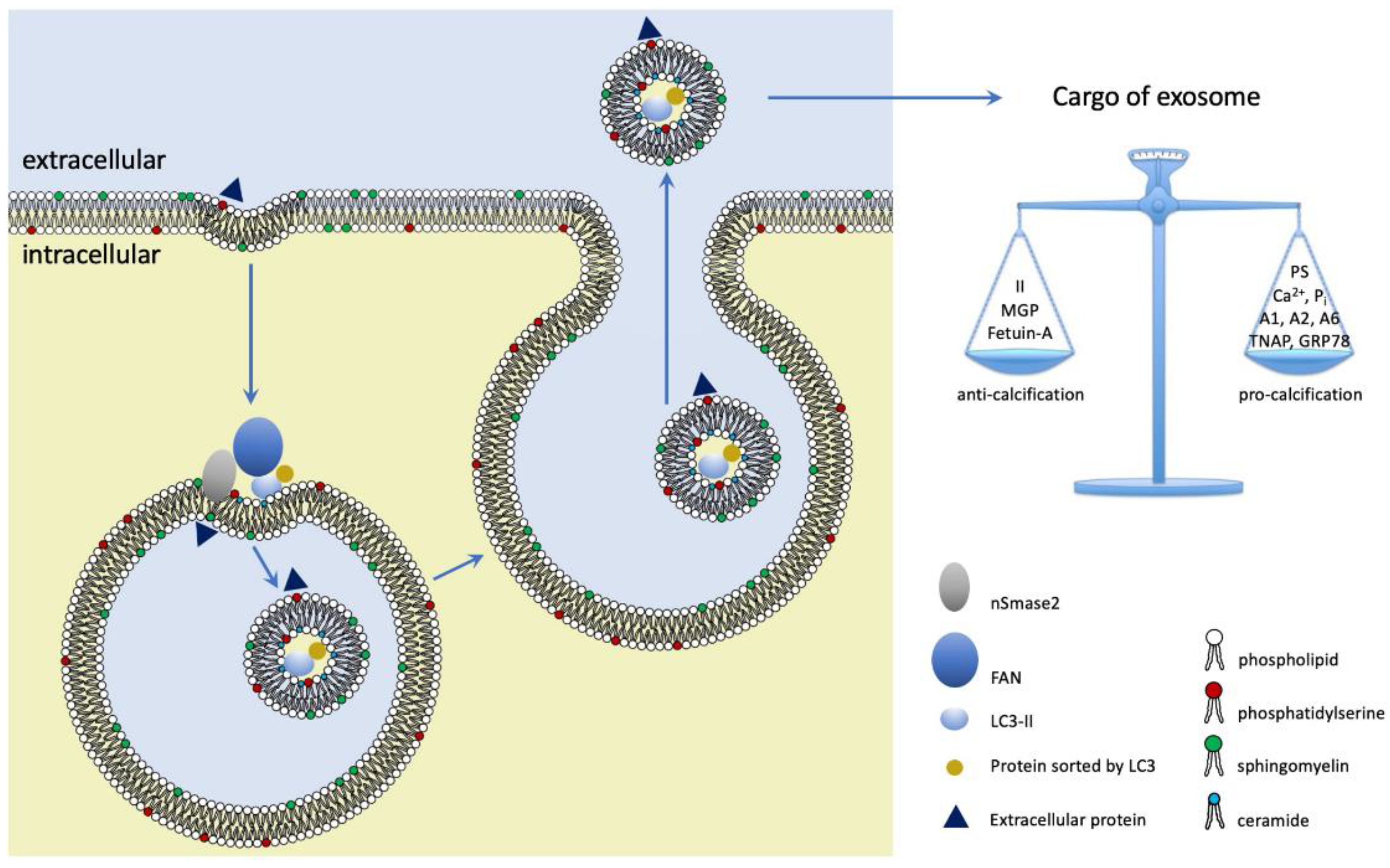
3. Extracellular Vesicles: Nomenclature, Structure and Biogenesis
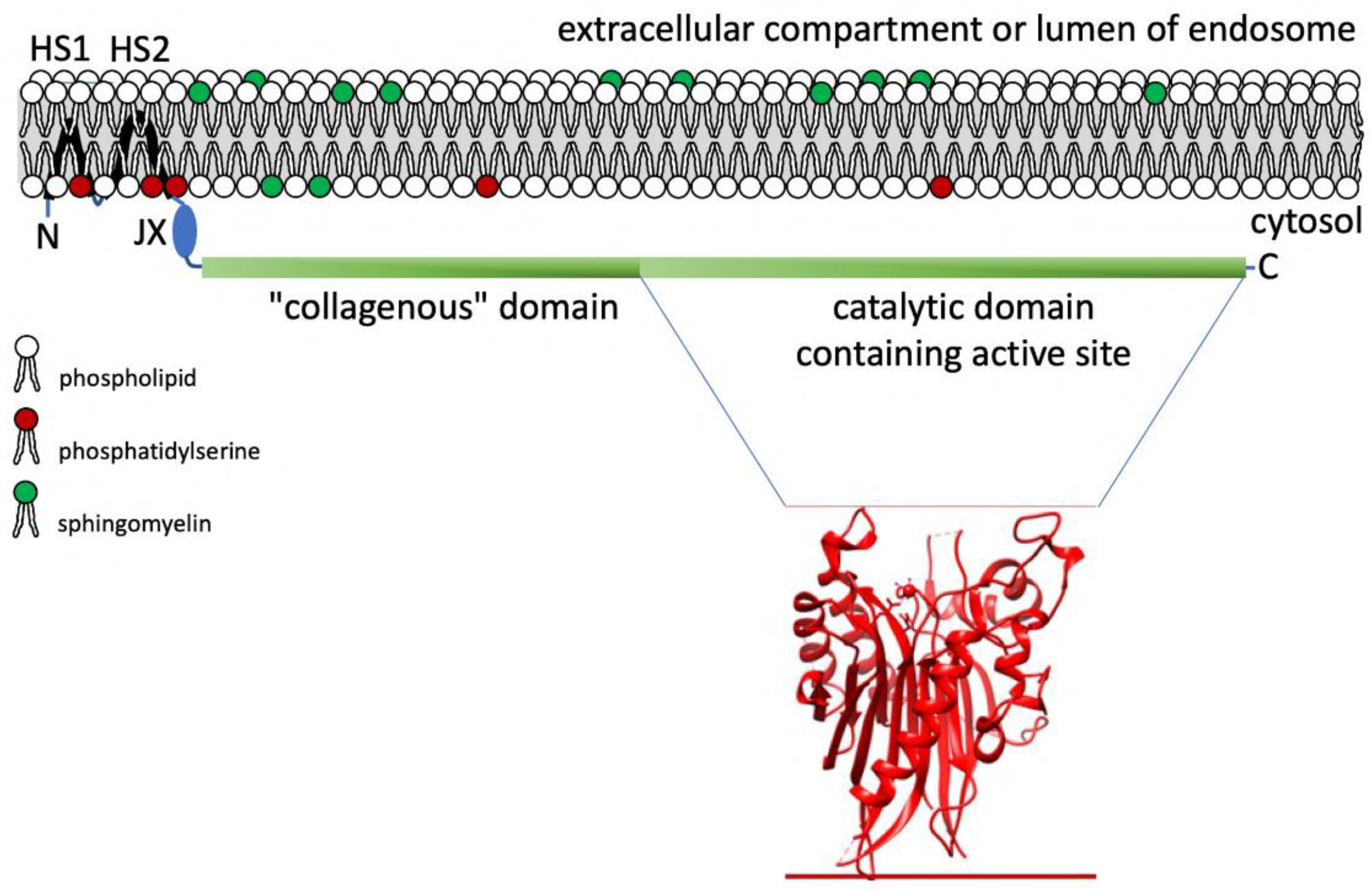
4. Neutral Sphingomyelinase 2: Structure and Function in Exosome Release
5. Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rennenberg, R.J.M.W.; Kessels, A.G.H.; Schurgers, L.J.; van Engelshoven, J.M.A.; de Leeuw, P.W.; Kroon, A.A. Vascular Calcifications as a Marker of Increased Cardiovascular Risk: A Meta-Analysis. Vasc. Health Risk Manag. 2009, 5, 185–197. [Google Scholar] [CrossRef] [PubMed]
- Demer, L.L.; Tintut, Y. Vascular Calcification: Pathobiology of a Multifaceted Disease. Circulation 2008, 117, 2938–2948. [Google Scholar] [CrossRef] [PubMed]
- Lindman, B.R.; Sukul, D.; Dweck, M.R.; Madhavan, M.V.; Arsenault, B.J.; Coylewright, M.; Merryman, W.D.; Newby, D.E.; Lewis, J.; Harrell, F.E., Jr.; et al. Evaluating Medical Therapy for Calcific Aortic Stenosis: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2021, 78, 2354–2376. [Google Scholar] [CrossRef]
- Brandenburg, V.; Al-Fakhri, N.; Nemeth, K.; Goettsch, C.; Schurgers, L.J.; Vermeer, C.; Hofbauer, L.C.; Schoppet, M. Calcification Inhibitors in Vascular Calciphylaxis Associated with Normal Renal Function. Thromb. Haemost. 2012, 108, 1241–1243. [Google Scholar] [CrossRef] [PubMed]
- Schinke, T.; Karsenty, G. Vascular Calcification--a Passive Process in Need of Inhibitors. Nephrol. Dial. Transplant. 2000, 15, 1272–1274. [Google Scholar] [CrossRef]
- Durham, A.L.; Speer, M.Y.; Scatena, M.; Giachelli, C.M.; Shanahan, C.M. Role of Smooth Muscle Cells in Vascular Calcification: Implications in Atherosclerosis and Arterial Stiffness. Cardiovasc. Res. 2018, 114, 590–600. [Google Scholar] [CrossRef]
- Jaminon, A.; Reesink, K.; Kroon, A.; Schurgers, L. The Role of Vascular Smooth Muscle Cells in Arterial Remodeling: Focus on Calcification-Related Processes. Int. J. Mol. Sci. 2019, 20, 5694. [Google Scholar] [CrossRef]
- Proudfoot, D.; Skepper, J.N.; Hegyi, L.; Bennett, M.R.; Shanahan, C.M.; Weissberg, P.L. Apoptosis Regulates Human Vascular Calcification In Vitro: Evidence for Initiation of Vascular Calcification by Apoptotic Bodies. Circ. Res. 2000, 87, 1055–1062. [Google Scholar] [CrossRef]
- Shanahan, C.M. Mechanisms of Vascular Calcification in CKD—Evidence for Premature Ageing? Nat. Rev. Nephrol. 2013, 9, 661–670. [Google Scholar] [CrossRef]
- Blaser, M.C.; Aikawa, E. Roles and Regulation of Extracellular Vesicles in Cardiovascular Mineral Metabolism. Front. Cardiovasc. Med. 2018, 5, 187. [Google Scholar] [CrossRef]
- Schurgers, L.J.; Akbulut, A.C.; Kaczor, D.M.; Halder, M.; Koenen, R.R.; Kramann, R. Initiation and Propagation of Vascular Calcification Is Regulated by a Concert of Platelet- and Smooth Muscle Cell-Derived Extracellular Vesicles. Front. Cardiovasc. Med. 2018, 5, 36. [Google Scholar] [CrossRef] [PubMed]
- Mas-Bargues, C.; Borrás, C.; Alique, M. The Contribution of Extracellular Vesicles from Senescent Endothelial and Vascular Smooth Muscle Cells to Vascular Calcification. Front. Cardiovasc. Med. 2022, 9, 854726. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Shan, S.-K.; Xu, F.; Zhong, J.-Y.; Wu, F.; Duan, J.-Y.; Guo, B.; Li, F.-X.-Z.; Wang, Y.; Zheng, M.-H.; et al. The Crosstalk between Endothelial Cells and Vascular Smooth Muscle Cells Aggravates High Phosphorus-Induced Arterial Calcification. Cell Death Dis. 2022, 13, 650. [Google Scholar] [CrossRef] [PubMed]
- Vajen, T.; Benedikter, B.J.; Heinzmann, A.C.A.; Vasina, E.M.; Henskens, Y.; Parsons, M.; Maguire, P.B.; Stassen, F.R.; Heemskerk, J.W.M.; Schurgers, L.J.; et al. Platelet Extracellular Vesicles Induce a pro-Inflammatory Smooth Muscle Cell Phenotype. J. Extracell. Vesicles 2017, 6, 1322454. [Google Scholar] [CrossRef] [PubMed]
- Shamseddine, A.A.; Airola, M.V.; Hannun, Y.A. Roles and Regulation of Neutral Sphingomyelinase-2 in Cellular and Pathological Processes. Adv. Biol. Regul. 2015, 57, 24–41. [Google Scholar] [CrossRef]
- Kapustin, A.N.; Chatrou, M.L.L.; Drozdov, I.; Zheng, Y.; Davidson, S.M.; Soong, D.; Furmanik, M.; Sanchis, P.; Rosales, R.T.M.D.; Hernandez, D.A.; et al. Vascular Smooth Muscle Cell Calcification Is Mediated by Regulated Exosome Secretion. Circ. Res. 2015, 116, 1312–1323. [Google Scholar] [CrossRef]
- Lallemand, T.; Rouahi, M.; Swiader, A.; Grazide, M.-H.; Geoffre, N.; Alayrac, P.; Recazens, E.; Coste, A.; Salvayre, R.; Negre-Salvayre, A.; et al. nSMase2 (Type 2-Neutral Sphingomyelinase) Deficiency or Inhibition by GW4869 Reduces Inflammation and Atherosclerosis in Apoe-/- Mice. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 1479–1492. [Google Scholar] [CrossRef]
- Grootaert, M.O.J.; Bennett, M.R. Vascular Smooth Muscle Cells in atherosclerosis: Time for a Reassessment. Cardiovasc. Res. 2021, 117, 2326–2339. [Google Scholar] [CrossRef]
- Petsophonsakul, P.; Furmanik, M.; Forsythe, R.; Dweck, M.; Schurink, G.W.; Natour, E.; Reutelingsperger, C.; Jacobs, M.; Mees, B.; Schurgers, L. Role of Vascular Smooth Muscle Cell Phenotypic Switching and Calcification in Aortic Aneurysm Formation. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 1351–1368. [Google Scholar] [CrossRef]
- Yap, C.; Mieremet, A.; de Vries, C.J.M.; Micha, D.; de Waard, V. Six Shades of Vascular Smooth Muscle Cells Illuminated by KLF4 (Krüppel-Like Factor 4). Arterioscler. Thromb. Vasc. Biol. 2021, 41, 2693–2707. [Google Scholar] [CrossRef]
- Schurgers, L.J.; Uitto, J.; Reutelingsperger, C.P. Vitamin K-Dependent Carboxylation of Matrix Gla-Protein: A Crucial Switch to Control Ectopic Mineralization. Trends Mol. Med. 2013, 19, 217–226. [Google Scholar] [CrossRef] [PubMed]
- Roumeliotis, S.; Dounousi, E.; Salmas, M.; Eleftheriadis, T.; Liakopoulos, V. Vascular Calcification in Chronic Kidney Disease: The Role of Vitamin K- Dependent Matrix Gla Protein. Front. Med. 2020, 7, 154. [Google Scholar] [CrossRef] [PubMed]
- Luo, G.; Ducy, P.; McKee, M.D.; Pinero, G.J.; Loyer, E.; Behringer, R.R.; Karsenty, G. Spontaneous Calcification of Arteries and Cartilage in Mice Lacking Matrix GLA Protein. Nature 1997, 386, 78–81. [Google Scholar] [CrossRef] [PubMed]
- Murshed, M.; Schinke, T.; McKee, M.D.; Karsenty, G. Extracellular Matrix Mineralization Is Regulated Locally; Different Roles of Two Gla-Containing Proteins. J. Cell Biol. 2004, 165, 625–630. [Google Scholar] [CrossRef]
- Popa, D.-S.; Bigman, G.; Rusu, M.E. The Role of Vitamin K in Humans: Implication in Aging and Age-Associated Diseases. Antioxidants 2021, 10, 566. [Google Scholar] [CrossRef]
- Liu, Y.-P.; Gu, Y.-M.; Thijs, L.; Knapen, M.H.J.; Salvi, E.; Citterio, L.; Petit, T.; Carpini, S.D.; Zhang, Z.; Jacobs, L.; et al. Inactive Matrix Gla Protein Is Causally Related to Adverse Health Outcomes: A Mendelian Randomization Study in a Flemish Population. Hypertension 2015, 65, 463–470. [Google Scholar] [CrossRef]
- Kapustin, A.N.; Davies, J.D.; Reynolds, J.L.; McNair, R.; Jones, G.T.; Sidibe, A.; Schurgers, L.J.; Skepper, J.N.; Proudfoot, D.; Mayr, M.; et al. Calcium Regulates Key Components of Vascular Smooth Muscle Cell-Derived Matrix Vesicles to Enhance Mineralization. Circ. Res. 2011, 109, e1–e12. [Google Scholar] [CrossRef]
- Onyedibe, K.I.; Wang, M.; Sintim, H.O. ENPP1, an Old Enzyme with New Functions, and Small Molecule Inhibitors-A STING in the Tale of ENPP1. Molecules 2019, 24, 4192. [Google Scholar] [CrossRef]
- Hamczyk, M.R.; Villa-Bellosta, R. Pyrophosphate Metabolism and Calcification. Aging 2018, 10, 3652–3653. [Google Scholar] [CrossRef]
- Hessle, L.; Johnson, K.A.; Anderson, H.C.; Narisawa, S.; Sali, A.; Goding, J.W.; Terkeltaub, R.; Millan, J.L. Tissue-Nonspecific Alkaline Phosphatase and Plasma Cell Membrane Glycoprotein-1 Are Central Antagonistic Regulators of Bone Mineralization. Proc. Natl. Acad. Sci. USA 2002, 99, 9445–9449. [Google Scholar] [CrossRef]
- Villa-Bellosta, R. Synthesis of Extracellular Pyrophosphate Increases in Vascular Smooth Muscle Cells During Phosphate-Induced Calcification. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 2137–2147. [Google Scholar] [CrossRef] [PubMed]
- Kapustin, A.N.; Shanahan, C.M. Calcium Regulation of Vascular Smooth Muscle Cell-Derived Matrix Vesicles. Trends Cardiovasc. Med. 2012, 22, 133–137. [Google Scholar] [CrossRef] [PubMed]
- Hutcheson, J.D.; Goettsch, C.; Bertazzo, S.; Maldonado, N.; Ruiz, J.L.; Goh, W.; Yabusaki, K.; Faits, T.; Bouten, C.; Franck, G.; et al. Genesis and Growth of Extracellular-Vesicle-Derived Microcalcification in Atherosclerotic Plaques. Nat. Mater. 2016, 15, 335–343. [Google Scholar] [CrossRef] [PubMed]
- Shanahan, C.M.; Crouthamel, M.H.; Kapustin, A.; Giachelli, C.M. Arterial Calcification in Chronic Kidney Disease: Key Roles for Calcium and Phosphate. Circ. Res. 2011, 109, 697–711. [Google Scholar] [CrossRef]
- Kapustin, A.N.; Shanahan, C.M. Emerging Roles for Vascular Smooth Muscle Cell Exosomes in Calcification and Coagulation. J. Physiol. 2016, 594, 2905–2914. [Google Scholar] [CrossRef]
- Shroff, R.; Long, D.A.; Shanahan, C. Mechanistic Insights into Vascular Calcification in CKD. J. Am. Soc. Nephrol. 2013, 24, 179–189. [Google Scholar] [CrossRef]
- Kapustin, A.N.; Schoppet, M.; Schurgers, L.J.; Reynolds, J.L.; McNair, R.; Heiss, A.; Jahnen-Dechent, W.; Hackeng, T.M.; Schlieper, G.; Harrison, P.; et al. Prothrombin Loading of Vascular Smooth Muscle Cell-Derived Exosomes Regulates Coagulation and Calcification. Arterioscler. Thromb. Vasc. Biol. 2017, 37, e22–e32. [Google Scholar] [CrossRef]
- Chen, N.X.; O’Neill, K.D.; Chen, X.; Moe, S.M. Annexin-Mediated Matrix Vesicle Calcification in Vascular Smooth Muscle Cells. J. Bone Miner. Res. 2008, 23, 1798–1805. [Google Scholar] [CrossRef]
- Goettsch, C.; Hutcheson, J.D.; Aikawa, M.; Iwata, H.; Pham, T.; Nykjaer, A.; Kjolby, M.; Rogers, M.; Michel, T.; Shibasaki, M.; et al. Sortilin Mediates Vascular Calcification via Its Recruitment into Extracellular Vesicles. J. Clin. Investig. 2016, 126, 1323–1336. [Google Scholar] [CrossRef]
- Rogers, M.A.; Buffolo, F.; Schlotter, F.; Atkins, S.K.; Lee, L.H.; Halu, A.; Blaser, M.C.; Tsolaki, E.; Higashi, H.; Luther, K.; et al. Annexin A1-Dependent Tethering Promotes Extracellular Vesicle Aggregation Revealed with Single-Extracellular Vesicle Analysis. Sci. Adv. 2020, 6, eabb1244. [Google Scholar] [CrossRef]
- Furmanik, M.; van Gorp, R.; Whitehead, M.; Ahmad, S.; Bordoloi, J.; Kapustin, A.; Schurgers, L.J.; Shanahan, C.M. Endoplasmic Reticulum Stress Mediates Vascular Smooth Muscle Cell Calcification via Increased Release of Grp78 (Glucose-Regulated Protein, 78 kDa)-Loaded Extracellular Vesicles. Arterioscler. Thromb. Vasc. Biol. 2021, 41, 898–914. [Google Scholar] [CrossRef] [PubMed]
- Bahram Sangani, N.; Gomes, A.R.; Curfs, L.M.G.; Reutelingsperger, C.P. The Role of Extracellular Vesicles during CNS Development. Prog. Neurobiol. 2021, 205, 102124. [Google Scholar] [CrossRef] [PubMed]
- Gomes, A.R.; Sangani, N.B.; Fernandes, T.G.; Diogo, M.M.; Curfs, L.M.G.; Reutelingsperger, C.P. Extracellular Vesicles in CNS Developmental Disorders. Int. J. Mol. Sci. 2020, 21, 9428. [Google Scholar] [CrossRef] [PubMed]
- Witwer, K.W.; Théry, C. Extracellular Vesicles or Exosomes? On Primacy, Precision, and Popularity Influencing a Choice of Nomenclature. J. Extracell. Vesicles 2019, 8, 1648167. [Google Scholar] [CrossRef] [PubMed]
- Théry, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G.K.; et al. Minimal Information for Studies of Extracellular Vesicles 2018 (MISEV2018): A Position Statement of the International Society for Extracellular Vesicles and Update of the MISEV2014 Guidelines. J. Extracell. Vesicles 2018, 7, 1535750. [Google Scholar] [CrossRef]
- Doyle, L.M.; Wang, M.Z. Overview of Extracellular Vesicles, Their Origin, Composition, Purpose, and Methods for Exosome Isolation and Analysis. Cells 2019, 8, 727. [Google Scholar] [CrossRef]
- Xu, X.; Lai, Y.; Hua, Z.-C. Apoptosis and Apoptotic Body: Disease Message and Therapeutic Target Potentials. Biosci. Rep. 2019, 39, BSR20180992. [Google Scholar] [CrossRef]
- Battistelli, M.; Falcieri, E. Apoptotic Bodies: Particular Extracellular Vesicles Involved in Intercellular Communication. Biology 2020, 9, 21. [Google Scholar] [CrossRef]
- Arandjelovic, S.; Ravichandran, K.S. Phagocytosis of Apoptotic Cells in Homeostasis. Nat. Immunol. 2015, 16, 907–917. [Google Scholar] [CrossRef]
- Cai, H.; Reinisch, K.; Ferro-Novick, S. Coats, Tethers, Rabs, and SNAREs Work Together to Mediate the Intracellular Destination of a Transport Vesicle. Dev. Cell 2007, 12, 671–682. [Google Scholar] [CrossRef]
- Tricarico, C.; Clancy, J.; D’Souza-Schorey, C. Biology and Biogenesis of Shed Microvesicles. Small GTPases 2017, 8, 220–232. [Google Scholar] [CrossRef] [PubMed]
- Rink, J.; Ghigo, E.; Kalaidzidis, Y.; Zerial, M. Rab Conversion as a Mechanism of Progression from Early to Late Endosomes. Cell 2005, 122, 735–749. [Google Scholar] [CrossRef] [PubMed]
- Poteryaev, D.; Datta, S.; Ackema, K.; Zerial, M.; Spang, A. Identification of the Switch in Early-to-Late Endosome Transition. Cell 2010, 141, 497–508. [Google Scholar] [CrossRef]
- Piper, R.C.; Katzmann, D.J. Biogenesis and Function of Multivesicular Bodies. Annu. Rev. Cell Dev. Biol. 2007, 23, 519–547. [Google Scholar] [CrossRef]
- Choezom, D.; Gross, J.C. Neutral Sphingomyelinase 2 Controls Exosome Secretion by Counteracting V-ATPase-Mediated Endosome Acidification. J. Cell Sci. 2022, 135, jcs259324. [Google Scholar] [CrossRef]
- Ostrowski, M.; Carmo, N.B.; Krumeich, S.; Fanget, I.; Raposo, G.; Savina, A.; Moita, C.F.; Schauer, K.; Hume, A.N.; Freitas, R.P.; et al. Rab27a and Rab27b Control Different Steps of the Exosome Secretion Pathway. Nat. Cell Biol. 2010, 12, 19–30. [Google Scholar] [CrossRef] [PubMed]
- Frankel, E.B.; Audhya, A. ESCRT-Dependent Cargo Sorting at Multivesicular Endosomes. Semin. Cell Dev. Biol. 2018, 74, 4–10. [Google Scholar] [CrossRef]
- Olmos, Y.; Carlton, J.G. The ESCRT Machinery: New Roles at New Holes. Curr. Opin. Cell Biol. 2016, 38, 1–11. [Google Scholar] [CrossRef]
- Stuffers, S.; Sem Wegner, C.; Stenmark, H.; Brech, A. Multivesicular Endosome Biogenesis in the Absence of ESCRTs. Traffic 2009, 10, 925–937. [Google Scholar] [CrossRef]
- Trajkovic, K.; Hsu, C.; Chiantia, S.; Rajendran, L.; Wenzel, D.; Wieland, F.; Schwille, P.; Brügger, B.; Simons, M. Ceramide Triggers Budding of Exosome Vesicles into Multivesicular Endosomes. Science 2008, 319, 1244–1247. [Google Scholar] [CrossRef]
- Larios, J.; Mercier, V.; Roux, A.; Gruenberg, J. ALIX- and ESCRT-III-Dependent Sorting of Tetraspanins to Exosomes. J. Cell Biol. 2020, 219, e201904113. [Google Scholar] [CrossRef] [PubMed]
- Leidal, A.M.; Huang, H.H.; Marsh, T.; Solvik, T.; Zhang, D.; Ye, J.; Kai, F.; Goldsmith, J.; Liu, J.Y.; Huang, Y.-H.; et al. The LC3-Conjugation Machinery Specifies the Loading of RNA-Binding Proteins into Extracellular Vesicles. Nat. Cell Biol. 2020, 22, 187–199. [Google Scholar] [CrossRef]
- Clarke, C.J.; Wu, B.X.; Hannun, Y.A. The Neutral Sphingomyelinase Family: Identifying Biochemical Connections. Adv. Enzym. Regul. 2011, 51, 51–58. [Google Scholar] [CrossRef]
- Xiang, H.; Jin, S.; Tan, F.; Xu, Y.; Lu, Y.; Wu, T. Physiological Functions and Therapeutic Applications of Neutral Sphingomyelinase and Acid Sphingomyelinase. Biomed. Pharmacother. 2021, 139, 111610. [Google Scholar] [CrossRef] [PubMed]
- Clarke, C.J.; Cloessner, E.A.; Roddy, P.L.; Hannun, Y.A. Neutral Sphingomyelinase 2 (nSMase2) Is the Primary Neutral Sphingomyelinase Isoform Activated by Tumour Necrosis Factor-α in MCF-7 Cells. Biochem. J. 2011, 435, 381–390. [Google Scholar] [CrossRef] [PubMed]
- Moylan, J.S.; Smith, J.D.; Wolf Horrell, E.M.; McLean, J.B.; Deevska, G.M.; Bonnell, M.R.; Nikolova-Karakashian, M.N.; Reid, M.B. Neutral Sphingomyelinase-3 Mediates TNF-Stimulated Oxidant Activity in Skeletal Muscle. Redox. Biol. 2014, 2, 910–920. [Google Scholar] [CrossRef] [PubMed]
- Tomiuk, S.; Zumbansen, M.; Stoffel, W. Characterization and Subcellular Localization of Murine and Human Magnesium-Dependent Neutral Sphingomyelinase. J. Biol. Chem. 2000, 275, 5710–5717. [Google Scholar] [CrossRef]
- Milhas, D.; Clarke, C.J.; Idkowiak-Baldys, J.; Canals, D.; Hannun, Y.A. Anterograde and Retrograde Transport of Neutral Sphingomyelinase-2 between the Golgi and the Plasma Membrane. Biochim. Biophys. Acta 2010, 1801, 1361–1374. [Google Scholar] [CrossRef]
- Hofmann, K.; Tomiuk, S.; Wolff, G.; Stoffel, W. Cloning and Characterization of the Mammalian Brain-Specific, Mg2+-Dependent Neutral Sphingomyelinase. Proc. Natl. Acad. Sci. USA 2000, 97, 5895–5900. [Google Scholar] [CrossRef]
- Wu, B.X.; Rajagopalan, V.; Roddy, P.L.; Clarke, C.J.; Hannun, Y.A. Identification and Characterization of Murine Mitochondria-Associated Neutral Sphingomyelinase (MA-nSMase), the Mammalian Sphingomyelin Phosphodiesterase 5. J. Biol. Chem. 2010, 285, 17993–18002. [Google Scholar] [CrossRef]
- Birbes, H.; El Bawab, S.; Hannun, Y.A.; Obeid, L.M. Selective Hydrolysis of a Mitochondrial Pool of Sphingomyelin Induces Apoptosis. FASEB J. 2001, 15, 2669–2679. [Google Scholar] [CrossRef] [PubMed]
- Fensome, A.C.; Josephs, M.; Katan, M.; Rodrigues-Lima, F. Biochemical Identification of a Neutral Sphingomyelinase 1 (NSM1)-like Enzyme as the Major NSM Activity in the DT40 B-Cell Line: Absence of a Role in the Apoptotic Response to Endoplasmic Reticulum Stress. Biochem. J 2002, 365, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Tani, M.; Hannun, Y.A. Analysis of Membrane Topology of Neutral Sphingomyelinase 2. FEBS Lett. 2007, 581, 1323–1328. [Google Scholar] [CrossRef]
- Airola, M.V.; Shanbhogue, P.; Shamseddine, A.A.; Guja, K.E.; Senkal, C.E.; Maini, R.; Bartke, N.; Wu, B.X.; Obeid, L.M.; Garcia-Diaz, M.; et al. Structure of Human nSMase2 Reveals an Interdomain Allosteric Activation Mechanism for Ceramide Generation. Proc. Natl. Acad. Sci. USA. 2017, 114, E5549–E5558. [Google Scholar] [CrossRef]
- Wu, B.X.; Clarke, C.J.; Matmati, N.; Montefusco, D.; Bartke, N.; Hannun, Y.A. Identification of Novel Anionic Phospholipid Binding Domains in Neutral Sphingomyelinase 2 with Selective Binding Preference. J. Biol. Chem. 2011, 286, 22362–22371. [Google Scholar] [CrossRef]
- Tepper, A.D.; Ruurs, P.; Wiedmer, T.; Sims, P.J.; Borst, J.; van Blitterswijk, W.J. Sphingomyelin Hydrolysis to Ceramide during the Execution Phase of Apoptosis Results from Phospholipid Scrambling and Alters Cell-Surface Morphology. J. Cell Biol. 2000, 150, 155–164. [Google Scholar] [CrossRef] [PubMed]
- Shanbhogue, P.; Hoffmann, R.M.; Airola, M.V.; Maini, R.; Hamelin, D.J.; Garcia-Diaz, M.; Burke, J.E.; Hannun, Y.A. The Juxtamembrane Linker in Neutral Sphingomyelinase-2 Functions as an Intramolecular Allosteric Switch That Activates the Enzyme. J. Biol. Chem. 2019, 294, 7488–7502. [Google Scholar] [CrossRef]
- Luberto, C.; Hassler, D.F.; Signorelli, P.; Okamoto, Y.; Sawai, H.; Boros, E.; Hazen-Martin, D.J.; Obeid, L.M.; Hannun, Y.A.; Smith, G.K. Inhibition of Tumor Necrosis Factor-Induced Cell Death in MCF7 by a Novel Inhibitor of Neutral Sphingomyelinase. J. Biol. Chem. 2002, 277, 41128–41139. [Google Scholar] [CrossRef]
- Nikolova-Karakashian, M.N.; Rozenova, K.A. Ceramide in Stress Response. Adv. Exp. Med. Biol. 2010, 688, 86–108. [Google Scholar]
- Stith, J.L.; Velazquez, F.N.; Obeid, L.M. Advances in Determining Signaling Mechanisms of Ceramide and Role in Disease. J. Lipid Res. 2019, 60, 913–918. [Google Scholar] [CrossRef]
- Kolmakova, A.; Kwiterovich, P.; Virgil, D.; Alaupovic, P.; Knight-Gibson, C.; Martin, S.F.; Chatterjee, S. Apolipoprotein C-I Induces Apoptosis in Human Aortic Smooth Muscle Cells via Recruiting Neutral Sphingomyelinase. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 264–269. [Google Scholar] [CrossRef] [PubMed]
- Liao, L.; Zhou, Q.; Song, Y.; Wu, W.; Yu, H.; Wang, S.; Chen, Y.; Ye, M.; Lu, L. Ceramide Mediates Ox-LDL-Induced Human Vascular Smooth Muscle Cell Calcification via p38 Mitogen-Activated Protein Kinase Signaling. PLoS ONE 2013, 8, e82379. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, N.S.; Goldschmidt-Arzi, M.; Sabanay, H.; Storch, J.; Levade, T.; Ribeiro, M.G.; Addadi, L.; Futerman, A.H. Accumulation of Ordered Ceramide-Cholesterol Domains in Farber Disease Fibroblasts. JIMD Rep. 2014, 12, 71–77. [Google Scholar] [PubMed]
- Kaltenegger, M.; Kremser, J.; Frewein, M.P.; Ziherl, P.; Bonthuis, D.J.; Pabst, G. Intrinsic Lipid Curvatures of Mammalian Plasma Membrane Outer Leaflet Lipids and Ceramides. Biochim. Biophys. Acta Biomembr. 2021, 1863, 183709. [Google Scholar] [CrossRef] [PubMed]
- Arenz, C.; Giannis, A. Synthesis of the First Selective Irreversible Inhibitor of Neutral Sphingomyelinase This Work Was Supported by Grants from the Fonds Der Chemischen Industrie. C.A. Is Grateful to the Land of Baden-Württemberg for a Scholarship from the Landesgraduiertenförderung. Angew. Chem. Int. Ed Engl. 2000, 39, 1440–1442. [Google Scholar]
- Verderio, C.; Gabrielli, M.; Giussani, P. Role of Sphingolipids in the Biogenesis and Biological Activity of Extracellular Vesicles. J. Lipid Res. 2018, 59, 1325–1340. [Google Scholar] [CrossRef]
- Poggio, M.; Hu, T.; Pai, C.-C.; Chu, B.; Belair, C.D.; Chang, A.; Montabana, E.; Lang, U.E.; Fu, Q.; Fong, L.; et al. Suppression of Exosomal PD-L1 Induces Systemic Anti-Tumor Immunity and Memory. Cell 2019, 177, 414–427.e13. [Google Scholar] [CrossRef]
- Kosaka, N.; Iguchi, H.; Yoshioka, Y.; Takeshita, F.; Matsuki, Y.; Ochiya, T. Secretory Mechanisms and Intercellular Transfer of microRNAs in Living Cells. J. Biol. Chem. 2010, 285, 17442–17452. [Google Scholar] [CrossRef]
- Yuyama, K.; Sun, H.; Mitsutake, S.; Igarashi, Y. Sphingolipid-Modulated Exosome Secretion Promotes Clearance of Amyloid-β by Microglia. J. Biol. Chem. 2012, 287, 10977–10989. [Google Scholar] [CrossRef]
- Li, J.; Liu, K.; Liu, Y.; Xu, Y.; Zhang, F.; Yang, H.; Liu, J.; Pan, T.; Chen, J.; Wu, M.; et al. Exosomes Mediate the Cell-to-Cell Transmission of IFN-α-Induced Antiviral Activity. Nat. Immunol. 2013, 14, 793–803. [Google Scholar] [CrossRef]
- Singh, R.; Pochampally, R.; Watabe, K.; Lu, Z.; Mo, Y.-Y. Exosome-Mediated Transfer of miR-10b Promotes Cell Invasion in Breast Cancer. Mol. Cancer 2014, 13, 256. [Google Scholar] [CrossRef] [PubMed]
- Asai, H.; Ikezu, S.; Tsunoda, S.; Medalla, M.; Luebke, J.; Haydar, T.; Wolozin, B.; Butovsky, O.; Kügler, S.; Ikezu, T. Depletion of Microglia and Inhibition of Exosome Synthesis Halt Tau Propagation. Nat. Neurosci. 2015, 18, 1584–1593. [Google Scholar] [CrossRef] [PubMed]
- Guo, B.B.; Bellingham, S.A.; Hill, A.F. The Neutral Sphingomyelinase Pathway Regulates Packaging of the Prion Protein into Exosomes. J. Biol. Chem. 2015, 290, 3455–3467. [Google Scholar] [CrossRef] [PubMed]
- Lang, J.K.; Young, R.F.; Ashraf, H.; Canty, J.M. Inhibiting Extracellular Vesicle Release from Human Cardiosphere Derived Cells with Lentiviral Knockdown of nSMase2 Differentially Effects Proliferation and Apoptosis in Cardiomyocytes, Fibroblasts and Endothelial Cells In Vitro. PLoS ONE 2016, 11, e0165926. [Google Scholar] [CrossRef]
- Dinkins, M.B.; Enasko, J.; Hernandez, C.; Wang, G.; Kong, J.; Helwa, I.; Liu, Y.; Terry, A.V., Jr.; Bieberich, E. Neutral Sphingomyelinase-2 Deficiency Ameliorates Alzheimer’s Disease Pathology and Improves Cognition in the 5XFAD Mouse. J. Neurosci. 2016, 36, 8653–8667. [Google Scholar] [CrossRef]
- Menck, K.; Sönmezer, C.; Worst, T.S.; Schulz, M.; Dihazi, G.H.; Streit, F.; Erdmann, G.; Kling, S.; Boutros, M.; Binder, C.; et al. Neutral Sphingomyelinases Control Extracellular Vesicles Budding from the Plasma Membrane. J. Extracell. Vesicles 2017, 6, 1378056. [Google Scholar] [CrossRef]
- Hitomi, K.; Okada, R.; Loo, T.M.; Miyata, K.; Nakamura, A.J.; Takahashi, A. DNA Damage Regulates Senescence-Associated Extracellular Vesicle Release via the Ceramide Pathway to Prevent Excessive Inflammatory Responses. Int. J. Mol. Sci. 2020, 21, 3720. [Google Scholar] [CrossRef]
- Aubin, I.; Adams, C.P.; Opsahl, S.; Septier, D.; Bishop, C.E.; Auge, N.; Salvayre, R.; Negre-Salvayre, A.; Goldberg, M.; Guénet, J.-L.; et al. A Deletion in the Gene Encoding Sphingomyelin Phosphodiesterase 3 (Smpd3) Results in Osteogenesis and Dentinogenesis Imperfecta in the Mouse. Nat. Genet. 2005, 37, 803–805. [Google Scholar] [CrossRef]
- Dickens, A.M.; Tovar-Y-Romo, L.B.; Yoo, S.-W.; Trout, A.L.; Bae, M.; Kanmogne, M.; Megra, B.; Williams, D.W.; Witwer, K.W.; Gacias, M.; et al. Astrocyte-Shed Extracellular Vesicles Regulate the Peripheral Leukocyte Response to Inflammatory Brain Lesions. Sci. Signal. 2017, 10, eaai7696. [Google Scholar] [CrossRef]
- Lecuyer, M.; Pathipati, P.; Faustino, J.; Vexler, Z.S. Neonatal Stroke Enhances Interaction of Microglia-Derived Extracellular Vesicles with Microglial Cells. Neurobiol. Dis. 2021, 157, 105431. [Google Scholar] [CrossRef]
- Rojas, C.; Sala, M.; Thomas, A.G.; Chaudhuri, A.D.; Yoo, S.-W.; Li, Z.; Dash, R.P.; Rais, R.; Haughey, N.J.; Nencka, R.; et al. A Novel and Potent Brain Penetrant Inhibitor of Extracellular Vesicle Release. Br. J. Pharmacol. 2019, 176, 3857–3870. [Google Scholar] [CrossRef]
- Tabatadze, N.; Savonenko, A.; Song, H.; Bandaru, V.V.R.; Chu, M.; Haughey, N.J. Inhibition of Neutral Sphingomyelinase-2 Perturbs Brain Sphingolipid Balance and Spatial Memory in Mice. J. Neurosci. Res. 2010, 88, 2940–2951. [Google Scholar] [CrossRef] [PubMed]
- Stoffel, W.; Hammels, I.; Jenke, B.; Schmidt-Soltau, I.; Niehoff, A. Neutral Sphingomyelinase 2 (SMPD3) Deficiency in Mice Causes Chondrodysplasia with Unimpaired Skeletal Mineralization. Am. J. Pathol. 2019, 189, 1831–1845. [Google Scholar] [CrossRef]
- Duivenvoorden, R.; Tang, J.; Cormode, D.P.; Mieszawska, A.J.; Izquierdo-Garcia, D.; Ozcan, C.; Otten, M.J.; Zaidi, N.; Lobatto, M.E.; van Rijs, S.M.; et al. A Statin-Loaded Reconstituted High-Density Lipoprotein Nanoparticle Inhibits Atherosclerotic Plaque Inflammation. Nat. Commun. 2014, 5, 3065. [Google Scholar] [CrossRef] [PubMed]
- Lameijer, M.; Binderup, T.; van Leent, M.M.T.; Senders, M.L.; Fay, F.; Malkus, J.; Sanchez-Gaytan, B.L.; Teunissen, A.J.P.; Karakatsanis, N.; Robson, P.; et al. Efficacy and Safety Assessment of a TRAF6-Targeted Nanoimmunotherapy in Atherosclerotic Mice and Non-Human Primates. Nat. Biomed. Eng. 2018, 2, 279–292. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
| Cell Type | Exosome Verification Method | Method to Demonstrate Role of nSMase2 | Examined Cargo of the Exosomes | Reference |
|---|---|---|---|---|
| Oli-neu | TEM | GW4869, spiroepoxide, siRNA | ProteoLipid Protein | [60] |
| HEK293 | WB (CD63) | GW4869, siRNA, overexpression | miR-16, miR-146a | [88] |
| Neuro2A | TEM, WB (Alix, Tsg101) | GW4869, siRNA | pro-Aβ fibrillogenesis activity | [89] |
| THP-1 | TEM, WB (CD63) | GW4869, spiroepoxide, shRNA | anti-viral activity | [90] |
| MDA-MB-231 | SEM, WB (CD63) | GW4869, overexpression | miR-106 | [91] |
| Primary human VSMCs | NTA, WB (CD9, CD63) | GW4869, spiroepoxide, siRNA | pro-calcifying activity | [16] |
| Primary murine microglia | IEM (Tsg101) | GW4869, siRNA | Tau46 | [92] |
| GT1-7 | TEM, WB (Tsg101, Flotillin-1) | GW4869, RNAi | Prion protein | [93] |
| Primary human cardiosphere-derived cells | TEM, NTA, WB (CD63, HSP70) | siRNA | pro-angiogenic and pro-survival activity | [94] |
| Primary mouse astrocytes | TNA, WB (Alix, Tsg101) | m-nSMase2fro/fro | Aβ oligomers | [95] |
| SKBR3 | TEM, NTA, WB (Alix, Tsg101, CD81) | GW4869, siRNA | Hsc70 | [96] |
| PC3 | TEM, NTA, WB (CD63) | CRISPR/Cas9 | PD-L1 | [87] |
| TIG-3 | TEM, NTA | siRNA, overexpression | none studied | [97] |
| HEK293T | TEM, WB (Alix, Tsg101, CD9) | GW4869, shRNA | LC3-II, SAFP, HNRNPK | [62] |
| Hela | NTA, WB (Alix, CD63, CD81, syntenin) | GW4869, siRNA | V-ATPase transmembrane subunit | [55] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pavlic, A.; Bahram Sangani, N.; Kerins, J.; Nicolaes, G.; Schurgers, L.; Reutelingsperger, C. Vascular Smooth Muscle Cell Neutral Sphingomyelinase 2 in the Release of Exosomes and Vascular Calcification. Int. J. Mol. Sci. 2022, 23, 9178. https://doi.org/10.3390/ijms23169178
Pavlic A, Bahram Sangani N, Kerins J, Nicolaes G, Schurgers L, Reutelingsperger C. Vascular Smooth Muscle Cell Neutral Sphingomyelinase 2 in the Release of Exosomes and Vascular Calcification. International Journal of Molecular Sciences. 2022; 23(16):9178. https://doi.org/10.3390/ijms23169178
Chicago/Turabian StylePavlic, Angelina, Nasim Bahram Sangani, Johanna Kerins, Gerry Nicolaes, Leon Schurgers, and Chris Reutelingsperger. 2022. "Vascular Smooth Muscle Cell Neutral Sphingomyelinase 2 in the Release of Exosomes and Vascular Calcification" International Journal of Molecular Sciences 23, no. 16: 9178. https://doi.org/10.3390/ijms23169178
APA StylePavlic, A., Bahram Sangani, N., Kerins, J., Nicolaes, G., Schurgers, L., & Reutelingsperger, C. (2022). Vascular Smooth Muscle Cell Neutral Sphingomyelinase 2 in the Release of Exosomes and Vascular Calcification. International Journal of Molecular Sciences, 23(16), 9178. https://doi.org/10.3390/ijms23169178