
TNF-α Affects Signature Cytokines of Th1 and Th17 T Cell Subsets through Differential Actions on TNFR1 and TNFR2

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
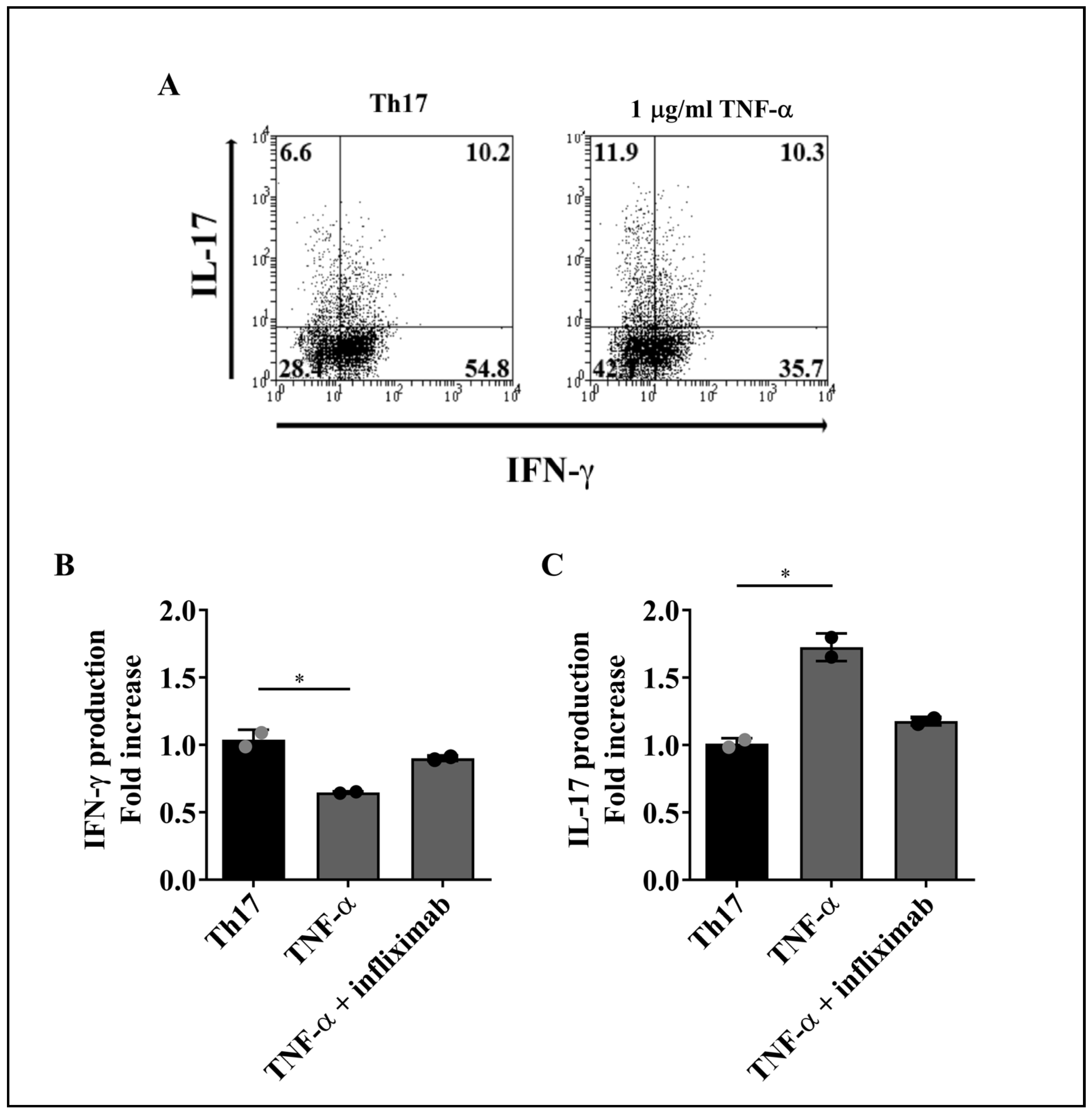
2.1. TNF-α Reduces IFN-γ and Increases IL-17 Production by Th1 and Th17 Cells
2.2. Reduced Production of IFN-γ by Th1 and Th17 Cells Is Not a Consequence of TNF-α-Mediated Apoptosis
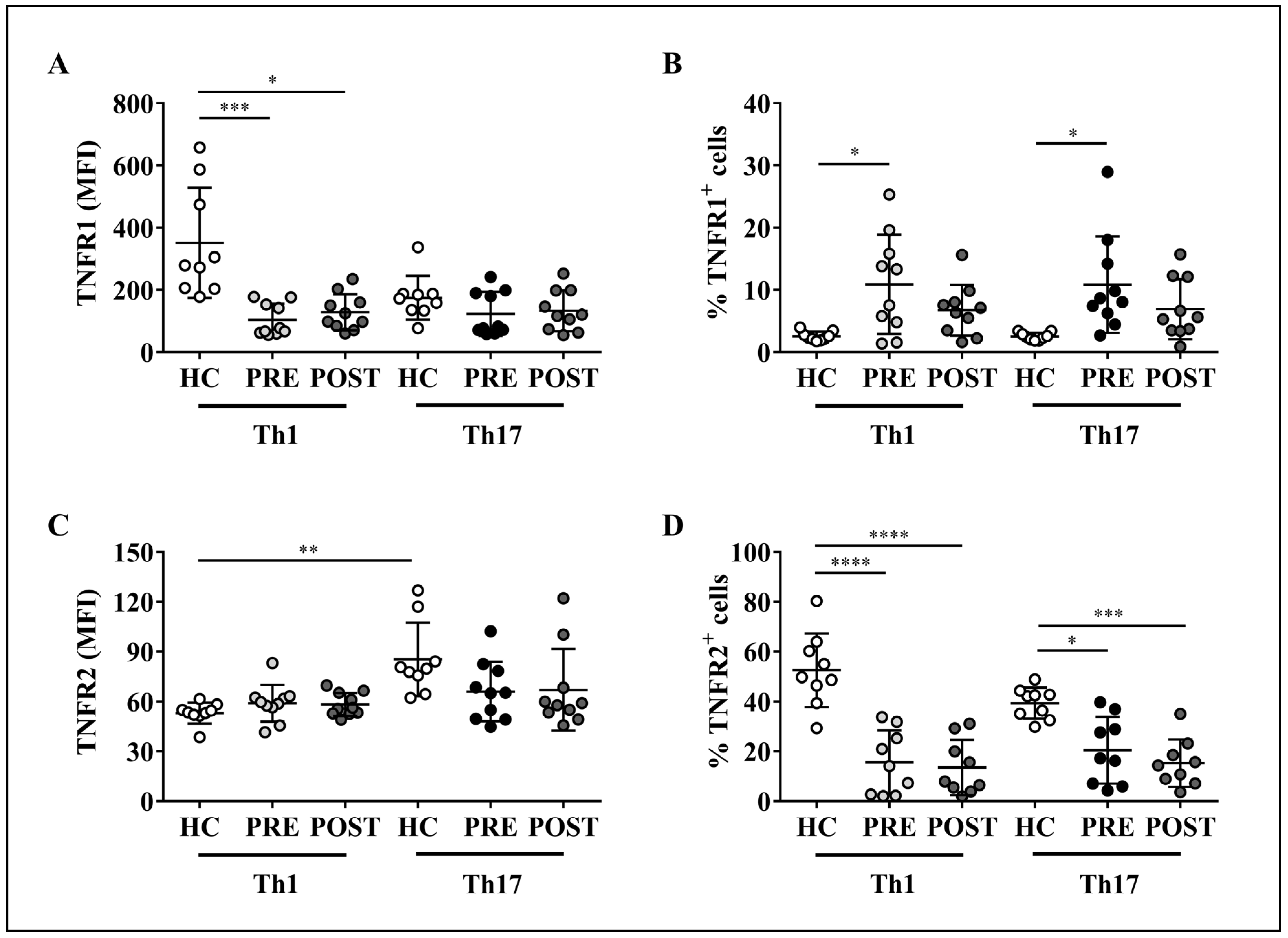
2.3. TNF-α Does Not Impact Membrane TNF-α Receptors Levels on Th1 and Th17 Cells
2.4. TNFR2 Mediates the Decrease in IFN-γ Production by Th1 and Th17 Cells
2.5. Engagement of TNFR1 and TNFR2 Enhances IL-17 Production by Th1 and Th17 Cells
2.6. Altered Expression of TNF-α Receptors on Th1 or Th17 Cells from RA Patients Is Not Modified by Adalimumab Treatment
3. Discussion
4. Materials and Methods
4.1. Healthy Donors and RA Patients
4.2. Buffy Coats, Blood Samples and PBMC
4.3. Cell Sorting
4.4. Cell Staining and Flow Cytometry for Transcription Factors and Intracellular Cytokines
4.5. Apoptosis Assay
4.6. Flow Cytometry for TNF-α Receptors
4.7. Blockade of TNF-α Receptors
4.8. Ex Vivo Staining of PBMCs from RA Patients
4.9. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Luckheeram, R.V.; Zhou, R.; Verma, A.D.; Xia, B. CD4(+)T cells: Differentiation and functions. Clin. Dev. Immunol. 2012, 2012, 925135. [Google Scholar] [CrossRef] [PubMed]
- Sallusto, F.; Lenig, D.; Mackay, C.R.; Lanzavecchia, A. Flexible programs of chemokine receptor expression on human polarized T helper 1 and 2 lymphocytes. J. Exp. Med. 1998, 187, 875–883. [Google Scholar] [CrossRef] [PubMed]
- Miossec, P.; Korn, T.; Kuchroo, V.K. Interleukin-17 and type 17 helper T cells. N. Engl. J. Med. 2009, 361, 888–898. [Google Scholar] [CrossRef]
- Hernandez-Santos, N.; Huppler, A.R.; Peterson, A.C.; Khader, S.A.; McKenna, K.C.; Gaffen, S.L. Th17 cells confer long-term adaptive immunity to oral mucosal Candida albicans infections. Mucosal Immunol. 2013, 6, 900–910. [Google Scholar] [CrossRef]
- Wu, Q.; Martin, R.J.; Rino, J.G.; Breed, R.; Torres, R.M.; Chu, H.W. IL-23-dependent IL-17 production is essential in neutrophil recruitment and activity in mouse lung defense against respiratory Mycoplasma pneumoniae infection. Microbes Infect. 2007, 9, 78–86. [Google Scholar] [CrossRef] [PubMed]
- Stadhouders, R.; Lubberts, E.; Hendriks, R.W. A cellular and molecular view of T helper 17 cell plasticity in autoimmunity. J. Autoimmun. 2018, 87, 1–15. [Google Scholar] [CrossRef]
- Smolen, J.S.; Aletaha, D.; Barton, A.; Burmester, G.R.; Emery, P.; Firestein, G.; Kavanaugh, A.; McInnes, I.B.; Solomon, D.H.; Strand, V.; et al. Rheumatoid arthritis. Nat. Rev. Dis. Primers. 2018, 4, 18001. [Google Scholar] [CrossRef]
- Firestein, G.S. Evolving concepts of rheumatoid arthritis. Nature 2003, 423, 356–361. [Google Scholar] [CrossRef]
- Dolhain, R.J.; van der Heiden, A.N.; ter Haar, N.T.; Breedveld, F.C.; Miltenburg, A.M. Shift toward T lymphocytes with a T helper 1 cytokine-secretion profile in the joints of patients with rheumatoid arthritis. Arthritis Rheum. 1996, 39, 1961–1969. [Google Scholar] [CrossRef]
- Jovanovic, D.V.; Di Battista, J.A.; Martel-Pelletier, J.; Jolicoeur, F.C.; He, Y.; Zhang, M.; Mineau, F.; Pelletier, J.-P. IL-17 stimulates the production and expression of proinflammatory cytokines, IL-beta and TNF-alpha, by human macrophages. J. Immunol. 1998, 160, 3513–3521. [Google Scholar]
- Chabaud, M.; Fossiez, F.; Taupin, J.L.; Miossec, P. Enhancing effect of IL-17 on IL-1-induced IL-6 and leukemia inhibitory factor production by rheumatoid arthritis synoviocytes and its regulation by Th2 cytokines. J. Immunol. 1998, 161, 409–414. [Google Scholar] [PubMed]
- Katz, Y.; Nadiv, O.; Beer, Y. Interleukin-17 enhances tumor necrosis factor alpha-induced synthesis of interleukins 1,6, and 8 in skin and synovial fibroblasts: A possible role as a "fine-tuning cytokine" in inflammation processes. Arthritis Rheum. 2001, 44, 2176–2184. [Google Scholar] [CrossRef]
- Chabaud, M.; Lubberts, E.; Joosten, L.; van Den Berg, W.; Miossec, P. IL-17 derived from juxta-articular bone and synovium contributes to joint degradation in rheumatoid arthritis. Arthritis Res. 2001, 3, 168–177. [Google Scholar] [CrossRef] [PubMed]
- Ronnelid, J.; Berg, L.; Rogberg, S.; Nilsson, A.; Albertsson, K.; Klareskog, L. Production of T cell cytokines at the single-cell level in patients with inflammatory arthritides: Enhanced activity in synovial fluid compared to blood. Br. J. Rheumat. 1998, 37, 7–14. [Google Scholar] [CrossRef]
- Milman, N.; Karsh, J.; Booth, R.A. Correlation of a multi-cytokine panel with clinical disease activity in patients with rheumatoid arthritis. Clin. Biochem. 2010, 43, 1309–1314. [Google Scholar] [CrossRef]
- Kim, E.Y.; Moudgil, K.D. Immunomodulation of autoimmune arthritis by pro-inflammatory cytokines. Cytokine 2017, 98, 87–96. [Google Scholar] [CrossRef]
- Harrington, L.E.; Hatton, R.D.; Mangan, P.R.; Turner, H.; Murphy, T.L.; Murphy, K.M.; Weaver, C.T. Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat. Immunol. 2005, 6, 1123–1132. [Google Scholar] [CrossRef]
- Annunziato, F.; Cosmi, L.; Santarlasci, V.; Maggi, L.; Liotta, F.; Mazzinghi, B.; Parente, E.; Fili, L.; Ferri, S.; Frosali, F.; et al. Phenotypic and functional features of human Th17 cells. J. Exp. Med. 2007, 204, 1849–1861. [Google Scholar] [CrossRef]
- Nistala, K.; Adams, S.; Cambrook, H.; Ursu, S.; Olivito, B.; de Jager, W.; Evans, J.G.; Cimaz, R.; Bajaj-Elliot, M.; Wadderburn, L.R. Th17 plasticity in human autoimmune arthritis is driven by the inflammatory environment. Proc. Natl. Acad. Sci. USA 2010, 107, 14751–14756. [Google Scholar] [CrossRef]
- Kennedy, A.; Fearon, U.; Veale, D.J.; Godson, C. Macrophages in synovial inflammation. Front. Immunol. 2011, 2, 52. [Google Scholar] [CrossRef]
- Brennan, F.M.; McInnes, I.B. Evidence that cytokines play a role in rheumatoid arthritis. J. Clin. Investig. 2008, 118, 3537–3545. [Google Scholar] [CrossRef] [PubMed]
- Feldmann, M.; Maini, R.N. Anti-TNF alpha therapy of rheumatoid arthritis: What have we learned? Annu. Rev. Immunol. 2001, 19, 163–196. [Google Scholar] [CrossRef] [PubMed]
- Faustman, D.; Davis, M. TNF receptor 2 pathway: Drug target for autoimmune diseases. Nat. Rev. Drug Discov. 2010, 9, 482–493. [Google Scholar] [CrossRef] [PubMed]
- MacEwan, D.J. TNF ligands and receptors—A matter of life and death. Br. J. Pharmacol. 2002, 135, 855–875. [Google Scholar] [CrossRef]
- Naude, P.J.; den Boer, J.A.; Luiten, P.G.; Eisel, U.L. Tumor necrosis factor receptor cross-talk. FEBS J. 2011, 278, 888–898. [Google Scholar] [CrossRef]
- Brennan, F.M.; Gibbons, D.L.; Mitchell, T.; Cope, A.P.; Maini, R.N.; Feldmann, M. Enhanced expression of tumor necrosis factor receptor mRNA and protein in mononuclear cells isolated from rheumatoid arthritis synovial joints. Eur. J. Immunol. 1992, 22, 1907–1912. [Google Scholar] [CrossRef]
- Raghav, S.K.; Gupta, B.; Agrawal, C.; Chaturvedi, V.P.; Das, H.R. Expression of TNF-alpha and related signaling molecules in the peripheral blood mononuclear cells of rheumatoid arthritis patients. Mediat. Inflamm. 2006, 2006, 12682. [Google Scholar] [CrossRef]
- Blüml, S.; Scheinecker, C.; Smolen, J.S.; Redlich, K. Targeting TNF receptors in rheumatoid arthritis. Int. Immunol. 2012, 24, 275–281. [Google Scholar] [CrossRef]
- Tada, Y.; Ho, A.; Koarada, S.; Morito, F.; Ushiyama, O.; Suzuki, N.; Kikuchi, Y.; Ohta, A.; Mak, T.W.; Nagasawa, K. Collagen-induced arthritis in TNF receptor-1-deficient mice: TNF receptor-2 can modulate arthritis in the absence of TNF receptor-1. Clin. Immunol. 2001, 99, 325–333. [Google Scholar] [CrossRef]
- Zhang, Y.H.; Heulsmann, A.; Tondravi, M.M.; Mukherjee, A.; Abu-Amer, Y. Tumor Necrosis Factor-a (TNF) Stimulates RANKL-induced Osteoclastogenesis via Coupling of TNF Type 1 Receptor and RANK Signaling Pathways. J. Biol. Chem. 2001, 276, 563–568. [Google Scholar] [CrossRef]
- Notley, C.A.; Inglis, J.J.; Alzabin, S.; McCann, F.E.; McNamee, K.E.; Williams, R.O. Blockade of tumor necrosis factor in collagen-induced arthritis reveals a novel immunoregulatory pathway for Th1 and Th17 cells. J. Exp. Med. 2008, 205, 2491–2497. [Google Scholar] [CrossRef] [PubMed]
- Kontoyiannis, D.; Pasparakis, M.; Pizarro, T.T.; Cominelli, F.; Kollias, G. Impaired On/Off Regulation of TNF Biosynthesis in Mice Lacking TNF AU-Rich Elements: Implications for Joint and Gut-Associated Immunopathologies. Immunity 1999, 10, 387–398. [Google Scholar] [CrossRef]
- Blüml, S.; Binder, N.B.; Niederreiter, B.; Polzer, K.; Hayer, S.; Tauber, S.; Schett, G.; Scheinecker, C.; Kollias, G.; Selzer, E.; et al. Antiinflammatory effects of tumor necrosis factor on hematopoietic cells in a murine model of erosive arthritis. Arthritis Rheum. 2010, 62, 1608–1619. [Google Scholar] [CrossRef]
- Annunziato, F.; Cosmi, L.; Liotta, F.; Maggi, E.; Romagnani, S. The phenotype of human Th17 cells and their precursors; the cytokines that mediate their differentiation and the role of Th17 cells in inflammation. Int. Immunol. 2008, 20, 1361–1368. [Google Scholar] [CrossRef] [PubMed]
- Cope, A.P.; Aderka, D.; Doherty, M.; Engelmann, H.; Gibbons, D.; Jones, A.C.; Engelmann, H.; Wallach, D.; Maini, R.N. Increased levels of soluble tumor necrosis factor receptors in the sera and synovial fluid of patients with rheumatic diseases. Arthritis Rheum. 1992, 35, 1160–1169. [Google Scholar] [CrossRef] [PubMed]
- Saxne, T.; Palladino, M.A.; Heinegård, D.; Talal, N.; Wollheim, F.A. Detection of tumor necrosis factor alpha but not tumor necrosis factor beta in rheumatoid arthritis synovial fluid and serum. Arthritis Rheum. 1988, 31, 1041–1045. [Google Scholar] [CrossRef]
- Aspalter, R.M.; Wolf, H.M.; Eibl, M.M. Chronic TNF-alpha exposure impairs TCR-signaling via TNF-RII but not TNF-RI. Cell. Immunol. 2005, 237, 55–67. [Google Scholar] [CrossRef]
- Acosta-Rodriguez, E.V.; Napolitani, G.; Lanzavecchia, A.; Sallusto, F. Interleukins 1β and 6 but not transforming growth factor-β are essential for the differentiation of interleukin 17-producing human T helper cells. Nat. Immunol. 2007, 8, 942–949. [Google Scholar] [CrossRef]
- Volpe, E.; Servant, N.; Zollinger, R.; Bogiatzi, S.I.; Hupé, P.; Barillot, E.; Soumelis, V. A critical function for transforming growth factor-β; interleukin 23 and proinflammatory cytokines in driving and modulating human TH-17 responses. Nat. Immunol. 2008, 9, 650–657. [Google Scholar] [CrossRef]
- Yang, L.; Anderson, D.E.; Baecher-Allan, C.; Hastings, W.D.; Bettelli, E.; Oukka, M.; Kuchroo, V.K.; Hafler, D.A. IL-21 and TGF-β are required for differentiation of human TH17 cells. Nature 2008, 454, 350–352. [Google Scholar] [CrossRef]
- Kamali, A.N.; Noorbakhsh, S.M.; Hamedifar, H.; Jadidi-Niaragh, F.; Yazdani, R.; Bautista, J.M.; Azizi, G. A role for Th1-like Th17 cells in the pathogenesis of inflammatory and autoimmune disorders. Mol. Immunol. 2019, 105, 107–115. [Google Scholar] [CrossRef] [PubMed]
- Aravena, O.; Pesce, B.; Soto, L.; Orrego, N.; Saburgo, F.; Wurmann, P.; Molina, M.C.; Alfaro, J.; Cuchacovich, M.; Aguillon, J.C.; et al. Anti-TNF therapy in patients with rheumatoid arthritis decreases Th1 and Th17 cell populations and expands IFN-γ-producing NK cell and regulatory T cell subsets. Immunobiology 2011, 216, 1256–1263. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.-Y.; Chen, Y.-M.; Chen, H.-H.; Hsieh, C.-W.; Lin, C.-C.; Lan, J.-L. Increasing levels of circulating Th17 cells and interleukin-17 in rheumatoid arthritis patients with an inadequate response to anti-TNF-α therapy. Arthritis Res. Ther. 2011, 13, R126. [Google Scholar] [CrossRef] [PubMed]
- Wright, H.L.; Bucknall, R.C.; Moots, R.J.; Edwards, S.W. Analysis of SF and plasma cytokines provides insights into the mechanisms of inflammatory arthritis and may predict response to therapy. Rheumatology 2012, 51, 451–459. [Google Scholar] [CrossRef]
- Schmidt, E.M.; Davies, M.; Mistry, P.; Green, P.; Giddins, G.; Feldmann, M.; Stoop, A.A.; Brennan, F.M. Selective blockade of tumor necrosis factor receptor I inhibits proinflammatory cytokine and chemokine production in human rheumatoid arthritis synovial membrane cell cultures. Arthritis Rheumatol. 2013, 65, 2262–2273. [Google Scholar] [CrossRef]
- Fischer, R.; Kontermann, R.E.; Pfizenmaier, K. Selective Targeting of TNF Receptors as a Novel Therapeutic Approach. Front. Cell Dev. Biol. 2020, 8, 401. [Google Scholar] [CrossRef]
- Grell, M.; Douni, E.; Wajant, H.; Lohden, M.; Clauss, M.; Maxeiner, B.; Georgopoulos, S.; Lesslauer, W.; Kollias, G.; Pfizenmaier, K.; et al. The transmembrane form of tumor-necrosis-factor is the prime activating ligand of the 80 kDa tumor-necrosis-factor receptor. Cell 1995, 83, 793–802. [Google Scholar] [CrossRef]
- Medvedev, A.E.; Espevik, T.; Ranges, G.; Sundan, A. Distinct roles of the two tumor necrosis factor (TNF) receptors in modulating TNF and lymphotoxin alpha effects. J. Biol. Chem. 1996, 271, 9778–9784. [Google Scholar] [CrossRef]
- Ware, C.F. Network communications: Lymphotoxins; LIGHT; and TNF. Annu. Rev. Immunol. 2005, 23, 787–819. [Google Scholar] [CrossRef]
- Calmon-Hamaty, F.; Combe, B.; Hahne, M.; Morel, J. Lymphotoxin α revisited: General features and implications in rheumatoid arthritis. Arthritis Res. Ther. 2011, 13, 232–236. [Google Scholar] [CrossRef]
- Rossol, M.; Schubert, K.; Meusch, U.; Schulz, A.; Biedermann, B.; Grosche, J.; Pierer, M.; Scholz, R.; Baerwald, C.; Thiel, A.; et al. Tumor necrosis factor receptor type I expression of CD4+ T cells in rheumatoid arthritis enables them to follow tumor necrosis factor gradients into the rheumatoid synovium. Arthritis Rheumatol. 2013, 65, 1468–1476. [Google Scholar] [CrossRef]
- Higuchi, M.; Aggarwal, B.B. TNF induces internalization of the p60 receptor and shedding of the p80 receptor. J. Immunol. 1994, 152, 3550–3558. [Google Scholar] [PubMed]
- Fischer, R.; Maier, O.; Naumer, M.; Krippner-Heidenreich, A.; Scheurich, P.; Pfizenmaier, K. Ligand-induced internalization of TNF receptor 2 mediated by a di-leucin motif is dispensable for activation of the NFκB pathway. Cell. Signal. 2011, 23, 161–170. [Google Scholar] [CrossRef] [PubMed]
- Mease, P.J. Adalimumab in the treatment of arthritis. Ther. Clin. Risk Manag. 2007, 3, 133–148. [Google Scholar] [CrossRef]
- Chanthaphavong, R.S.; Loughran, P.A.; Lee, T.Y.; Scott, M.J.; Billiar, T.R. A role for cGMP in inducible nitric-oxide synthase (iNOS)-induced tumor necrosis factor (TNF) α-converting enzyme (TACE/ADAM17) activation, translocation, and TNF receptor 1 (TNFR1) shedding in hepatocytes. J. Biol. Chem. 2012, 287, 35887–35898. [Google Scholar] [CrossRef]
- Aderka, D.; Engelmann, H.; Maor, Y.; Brakebusch, C.; Wallach, D. Stabilization of the bioactivity of tumor necrosis factor by its soluble receptors. J. Exp. Med. 1992, 175, 323–329. [Google Scholar] [CrossRef]
- Heilig, B.; Wermann, M.; Gallati, H.; Brockhaus, M.; Berke, B.; Egen, O.; Pezzutto, A.; Hustein, W. Elevated TNF receptor plasma concentrations in patients with rheumatoid arthritis. Clin. Investig. 1992, 70, 22–27. [Google Scholar] [CrossRef]
- Lopatnikova, J.A.; Golikova, E.A.; Shkaruba, N.S.; Sizikov, A.E.; Sennikov, S.V. Analysis of the levels of tumour necrosis factor (TNF), autoantibodies to TNF, and soluble TNF receptors in patients with rheumatoid arthritis. Scand. J. Rheumatol. 2013, 42, 429–432. [Google Scholar] [CrossRef]
- Yap, H.-Y.; Tee, S.Z.-Y.; Wong, M.M.-T.; Chow, S.-K.; Peh, S.-C.; Teow, S.-Y. Pathogenic Role of Immune Cells in Rheumatoid Arthritis: Implications in Clinical Treatment and Biomarker Development. Cells 2018, 7, 161. [Google Scholar] [CrossRef]
- van de Putte, L.B.A.; Atkins, C.; Malaise, M.; Sany, J.; Russell, A.S.; van Riel, P.L.C.M.; Settas, L.; Bijlsma, J.W.; Todesco, S.; Dougados, M.; et al. Efficacy and safety of adalimumab as monotherapy in patients with rheumatoid arthritis for whom previous disease modifying antirheumatic drug treatment has failed. Ann. Rheum. Dis. 2004, 63, 508–516. [Google Scholar] [CrossRef]
- Buch, M.H.; Bingham, S.J.; Bryer, D.; Emery, P. Long-term infliximab treatment in rheumatoid arthritis: Subsequent outcome of initial responders. Rheumatology 2007, 46, 1153–1156. [Google Scholar] [CrossRef] [PubMed]
- Alzabin, S.; Abraham, S.M.; Taher, T.E.; Palfreeman, A.; Hull, D.; McNamee, K.; Jawad, A.; Pathan, E.; Kinderlerer, A.; Taylor, P.C.; et al. Incomplete response of inflammatory arthritis to TNFα blockade is associated with the Th17 pathway. Ann. Rheum. Dis. 2012, 71, 1741–1748. [Google Scholar] [CrossRef]
- Faustman, D.; Davis, M. TNF Receptor 2 and Disease: Autoimmunity and Regenerative Medicine. Front. Immunol. 2013, 4, 478. [Google Scholar] [CrossRef] [PubMed]
- Aletaha, D.; Neogi, T.; Silman, A.J.; Funovits, J.; Felson, D.T.; Bingham, C.O., 3rd; Birnbaum, N.S.; Burmester, G.R.; Bykerk, V.P.; Cohen, M.D.; et al. Rheumatoid arthritis classification criteria: An American College of Rheumatology/European League Against Rheumatism collaborative initiative. Arthritis Rheum. 2010, 62, 2569–2581. [Google Scholar] [CrossRef] [PubMed]
- Campbell, J.D.M.; Foerster, A.; Lasmanowicz, V.; Niemöller, M.; Scheffold, A.; Fahrendorff, M.; Rauser, G.; Assenmacher, M.; Richter, A. Rapid detection; enrichment and propagation of specific T cell subsets based on cytokine secretion. Clin. Exp. Immunol. 2010, 163, 1–10. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pesce, B.; Ribeiro, C.H.; Larrondo, M.; Ramos, V.; Soto, L.; Catalán, D.; Aguillón, J.C. TNF-α Affects Signature Cytokines of Th1 and Th17 T Cell Subsets through Differential Actions on TNFR1 and TNFR2. Int. J. Mol. Sci. 2022, 23, 9306. https://doi.org/10.3390/ijms23169306
Pesce B, Ribeiro CH, Larrondo M, Ramos V, Soto L, Catalán D, Aguillón JC. TNF-α Affects Signature Cytokines of Th1 and Th17 T Cell Subsets through Differential Actions on TNFR1 and TNFR2. International Journal of Molecular Sciences. 2022; 23(16):9306. https://doi.org/10.3390/ijms23169306
Chicago/Turabian StylePesce, Bárbara, Carolina H. Ribeiro, Milton Larrondo, Verónica Ramos, Lilian Soto, Diego Catalán, and Juan Carlos Aguillón. 2022. "TNF-α Affects Signature Cytokines of Th1 and Th17 T Cell Subsets through Differential Actions on TNFR1 and TNFR2" International Journal of Molecular Sciences 23, no. 16: 9306. https://doi.org/10.3390/ijms23169306
APA StylePesce, B., Ribeiro, C. H., Larrondo, M., Ramos, V., Soto, L., Catalán, D., & Aguillón, J. C. (2022). TNF-α Affects Signature Cytokines of Th1 and Th17 T Cell Subsets through Differential Actions on TNFR1 and TNFR2. International Journal of Molecular Sciences, 23(16), 9306. https://doi.org/10.3390/ijms23169306