
Involvement of Ferroptosis in Diabetes-Induced Liver Pathology

 , ,
, ,
Abstract
:
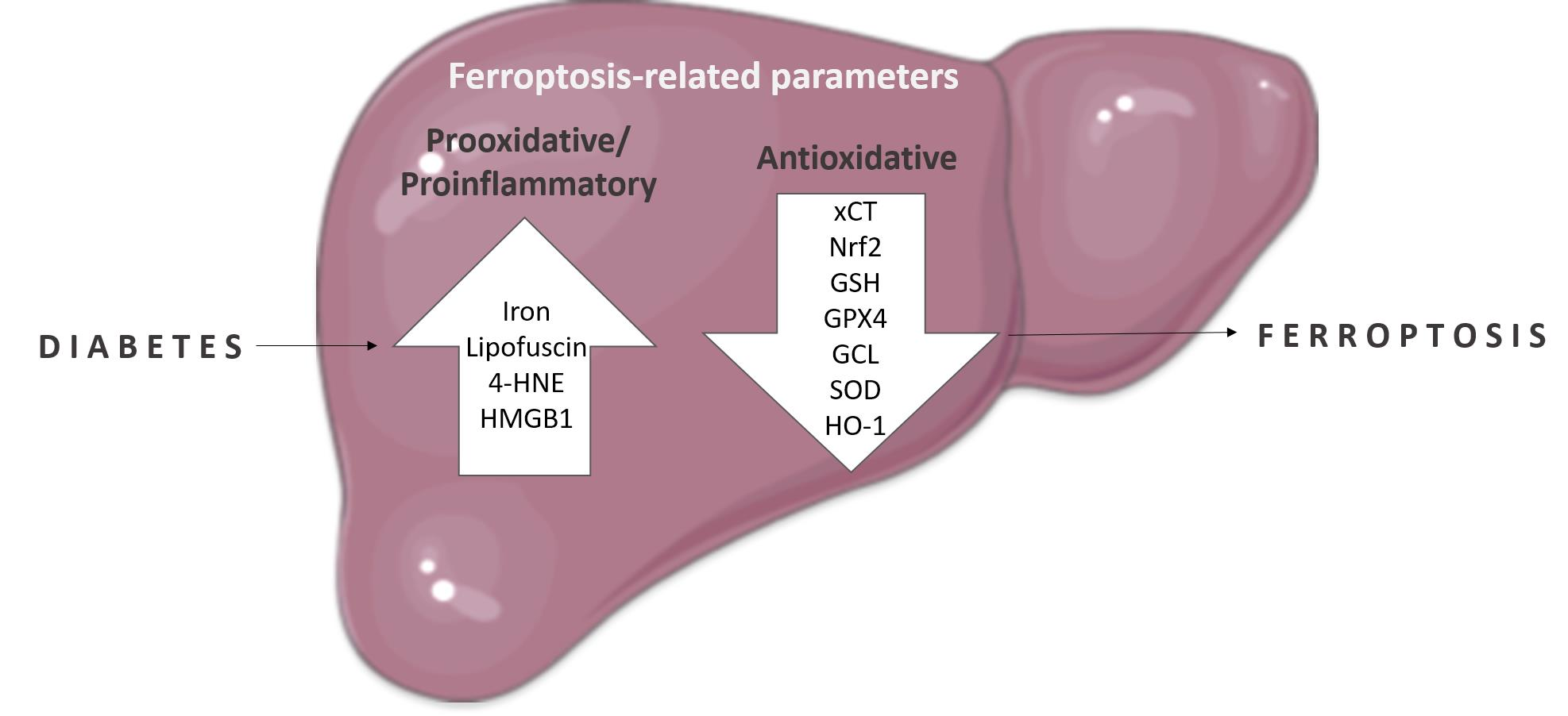
1. Introduction
2. Results
2.1. Fer-1 Attenuates Diabetes-Induced Liver Damage
2.2. Fer-1 Weakens Lipid Peroxidation in Diabetic Liver
2.3. Fer-1 Improves Diabetes-Induced Attenuation of GSH-Related Antioxidant Defense in the Liver
2.4. Fer-1 Re-Establishes Diabetes-Induced Disturbances in Hepatic Nrf2 Signaling
2.5. Fer-1 Abrogates Diabetes-Induced Activation of HMGB1 and Increase in Inflammatory Cytokines
3. Discussion
4. Materials and Methods
4.1. Experimental Design
4.2. Biochemical Serum Analysis
4.3. Microscopic Examination
4.3.1. Histological, Morphometric, and Stereological Analyses
4.3.2. Iron Staining
4.3.3. Lipofuscin Detection
4.3.4. Immunohistochemistry
4.4. Analysis of GSH Content and Activities of Antioxidative Defense Enzymes
4.5. SDS-Polyacrylamide Gel Electrophoresis (PAGE) and Western Blot Analysis
4.6. ELISA Assay for Measurement of TNF-α and IL-6
4.7. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bell, D.S.; Allbright, E. The multifaceted associations of hepatobiliary disease and diabetes. Endocr. Pract. Off. J. Am. Coll. Endocrinol. Am. Assoc. Clin. Endocrinol. 2007, 13, 300–312. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, J.; Nazratun Nafizah, A.H.; Zariyantey, A.H.; Budin, S.B. Mechanisms of diabetes-induced liver damage: The role of oxidative stress and inflammation. Sultan Qaboos Univ. Med. J. 2016, 16, e132–e141. [Google Scholar] [CrossRef] [PubMed]
- Marangiello, R.; Giorgetti, R. A case of glycogenosis in a patient with insulin dependent diabetes. Minerva Pediatrica 1996, 48, 279–281. [Google Scholar] [PubMed]
- Chatila, R.; West, A.B. Hepatomegaly and abnormal liver tests due to glycogenosis in adults with diabetes. Medicine 1996, 75, 327–333. [Google Scholar] [CrossRef] [PubMed]
- Luedde, T.; Kaplowitz, N.; Schwabe, R.F. Cell death and cell death responses in liver disease: Mechanisms and clinical relevance. Gastroenterology 2014, 147, 765–783.e764. [Google Scholar] [CrossRef] [PubMed]
- Grigorov, I.; Bogojevic, D.; Jovanovic, S.; Petrovic, A.; Ivanovic-Matic, S.; Zolotarevski, L.; Poznanovic, G.; Martinovic, V. Hepatoprotective effects of melatonin against pronecrotic cellular events in streptozotocin-induced diabetic rats. J. Physiol. Biochem. 2014, 70, 441–450. [Google Scholar] [CrossRef] [PubMed]
- Seven, A.; Guzel, S.; Seymen, O.; Civelek, S.; Bolayirli, M.; Uncu, M.; Burcak, G. Effects of vitamin e supplementation on oxidative stress in streptozotocin induced diabetic rats: Investigation of liver and plasma. Yonsei Med. J. 2004, 45, 703–710. [Google Scholar] [CrossRef]
- Petrovic, A.; Bogojevic, D.; Korac, A.; Golic, I.; Jovanovic-Stojanov, S.; Martinovic, V.; Ivanovic-Matic, S.; Stevanovic, J.; Poznanovic, G.; Grigorov, I. Oxidative stress-dependent contribution of hmgb1 to the interplay between apoptosis and autophagy in diabetic rat liver. J. Physiol. Biochem. 2017, 73, 511–521. [Google Scholar] [CrossRef]
- Johansen, J.S.; Harris, A.K.; Rychly, D.J.; Ergul, A. Oxidative stress and the use of antioxidants in diabetes: Linking basic science to clinical practice. Cardiovasc. Diabetol. 2005, 4, 5. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef]
- Feng, X.; Wang, S.; Sun, Z.; Dong, H.; Yu, H.; Huang, M.; Gao, X. Ferroptosis enhanced diabetic renal tubular injury via hif-1alpha/ho-1 pathway in db/db mice. Front. Endocrinol. 2021, 12, 626390. [Google Scholar] [CrossRef]
- Kim, S.; Kang, S.W.; Joo, J.; Han, S.H.; Shin, H.; Nam, B.Y.; Park, J.; Yoo, T.H.; Kim, G.; Lee, P.; et al. Characterization of ferroptosis in kidney tubular cell death under diabetic conditions. Cell Death Dis. 2021, 12, 160. [Google Scholar] [CrossRef]
- Li, S.; Zheng, L.; Zhang, J.; Liu, X.; Wu, Z. Inhibition of ferroptosis by up-regulating nrf2 delayed the progression of diabetic nephropathy. Free. Radic. Biol. Med. 2021, 162, 435–449. [Google Scholar] [CrossRef]
- Hao, L.; Mi, J.; Song, L.; Guo, Y.; Li, Y.; Yin, Y.; Zhang, C. Slc40a1 mediates ferroptosis and cognitive dysfunction in type 1 diabetes. Neuroscience 2021, 463, 216–226. [Google Scholar] [CrossRef]
- Ma, H.; Wang, X.; Zhang, W.; Li, H.; Zhao, W.; Sun, J.; Yang, M. Melatonin suppresses ferroptosis induced by high glucose via activation of the nrf2/ho-1 signaling pathway in type 2 diabetic osteoporosis. Oxid. Med. Cell. Longev. 2020, 2020, 9067610. [Google Scholar] [CrossRef]
- Luo, E.F.; Li, H.X.; Qin, Y.H.; Qiao, Y.; Yan, G.L.; Yao, Y.Y.; Li, L.Q.; Hou, J.T.; Tang, C.C.; Wang, D. Role of ferroptosis in the process of diabetes-induced endothelial dysfunction. World J. Diabetes 2021, 12, 124–137. [Google Scholar] [CrossRef]
- Zhu, Z.; Duan, P.; Song, H.; Zhou, R.; Chen, T. Downregulation of circular rna psen1 ameliorates ferroptosis of the high glucose treated retinal pigment epithelial cells via mir-200b-3p/cofilin-2 axis. Bioengineered 2021, 12, 12555–12567. [Google Scholar] [CrossRef]
- Stancic, A.; Saksida, T.; Markelic, M.; Vucetic, M.; Grigorov, I.; Martinovic, V.; Gajic, D.; Ivanovic, A.; Velickovic, K.; Savic, N.; et al. Ferroptosis as a novel determinant of beta-cell death in diabetic conditions. Oxid. Med. Cell. Longev. 2022, 2022, 3873420. [Google Scholar] [CrossRef]
- Zilka, O.; Shah, R.; Li, B.; Friedmann Angeli, J.P.; Griesser, M.; Conrad, M.; Pratt, D.A. On the mechanism of cytoprotection by ferrostatin-1 and liproxstatin-1 and the role of lipid peroxidation in ferroptotic cell death. ACS Cent. Sci. 2017, 3, 232–243. [Google Scholar] [CrossRef]
- Martin-Sanchez, D.; Ruiz-Andres, O.; Poveda, J.; Carrasco, S.; Cannata-Ortiz, P.; Sanchez-Niño, M.D.; Ruiz Ortega, M.; Egido, J.; Linkermann, A.; Ortiz, A.; et al. Ferroptosis, but not necroptosis, is important in nephrotoxic folic acid-induced aki. J. Am. Soc. Nephrol. 2017, 28, 218–229. [Google Scholar] [CrossRef]
- Jiang, X.; Stockwell, B.R.; Conrad, M. Ferroptosis: Mechanisms, biology and role in disease. Nat. Rev. Mol. Cell Biol. 2021, 22, 266–282. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Feng, Y.; Li, H.; Chen, X.; Wang, G.; Xu, S.; Li, Y.; Zhao, L. Ferrostatin-1 alleviates lipopolysaccharide-induced acute lung injury via inhibiting ferroptosis. Cell. Mol. Biol. Lett. 2020, 25, 10. [Google Scholar] [CrossRef] [PubMed]
- Yang, N.; Shang, Y. Ferrostatin-1 and 3-methyladenine ameliorate ferroptosis in ova-induced asthma model and in il-13-challenged beas-2b cells. Oxid. Med. Cell. Longev. 2022, 2022, 9657933. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; An, P.; Xie, E.; Wu, Q.; Fang, X.; Gao, H.; Zhang, Z.; Li, Y.; Wang, X.; Zhang, J.; et al. Characterization of ferroptosis in murine models of hemochromatosis. Hepatology 2017, 66, 449–465. [Google Scholar] [CrossRef]
- Aboulmagd, Y.M.; El-Bahy, A.A.Z.; Menze, E.T.; Azab, S.S.; El-Demerdash, E. Role of linagliptin in preventing the pathological progression of hepatic fibrosis in high fat diet and streptozotocin-induced diabetic obese rats. Eur. J. Pharmacol. 2020, 881, 173224. [Google Scholar] [CrossRef]
- Bataller, R.; Brenner, D.A. Liver fibrosis. J. Clin. Investig. 2005, 115, 209–218. [Google Scholar] [CrossRef]
- Lomonaco, R.; Godinez Leiva, E.; Bril, F.; Shrestha, S.; Mansour, L.; Budd, J.; Portillo Romero, J.; Schmidt, S.; Chang, K.L.; Samraj, G.; et al. Advanced liver fibrosis is common in patients with type 2 diabetes followed in the outpatient setting: The need for systematic screening. Diabetes Care 2021, 44, 399–406. [Google Scholar] [CrossRef]
- Miyaoka, Y.; Miyajima, A. To divide or not to divide: Revisiting liver regeneration. Cell Div. 2013, 8, 8. [Google Scholar] [CrossRef]
- Martin, N.C.; McCullough, C.T.; Bush, P.G.; Sharp, L.; Hall, A.C.; Harrison, D.J. Functional analysis of mouse hepatocytes differing in DNA content: Volume, receptor expression, and effect of ifngamma. J. Cell. Physiol. 2002, 191, 138–144. [Google Scholar] [CrossRef]
- Yamada, T.; Sogawa, K.; Kim, J.K.; Izumi, K.; Suzuki, Y.; Muramatsu, Y.; Sumida, T.; Hamakawa, H.; Matsumoto, K. Increased polyploidy, delayed mitosis and reduced protein phosphatase-1 activity associated with excess copper in the long evans cinnamon rat. Res. Commun. Mol. Pathol. Pharmacol. 1998, 99, 283–304. [Google Scholar]
- Gorla, G.R.; Malhi, H.; Gupta, S. Polyploidy associated with oxidative injury attenuates proliferative potential of cells. J. Cell Sci. 2001, 114, 2943–2951. [Google Scholar] [CrossRef]
- Lucchesi, A.N.; Freitas, N.T.; Cassettari, L.L.; Marques, S.F.; Spadella, C.T. Diabetes mellitus triggers oxidative stress in the liver of alloxan-treated rats: A mechanism for diabetic chronic liver disease. Acta Cir. Bras. 2013, 28, 502–508. [Google Scholar] [CrossRef]
- Soto-Urquieta, M.G.; Lopez-Briones, S.; Perez-Vazquez, V.; Saavedra-Molina, A.; Gonzalez-Hernandez, G.A.; Ramirez-Emiliano, J. Curcumin restores mitochondrial functions and decreases lipid peroxidation in liver and kidneys of diabetic db/db mice. Biol. Res. 2014, 47, 74. [Google Scholar] [CrossRef]
- Ahmed Mobasher, M.; Galal El-Tantawi, H.; Samy El-Said, K. Metformin ameliorates oxidative stress induced by diabetes mellitus and hepatocellular carcinoma in rats. Rep. Biochem. Mol. Biol. 2020, 9, 115–128. [Google Scholar] [CrossRef]
- Halliwell, B. Reactive species and antioxidants. Redox biology is a fundamental theme of aerobic life. Plant Physiol. 2006, 141, 312–322. [Google Scholar] [CrossRef]
- Altamura, S.; Mudder, K.; Schlotterer, A.; Fleming, T.; Heidenreich, E.; Qiu, R.; Hammes, H.P.; Nawroth, P.; Muckenthaler, M.U. Iron aggravates hepatic insulin resistance in the absence of inflammation in a novel db/db mouse model with iron overload. Mol. Metab. 2021, 51, 101235. [Google Scholar] [CrossRef]
- Lee, H.; Zandkarimi, F.; Zhang, Y.; Meena, J.K.; Kim, J.; Zhuang, L.; Tyagi, S.; Ma, L.; Westbrook, T.F.; Steinberg, G.R.; et al. Energy-stress-mediated ampk activation inhibits ferroptosis. Nat. Cell Biol. 2020, 22, 225–234. [Google Scholar] [CrossRef]
- Catala, A. An overview of lipid peroxidation with emphasis in outer segments of photoreceptors and the chemiluminescence assay. Int. J. Biochem. Cell Biol. 2006, 38, 1482–1495. [Google Scholar] [CrossRef]
- Goedeke, L.; Bates, J.; Vatner, D.F.; Perry, R.J.; Wang, T.; Ramirez, R.; Li, L.; Ellis, M.W.; Zhang, D.; Wong, K.E.; et al. Acetyl-coa carboxylase inhibition reverses nafld and hepatic insulin resistance but promotes hypertriglyceridemia in rodents. Hepatology 2018, 68, 2197–2211. [Google Scholar] [CrossRef]
- Seibt, T.M.; Proneth, B.; Conrad, M. Role of gpx4 in ferroptosis and its pharmacological implication. Free. Radic. Biol. Med. 2019, 133, 144–152. [Google Scholar] [CrossRef]
- Yant, L.J.; Ran, Q.; Rao, L.; Van Remmen, H.; Shibatani, T.; Belter, J.G.; Motta, L.; Richardson, A.; Prolla, T.A. The selenoprotein gpx4 is essential for mouse development and protects from radiation and oxidative damage insults. Free. Radic. Biol. Med. 2003, 34, 496–502. [Google Scholar] [CrossRef]
- Yang, W.S.; SriRamaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B.; et al. Regulation of ferroptotic cancer cell death by gpx4. Cell 2014, 156, 317–331. [Google Scholar] [CrossRef] [PubMed]
- Carlson, B.A.; Tobe, R.; Yefremova, E.; Tsuji, P.A.; Hoffmann, V.J.; Schweizer, U.; Gladyshev, V.N.; Hatfield, D.L.; Conrad, M. Glutathione peroxidase 4 and vitamin e cooperatively prevent hepatocellular degeneration. Redox Biol. 2016, 9, 22–31. [Google Scholar] [CrossRef] [PubMed]
- Conrad, M.; Schneider, M.; Seiler, A.; Bornkamm, G.W. Physiological role of phospholipid hydroperoxide glutathione peroxidase in mammals. Biol. Chem. 2007, 388, 1019–1025. [Google Scholar] [CrossRef]
- Imai, H.; Matsuoka, M.; Kumagai, T.; Sakamoto, T.; Koumura, T. Lipid peroxidation-dependent cell death regulated by gpx4 and ferroptosis. Curr. Top. Microbiol. Immunol. 2017, 403, 143–170. [Google Scholar]
- Bao, Y.; Jemth, P.; Mannervik, B.; Williamson, G. Reduction of thymine hydroperoxide by phospholipid hydroperoxide glutathione peroxidase and glutathione transferases. FEBS Lett. 1997, 410, 210–212. [Google Scholar] [CrossRef]
- Bartsch, H.; Nair, J.; Owen, R.W. Exocyclic DNA adducts as oxidative stress markers in colon carcinogenesis: Potential role of lipid peroxidation, dietary fat and antioxidants. Biol. Chem. 2002, 383, 915–921. [Google Scholar] [CrossRef]
- Traverso, N.; Menini, S.; Odetti, P.; Pronzato, M.A.; Cottalasso, D.; Marinari, U.M. Diabetes impairs the enzymatic disposal of 4-hydroxynonenal in rat liver. Free Radic. Biol. Med. 2002, 32, 350–359. [Google Scholar] [CrossRef]
- Gao, Z.; Sui, J.; Fan, R.; Qu, W.; Dong, X.; Sun, D. Emodin protects against acute pancreatitis-associated lung injury by inhibiting nlpr3 inflammasome activation via nrf2/ho-1 signaling. Drug Des. Dev. Ther. 2020, 14, 1971–1982. [Google Scholar] [CrossRef]
- Yang, B.; Bai, Y.; Yin, C.; Qian, H.; Xing, G.; Wang, S.; Li, F.; Bian, J.; Aschner, M.; Lu, R. Activation of autophagic flux and the nrf2/are signaling pathway by hydrogen sulfide protects against acrylonitrile-induced neurotoxicity in primary rat astrocytes. Arch. Toxicol. 2018, 92, 2093–2108. [Google Scholar] [CrossRef]
- He, C.H.; Gong, P.; Hu, B.; Stewart, D.; Choi, M.E.; Choi, A.M.; Alam, J. Identification of activating transcription factor 4 (atf4) as an nrf2-interacting protein. Implication for heme oxygenase-1 gene regulation. J. Biol. Chem. 2001, 276, 20858–20865. [Google Scholar] [CrossRef] [PubMed]
- Keum, Y.S. Regulation of nrf2-mediated phase ii detoxification and anti-oxidant genes. Biomol. Ther. 2012, 20, 144–151. [Google Scholar] [CrossRef] [PubMed]
- Montes, S.; Juarez-Rebollar, D.; Nava-Ruiz, C.; Sanchez-Garcia, A.; Heras-Romero, Y.; Rios, C.; Mendez-Armenta, M. Immunohistochemical study of nrf2-antioxidant response element as indicator of oxidative stress induced by cadmium in developing rats. Oxid. Med. Cell. Longev. 2015, 2015, 570650. [Google Scholar] [CrossRef] [PubMed]
- Behl, T.; Kaur, I.; Sehgal, A.; Sharma, E.; Kumar, A.; Grover, M.; Bungau, S. Unfolding nrf2 in diabetes mellitus. Mol. Biol. Rep. 2021, 48, 927–939. [Google Scholar] [CrossRef]
- David, J.A.; Rifkin, W.J.; Rabbani, P.S.; Ceradini, D.J. The nrf2/keap1/are pathway and oxidative stress as a therapeutic target in type ii diabetes mellitus. J. Diabetes Res. 2017, 2017, 4826724. [Google Scholar] [CrossRef]
- Rappaport, A.M.; Borowy, Z.J.; Lougheed, W.M.; Lotto, W.N. Subdivision of hexagonal liver lobules into a structural and functional unit; role in hepatic physiology and pathology. Anat. Rec. 1954, 119, 11–33. [Google Scholar] [CrossRef]
- Jin, X.; Gong, L.; Peng, Y.; Li, L.; Liu, G. Enhancer-bound nrf2 licenses hif-1alpha transcription under hypoxia to promote cisplatin resistance in hepatocellular carcinoma cells. Aging 2020, 13, 364–375. [Google Scholar] [CrossRef]
- Potteti, H.R.; Noone, P.M.; Tamatam, C.R.; Ankireddy, A.; Noel, S.; Rabb, H.; Reddy, S.P. Nrf2 mediates hypoxia-inducible hif1alpha activation in kidney tubular epithelial cells. Am. J. Physiol. Ren. Physiol. 2021, 320, F464–F474. [Google Scholar] [CrossRef]
- Soto-Gutierrez, A.; Gough, A.; Vernetti, L.A.; Taylor, D.L.; Monga, S.P. Pre-clinical and clinical investigations of metabolic zonation in liver diseases: The potential of microphysiology systems. Exp. Biol. Med. 2017, 242, 1605–1616. [Google Scholar] [CrossRef]
- Cunningham, R.P.; Porat-Shliom, N. Liver zonation—Revisiting old questions with new technologies. Front. Physiol. 2021, 12, 732929. [Google Scholar] [CrossRef]
- Lu, S.C. Regulation of glutathione synthesis. Mol. Asp. Med. 2009, 30, 42–59. [Google Scholar] [CrossRef]
- Chen, Y.; Shertzer, H.G.; Schneider, S.N.; Nebert, D.W.; Dalton, T.P. Glutamate cysteine ligase catalysis: Dependence on atp and modifier subunit for regulation of tissue glutathione levels. J. Biol. Chem. 2005, 280, 33766–33774. [Google Scholar] [CrossRef]
- Yang, Y.; Dieter, M.Z.; Chen, Y.; Shertzer, H.G.; Nebert, D.W.; Dalton, T.P. Initial characterization of the glutamate-cysteine ligase modifier subunit gclm(−/−) knockout mouse. Novel model system for a severely compromised oxidative stress response. J. Biol. Chem. 2002, 277, 49446–49452. [Google Scholar] [CrossRef]
- Kubo, Y.; Wruck, C.J.; Fragoulis, A.; Drescher, W.; Pape, H.C.; Lichte, P.; Fischer, H.; Tohidnezhad, M.; Hildebrand, F.; Pufe, T.; et al. Role of nrf2 in fracture healing: Clinical aspects of oxidative stress. Calcif. Tissue Int. 2019, 105, 341–352. [Google Scholar] [CrossRef]
- Shan, Y.; Li, J.; Zhu, A.; Kong, W.; Ying, R.; Zhu, W. Ginsenoside rg3 ameliorates acute pancreatitis by activating the nrf2/ho1mediated ferroptosis pathway. Int. J. Mol. Med. 2022, 50, 89. [Google Scholar] [CrossRef]
- Chen, R.; Kang, R.; Tang, D. The mechanism of hmgb1 secretion and release. Exp. Mol. Med. 2022, 54, 91–102. [Google Scholar] [CrossRef]
- Yu, Y.; Tang, D.; Kang, R. Oxidative stress-mediated hmgb1 biology. Front. Physiol. 2015, 6, 93. [Google Scholar] [CrossRef]
- Wang, Y.; Zhong, J.; Zhang, X.; Liu, Z.; Yang, Y.; Gong, Q.; Ren, B. The role of hmgb1 in the pathogenesis of type 2 diabetes. J. Diabetes Res. 2016, 2016, 2543268. [Google Scholar] [CrossRef]
- Wen, Q.; Liu, J.; Kang, R.; Zhou, B.; Tang, D. The release and activity of hmgb1 in ferroptosis. Biochem. Biophys. Res. Commun. 2019, 510, 278–283. [Google Scholar] [CrossRef]
- Wu, Y.; Zhao, Y.; Yang, H.Z.; Wang, Y.J.; Chen, Y. Hmgb1 regulates ferroptosis through nrf2 pathway in mesangial cells in response to high glucose. Biosci. Rep. 2021, 41, BSR20202924. [Google Scholar] [CrossRef]
- Ye, F.; Chai, W.; Xie, M.; Yang, M.; Yu, Y.; Cao, L.; Yang, L. Hmgb1 regulates erastin-induced ferroptosis via ras-jnk/p38 signaling in hl-60/nras(q61l) cells. Am. J. Cancer Res. 2019, 9, 730–739. [Google Scholar] [PubMed]
- Velickovic, N.; Teofilovic, A.; Ilic, D.; Djordjevic, A.; Vojnovic Milutinovic, D.; Petrovic, S.; Preitner, F.; Tappy, L.; Matic, G. Modulation of hepatic inflammation and energy-sensing pathways in the rat liver by high-fructose diet and chronic stress. Eur. J. Nutr. 2019, 58, 1829–1845. [Google Scholar] [CrossRef] [PubMed]
- Jovanovic Stojanov, S.; Martinovic, V.; Bogojevic, D.; Poznanovic, G.; Petrovic, A.; Ivanovic-Matic, S.; Grigorov, I. Modulation of diabetes-related liver injury by the hmgb1/tlr4 inflammatory pathway. J. Physiol. Biochem. 2018, 74, 345–358. [Google Scholar] [CrossRef] [PubMed]
- Koko, V.; Djordjeviae, J.; Cvijiae, G.; Davidoviae, V. Effect of acute heat stress on rat adrenal glands: A morphological and stereological study. J. Exp. Biol. 2004, 207, 4225–4230. [Google Scholar] [CrossRef]
- Krishna, M. Role of special stains in diagnostic liver pathology. Clin. Liver Dis. 2013, 2, S8–S10. [Google Scholar] [CrossRef]
- Arjmand, A.; Tsipouras, M.G.; Tzallas, A.T.; Forlano, R.; Manousou, P.; Giannakeas, N. Quantification of liver fibrosis—A comparative study. Appl. Sci. 2020, 10, 447. [Google Scholar] [CrossRef]
- Meguro, R.; Asano, Y.; Odagiri, S.; Li, C.; Iwatsuki, H.; Shoumura, K. Nonheme-iron histochemistry for light and electron microscopy: A historical, theoretical and technical review. Arch. Histol. Cytol. 2007, 70, 1–19. [Google Scholar] [CrossRef]
- Paglia, D.E.; Valentine, W.N. Studies on the quantitative and qualitative characterization of erythrocyte glutathione peroxidase. J. Lab. Clin. Med. 1967, 70, 158–169. [Google Scholar]
- Habig, W.H.; Pabst, M.J.; Jakoby, W.B. Glutathione s-transferases. The first enzymatic step in mercapturic acid formation. J. Biol. Chem. 1974, 249, 7130–7139. [Google Scholar] [CrossRef]
- Glatzle, D.; Vuilleumier, J.P.; Weber, F.; Decker, K. Glutathione reductase test with whole blood, a convenient procedure for the assessment of the riboflavin status in humans. Experientia 1974, 30, 665–667. [Google Scholar] [CrossRef]
- Griffith, O.W. Determination of glutathione and glutathione disulfide using glutathione reductase and 2-vinylpyridine. Anal. Biochem. 1980, 106, 207–212. [Google Scholar] [CrossRef]
- Misra, H.P.; Fridovich, I. The role of superoxide anion in the autoxidation of epinephrine and a simple assay for superoxide dismutase. J. Biol. Chem. 1972, 247, 3170–3175. [Google Scholar] [CrossRef]
- Lowry, O.H.; Rosebrough, N.J.; Farr, A.L.; Randall, R.J. Protein measurement with the folin phenol reagent. J. Biol. Chem. 1951, 193, 265–275. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ctrl | DM | DM + Fer-1 | ANOVA | |
|---|---|---|---|---|
| Body mass (g) | 26.8 ± 0.9 | 23.0 ± 0.8 * | 23.3 ± 1.4 | F(2) = 4.17 p = 0.0300 |
| Blood glucose (mmol/L) | 9.0 ± 0.4 | 16.8 ± 1.7 ** | 13.9 ± 1.5 | F(2) = 7.34 p = 0.0043 |
| Serum ALT (U/L) | 57.2 ± 5.6 | 76.2 ± 2.5 * | 61.1 ± 5.2 # | F(2) = 4.08 p = 0.0472 |
| Serum TG (mmol/L) | 1.548 ± 0.02 | 1.7 ± 0.05 * | 1.5 ± 0.05 # | F(2) = 7.78 p = 0.0068 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stancic, A.; Velickovic, K.; Markelic, M.; Grigorov, I.; Saksida, T.; Savic, N.; Vucetic, M.; Martinovic, V.; Ivanovic, A.; Otasevic, V. Involvement of Ferroptosis in Diabetes-Induced Liver Pathology. Int. J. Mol. Sci. 2022, 23, 9309. https://doi.org/10.3390/ijms23169309
Stancic A, Velickovic K, Markelic M, Grigorov I, Saksida T, Savic N, Vucetic M, Martinovic V, Ivanovic A, Otasevic V. Involvement of Ferroptosis in Diabetes-Induced Liver Pathology. International Journal of Molecular Sciences. 2022; 23(16):9309. https://doi.org/10.3390/ijms23169309
Chicago/Turabian StyleStancic, Ana, Ksenija Velickovic, Milica Markelic, Ilijana Grigorov, Tamara Saksida, Nevena Savic, Milica Vucetic, Vesna Martinovic, Andjelija Ivanovic, and Vesna Otasevic. 2022. "Involvement of Ferroptosis in Diabetes-Induced Liver Pathology" International Journal of Molecular Sciences 23, no. 16: 9309. https://doi.org/10.3390/ijms23169309