
Expression of the Calcium-Binding Protein CALB1 Is Induced and Controls Intracellular Ca2+ Levels in Senescent Cells


 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
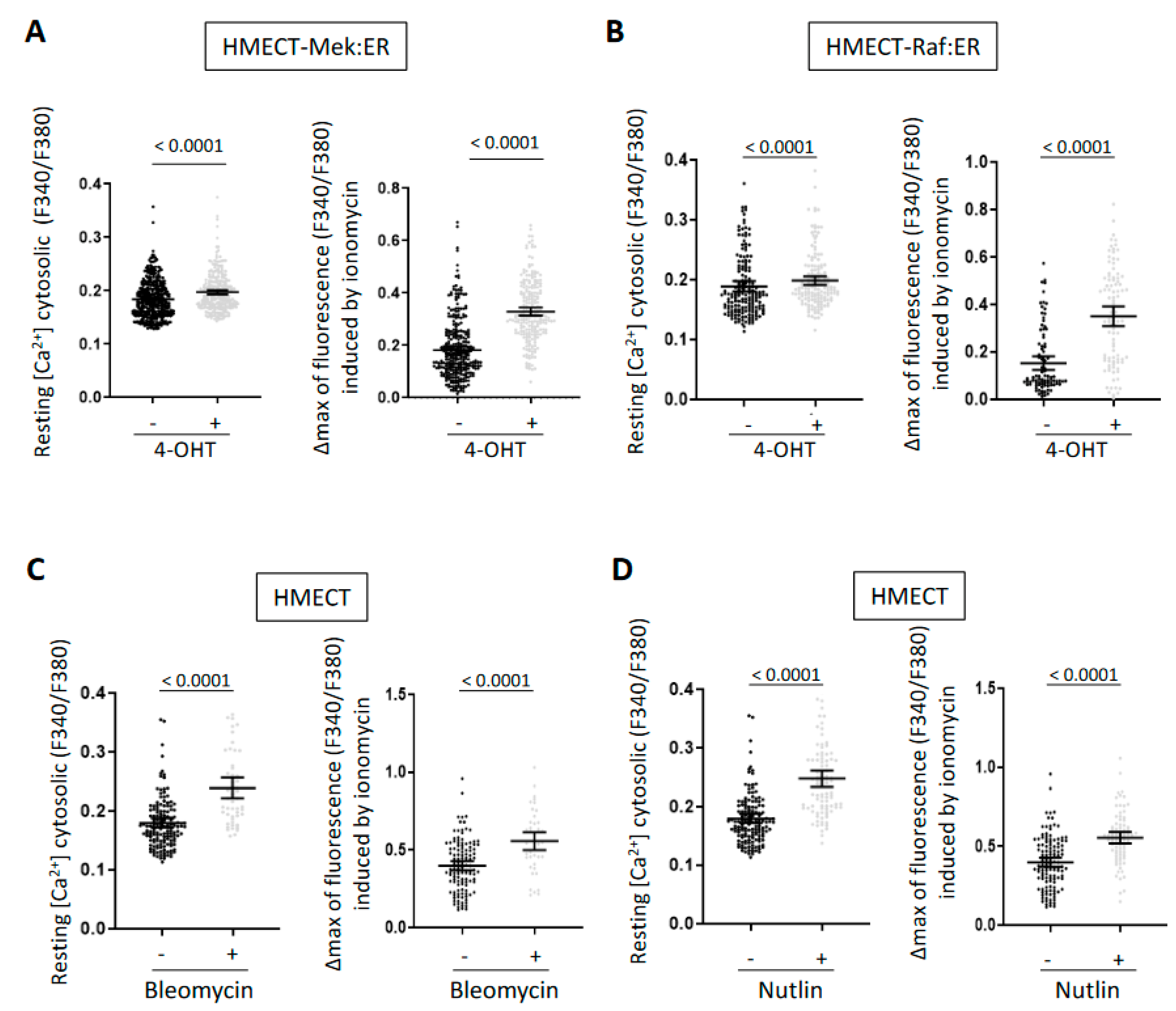
2.1. A Rise in Intracellular Calcium Content Is a Common Mark of Senescence in HMECT
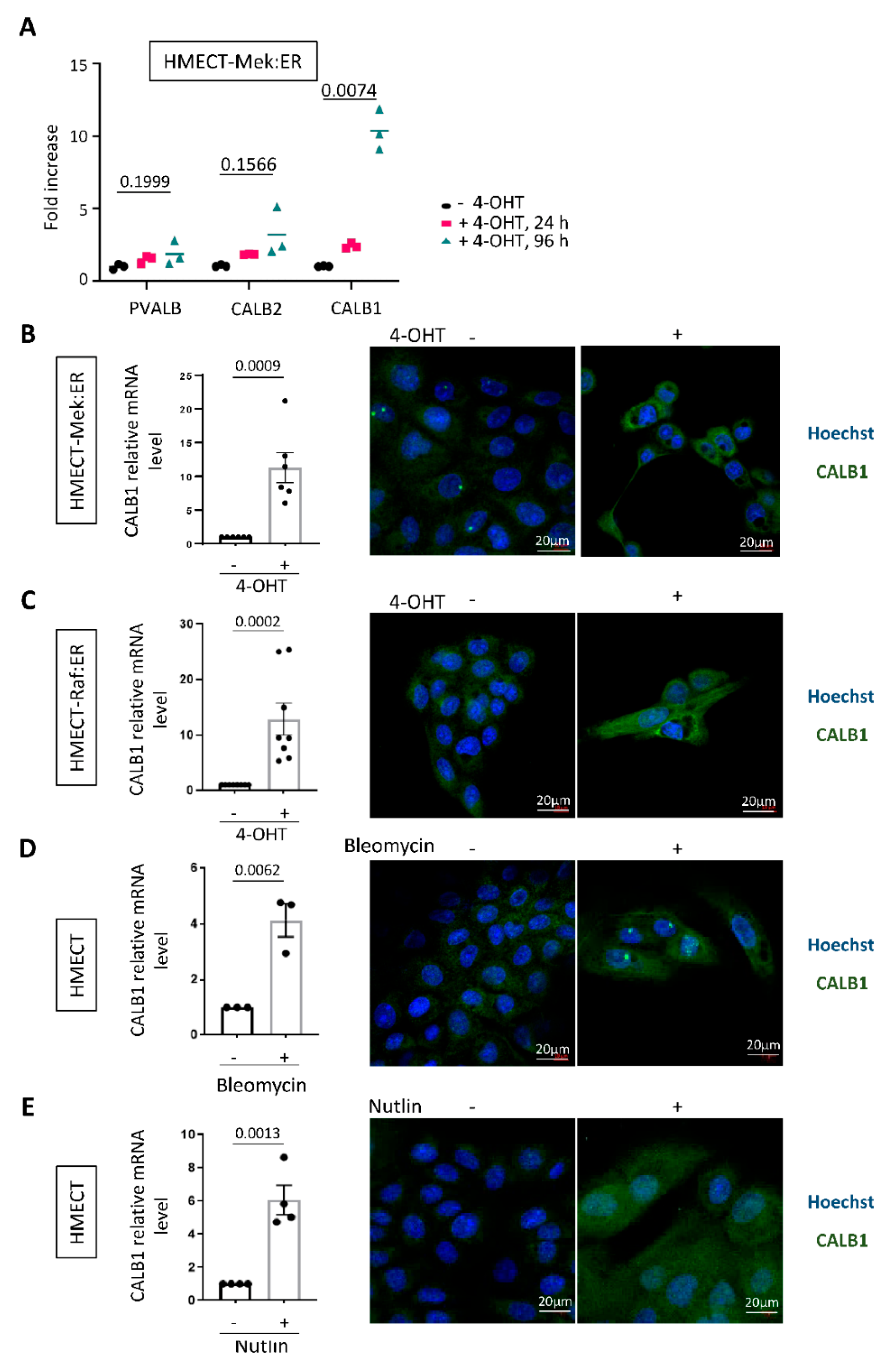
2.2. CALB1 Expression Is Induced in Senescent HMECT
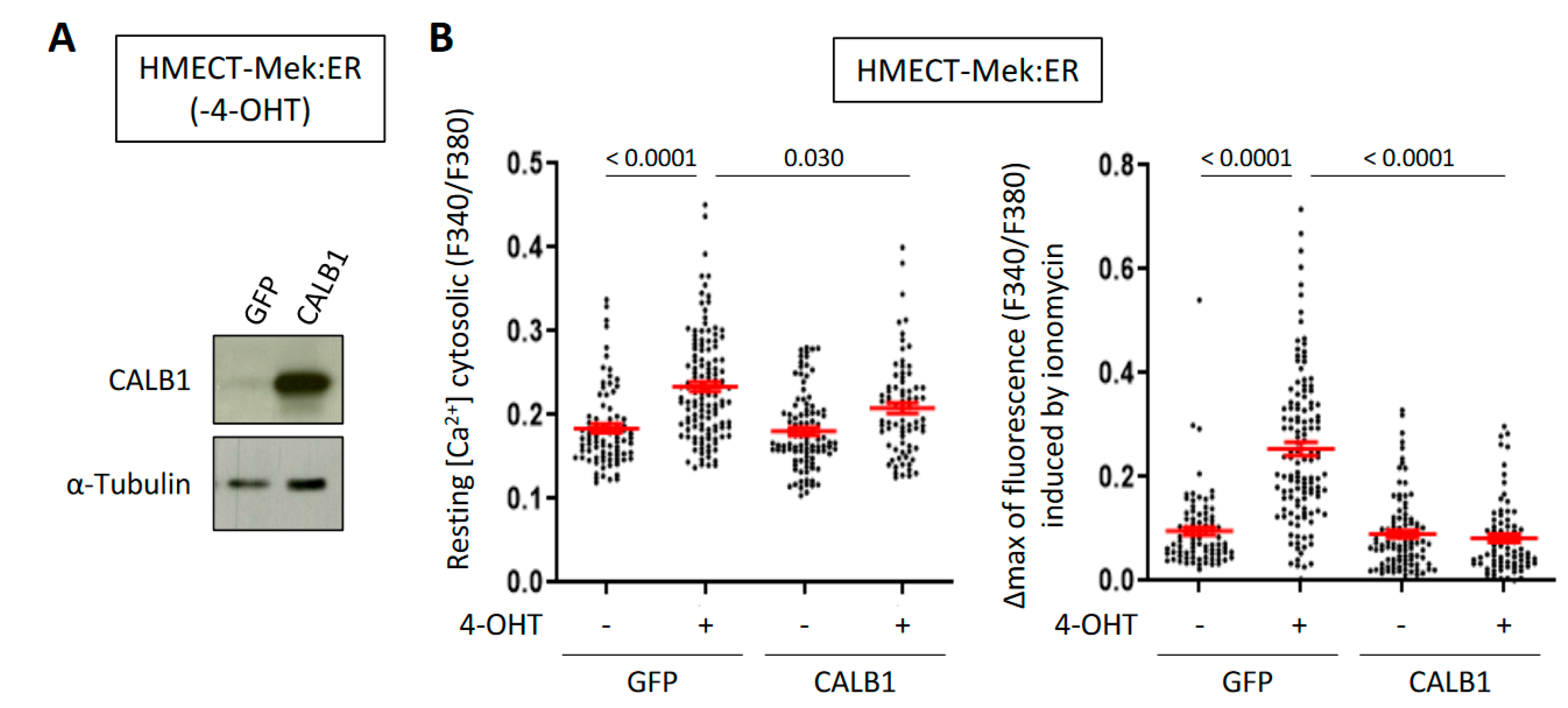
2.3. CALB1 Expression Is Induced by the Calcium-Dependent Calcineurin/NFAT Pathway and Its Constitutive Expression Limits Intracellular Ca2+ Accumulation upon OIS in HMECT
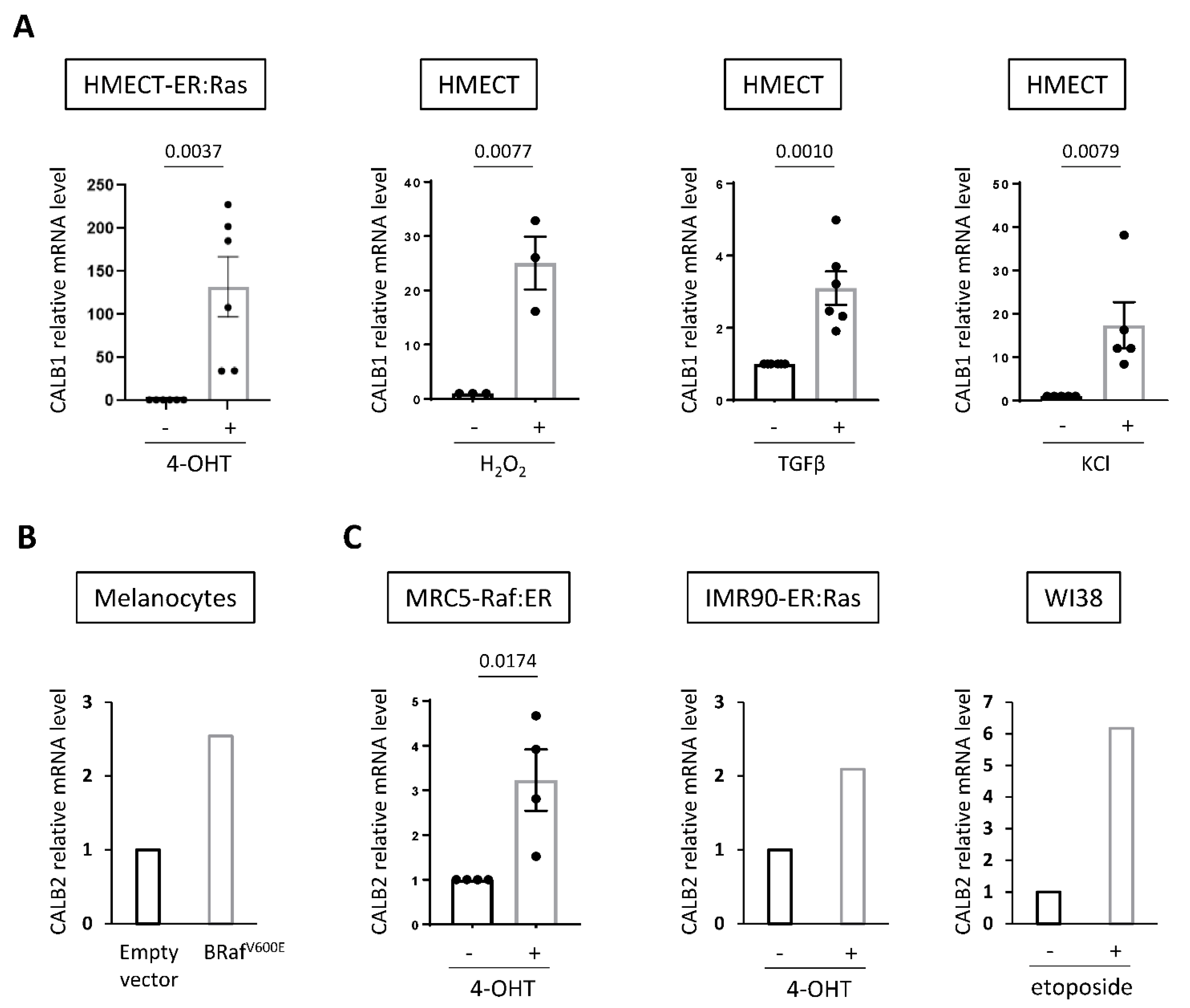
2.4. Increased Expression of Calbindins Is a Frequent Mark of Senescent Cells
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Reagents
4.2. Vectors, Transfection, and Infection
4.3. RNA Extraction, Reverse Transcription and Real-Time Quantitative PCR
4.4. Western Blot
4.5. Immunofluorescence Staining
4.6. Calcium Imaging
4.7. Statistical Analyses
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gorgoulis, V.; Adams, P.D.; Alimonti, A.; Bennett, D.C.; Bischof, O.; Bishop, C.; Campisi, J.; Collado, M.; Evangelou, K.; Ferbeyre, G.; et al. Cellular Senescence: Defining a Path Forward. Cell 2019, 179, 813–827. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Segura, A.; Nehme, J.; Demaria, M. Hallmarks of Cellular Senescence. Trends Cell Biol. 2018, 28, 436–453. [Google Scholar] [CrossRef] [PubMed]
- Kumari, R.; Jat, P. Mechanisms of Cellular Senescence: Cell Cycle Arrest and Senescence Associated Secretory Phenotype. Front Cell Dev. Biol. 2021, 9, 645593. [Google Scholar] [CrossRef] [PubMed]
- Farfariello, V.; Iamshanova, O.; Germain, E.; Fliniaux, I.; Prevarskaya, N. Calcium homeostasis in cancer: A focus on senescence. Biochim. Biophys. Acta 2015, 1853, 1974–1979. [Google Scholar] [CrossRef] [PubMed]
- Martin, N.; Bernard, D. Calcium signaling and cellular senescence. Cell Calcium 2018, 70, 16–23. [Google Scholar] [CrossRef]
- Borodkina, A.V.; Shatrova, A.N.; Deryabin, P.I.; Griukova, A.A.; Abushik, P.A.; Antonov, S.M.; Nikolsky, N.N.; Burova, E.B. Calcium alterations signal either to senescence or to autophagy induction in stem cells upon oxidative stress. Aging (Albany NY) 2016, 8, 3400–3418. [Google Scholar] [CrossRef]
- Wu, X.; Nguyen, B.C.; Dziunycz, P.; Chang, S.; Brooks, Y.; Lefort, K.; Hofbauer, G.F.; Dotto, G.P. Opposing roles for calcineurin and ATF3 in squamous skin cancer. Nature 2010, 465, 368–372. [Google Scholar] [CrossRef]
- Ma, X.; Warnier, M.; Raynard, C.; Ferrand, M.; Kirsh, O.; Defossez, P.A.; Martin, N.; Bernard, D. The nuclear receptor RXRA controls cellular senescence by regulating calcium signaling. Aging Cell 2018, 17, e12831. [Google Scholar] [CrossRef]
- Wiel, C.; Lallet-Daher, H.; Gitenay, D.; Gras, B.; Le Calve, B.; Augert, A.; Ferrand, M.; Prevarskaya, N.; Simonnet, H.; Vindrieux, D.; et al. Endoplasmic reticulum calcium release through ITPR2 channels leads to mitochondrial calcium accumulation and senescence. Nat. Commun. 2014, 5, 3792. [Google Scholar] [CrossRef]
- Ziegler, D.V.; Vindrieux, D.; Goehrig, D.; Jaber, S.; Collin, G.; Griveau, A.; Wiel, C.; Bendridi, N.; Djebali, S.; Farfariello, V.; et al. Calcium channel ITPR2 and mitochondria-ER contacts promote cellular senescence and aging. Nat. Commun. 2021, 12, 720. [Google Scholar] [CrossRef]
- Farfariello, V.; Gordienko, D.V.; Mesilmany, L.; Touil, Y.; Germain, E.; Fliniaux, I.; Desruelles, E.; Gkika, D.; Roudbaraki, M.; Shapovalov, G.; et al. TRPC3 shapes the ER-mitochondria Ca(2+) transfer characterizing tumour-promoting senescence. Nat. Commun. 2022, 13, 956. [Google Scholar] [CrossRef] [PubMed]
- Schwaller, B. Cytosolic Ca2+ buffers. Cold Spring Harb. Perspect. Biol. 2010, 2, a004051. [Google Scholar] [CrossRef] [PubMed]
- Gitenay, D.; Wiel, C.; Lallet-Daher, H.; Vindrieux, D.; Aubert, S.; Payen, L.; Simonnet, H.; Bernard, D. Glucose metabolism and hexosamine pathway regulate oncogene-induced senescence. Cell Death. Dis. 2014, 5, e1089. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lallet-Daher, H.; Wiel, C.; Gitenay, D.; Navaratnam, N.; Augert, A.; Le Calve, B.; Verbeke, S.; Carling, D.; Aubert, S.; Vindrieux, D.; et al. Potassium Channel KCNA1 Modulates Oncogene-Induced Senescence and Transformation. Cancer Res. 2013, 73, 5253–5265. [Google Scholar] [CrossRef]
- Wiel, C.; Augert, A.; Vincent, D.F.; Gitenay, D.; Vindrieux, D.; Le Calve, B.; Arfi, V.; Lallet-Daher, H.; Reynaud, C.; Treilleux, I.; et al. Lysyl oxidase activity regulates oncogenic stress response and tumorigenesis. Cell Death. Dis. 2013, 4, e855. [Google Scholar] [CrossRef]
- Giorgi, C.; Danese, A.; Missiroli, S.; Patergnani, S.; Pinton, P. Calcium Dynamics as a Machine for Decoding Signals. Trends Cell Biol. 2018, 28, 258–273. [Google Scholar] [CrossRef]
- Warnier, M.; Flaman, J.M.; Chouabe, C.; Wiel, C.; Gras, B.; Griveau, A.; Blanc, E.; Foy, J.P.; Mathot, P.; Saintigny, P.; et al. The SCN9A channel and plasma membrane depolarization promote cellular senescence through Rb pathway. Aging Cell 2018, 17, e12736. [Google Scholar] [CrossRef]
- Hajibeigi, A.; Dioum, E.M.; Guo, J.; Oz, O.K. Identification of novel regulatory NFAT and TFII-I binding elements in the calbindin-D28k promoter in response to serum deprivation. Biochem. Biophys. Res. Commun. 2015, 465, 414–420. [Google Scholar] [CrossRef]
- Cipriano, R.; Kan, C.E.; Graham, J.; Danielpour, D.; Stampfer, M.; Jackson, M.W. TGF-beta signaling engages an ATM-CHK2-p53-independent RAS-induced senescence and prevents malignant transformation in human mammary epithelial cells. Proc. Natl. Acad. Sci. USA 2011, 108, 8668–8673. [Google Scholar] [CrossRef]
- Dasari, A.; Bartholomew, J.N.; Volonte, D.; Galbiati, F. Oxidative stress induces premature senescence by stimulating caveolin-1 gene transcription through p38 mitogen-activated protein kinase/Sp1-mediated activation of two GC-rich promoter elements. Cancer Res. 2006, 66, 10805–10814. [Google Scholar] [CrossRef]
- Vindrieux, D.; Augert, A.; Girard, C.A.; Gitenay, D.; Lallet-Daher, H.; Wiel, C.; Le Calve, B.; Gras, B.; Ferrand, M.; Verbeke, S.; et al. PLA2R1 mediates tumor suppression by activating JAK2. Cancer Res. 2013, 73, 6334–6345. [Google Scholar] [CrossRef]
- Pawlikowski, J.S.; McBryan, T.; van, T.J.; Drotar, M.E.; Hewitt, R.N.; Maier, A.B.; King, A.; Blyth, K.; Wu, H.; Adams, P.D. Wnt signaling potentiates nevogenesis. Proc. Natl. Acad. Sci. USA 2013, 110, 16009–16014. [Google Scholar] [CrossRef] [PubMed]
- Acosta, J.C.; Banito, A.; Wuestefeld, T.; Georgilis, A.; Janich, P.; Morton, J.P.; Athineos, D.; Kang, T.W.; Lasitschka, F.; Andrulis, M.; et al. A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat. Cell Biol. 2013, 15, 978–990. [Google Scholar] [CrossRef] [PubMed]
- Contrepois, K.; Coudereau, C.; Benayoun, B.A.; Schuler, N.; Roux, P.F.; Bischof, O.; Courbeyrette, R.; Carvalho, C.; Thuret, J.Y.; Ma, Z.; et al. Histone variant H2A.J accumulates in senescent cells and promotes inflammatory gene expression. Nat. Commun. 2017, 8, 14995. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, D.A.; Clark, R.R.; Bartling, T.R.; Trebak, M.; Melendez, J.A. Redox control of the senescence regulator interleukin-1alpha and the secretory phenotype. J. Biol. Chem. 2013, 288, 32149–32159. [Google Scholar] [CrossRef]
- Ito, N.; Ruegg, U.T.; Takeda, S. ATP-Induced Increase in Intracellular Calcium Levels and Subsequent Activation of mTOR as Regulators of Skeletal Muscle Hypertrophy. Int. J. Mol. Sci. 2018, 19, 2804. [Google Scholar] [CrossRef]
- Murakami, T.; Ockinger, J.; Yu, J.; Byles, V.; McColl, A.; Hofer, A.M.; Horng, T. Critical role for calcium mobilization in activation of the NLRP3 inflammasome. Proc. Natl. Acad. Sci. USA 2012, 109, 11282–11287. [Google Scholar] [CrossRef]
- Sankar, N.; deTombe, P.P.; Mignery, G.A. Calcineurin-NFATc regulates type 2 inositol 1,4,5-trisphosphate receptor (InsP3R2) expression during cardiac remodeling. J. Biol. Chem. 2014, 289, 6188–6198. [Google Scholar] [CrossRef]
- Manda, K.R.; Tripathi, P.; Hsi, A.C.; Ning, J.; Ruzinova, M.B.; Liapis, H.; Bailey, M.; Zhang, H.; Maher, C.A.; Humphrey, P.A.; et al. NFATc1 promotes prostate tumorigenesis and overcomes PTEN loss-induced senescence. Oncogene 2016, 35, 3282–3292. [Google Scholar] [CrossRef]
- Cao, L.Q.; Wang, Y.N.; Liang, M.; Pan, M.Z. CALB1 enhances the interaction between p53 and MDM2, and inhibits the senescence of ovarian cancer cells. Mol. Med. Rep. 2019, 19, 5097–5104. [Google Scholar] [CrossRef]
- Guo, Q.; Christakos, S.; Robinson, N.; Mattson, M.P. Calbindin D28k blocks the proapoptotic actions of mutant presenilin 1: Reduced oxidative stress and preserved mitochondrial function. Proc. Natl. Acad. Sci. USA 1998, 95, 3227–3232. [Google Scholar] [CrossRef] [PubMed]
- Bobay, B.G.; Stewart, A.L.; Tucker, A.T.; Thompson, R.J.; Varney, K.M.; Cavanagh, J. Structural insights into the calcium-dependent interaction between calbindin-D28K and caspase-3. FEBS Lett. 2012, 586, 3582–3589. [Google Scholar] [CrossRef]
- Bellido, T.; Huening, M.; Raval-Pandya, M.; Manolagas, S.C.; Christakos, S. Calbindin-D28k is expressed in osteoblastic cells and suppresses their apoptosis by inhibiting caspase-3 activity. J. Biol. Chem. 2000, 275, 26328–26332. [Google Scholar] [CrossRef]
- Jochems, F.; Thijssen, B.; De Conti, G.; Jansen, R.; Pogacar, Z.; Groot, K.; Wang, L.; Schepers, A.; Wang, C.; Jin, H.; et al. The Cancer SENESCopedia: A delineation of cancer cell senescence. Cell Rep. 2021, 36, 109441. [Google Scholar] [CrossRef] [PubMed]
- Maida, Y.; Yasukawa, M.; Furuuchi, M.; Lassmann, T.; Possemato, R.; Okamoto, N.; Kasim, V.; Hayashizaki, Y.; Hahn, W.C.; Masutomi, K. An RNA-dependent RNA polymerase formed by TERT and the RMRP RNA. Nature 2009, 461, 230–235. [Google Scholar] [CrossRef] [PubMed]
- Jensen, K.J.; Moyer, C.B.; Janes, K.A. Network Architecture Predisposes an Enzyme to Either Pharmacologic or Genetic Targeting. Cell Syst. 2016, 2, 112–121. [Google Scholar] [CrossRef] [PubMed]
- Young, A.R.; Narita, M.; Ferreira, M.; Kirschner, K.; Sadaie, M.; Darot, J.F.; Tavare, S.; Arakawa, S.; Shimizu, S.; Watt, F.M.; et al. Autophagy mediates the mitotic senescence transition. Genes Dev. 2009, 23, 798–803. [Google Scholar] [CrossRef]
- Monticelli, S.; Rao, A. NFAT1 and NFAT2 are positive regulators of IL-4 gene transcription. Eur. J. Immunol. 2002, 32, 2971–2978. [Google Scholar] [CrossRef]
- Raynard, C.; Ma, X.; Huna, A.; Tessier, N.; Massemin, A.; Zhu, K.; Flaman, J.M.; Moulin, F.; Goehrig, D.; Medard, J.J.; et al. NF-kappaB-dependent secretome of senescent cells can trigger neuroendocrine transdifferentiation of breast cancer cells. Aging Cell 2022, 21, e13632. [Google Scholar] [CrossRef]
- Lim, D.; Mapelli, L.; Canonico, P.L.; Moccia, F.; Genazzani, A.A. Neuronal Activity-Dependent Activation of Astroglial Calcineurin in Mouse Primary Hippocampal Cultures. Int. J. Mol. Sci. 2018, 19, 2997. [Google Scholar] [CrossRef]
- Sanchez-Tecuatl, M.; Vargaz-Guadarrama, A.; Ramirez-Cortes, J.M..; Gomez-Gil, P.; Moccia, F.; Berra-Romani, R. Automated Intracellular Calcium Profiles Extraction from Endothelial Cells Using Digital Fluorescence Images. Int. J. Mol. Sci. 2018, 19, 3440. [Google Scholar]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Raynard, C.; Tessier, N.; Huna, A.; Warnier, M.; Flaman, J.-M.; Van Coppenolle, F.; Ducreux, S.; Martin, N.; Bernard, D. Expression of the Calcium-Binding Protein CALB1 Is Induced and Controls Intracellular Ca2+ Levels in Senescent Cells. Int. J. Mol. Sci. 2022, 23, 9376. https://doi.org/10.3390/ijms23169376
Raynard C, Tessier N, Huna A, Warnier M, Flaman J-M, Van Coppenolle F, Ducreux S, Martin N, Bernard D. Expression of the Calcium-Binding Protein CALB1 Is Induced and Controls Intracellular Ca2+ Levels in Senescent Cells. International Journal of Molecular Sciences. 2022; 23(16):9376. https://doi.org/10.3390/ijms23169376
Chicago/Turabian StyleRaynard, Clotilde, Nolwenn Tessier, Anda Huna, Marine Warnier, Jean-Michel Flaman, Fabien Van Coppenolle, Sylvie Ducreux, Nadine Martin, and David Bernard. 2022. "Expression of the Calcium-Binding Protein CALB1 Is Induced and Controls Intracellular Ca2+ Levels in Senescent Cells" International Journal of Molecular Sciences 23, no. 16: 9376. https://doi.org/10.3390/ijms23169376
APA StyleRaynard, C., Tessier, N., Huna, A., Warnier, M., Flaman, J.-M., Van Coppenolle, F., Ducreux, S., Martin, N., & Bernard, D. (2022). Expression of the Calcium-Binding Protein CALB1 Is Induced and Controls Intracellular Ca2+ Levels in Senescent Cells. International Journal of Molecular Sciences, 23(16), 9376. https://doi.org/10.3390/ijms23169376