Serotonin 5-HT6 Receptor Ligands and Butyrylcholinesterase Inhibitors Displaying Antioxidant Activity—Design, Synthesis and Biological Evaluation of Multifunctional Agents against Alzheimer’s Disease

 , , , , , ,
, , , , , ,
Abstract
:1. Introduction
2. Results and Discussion
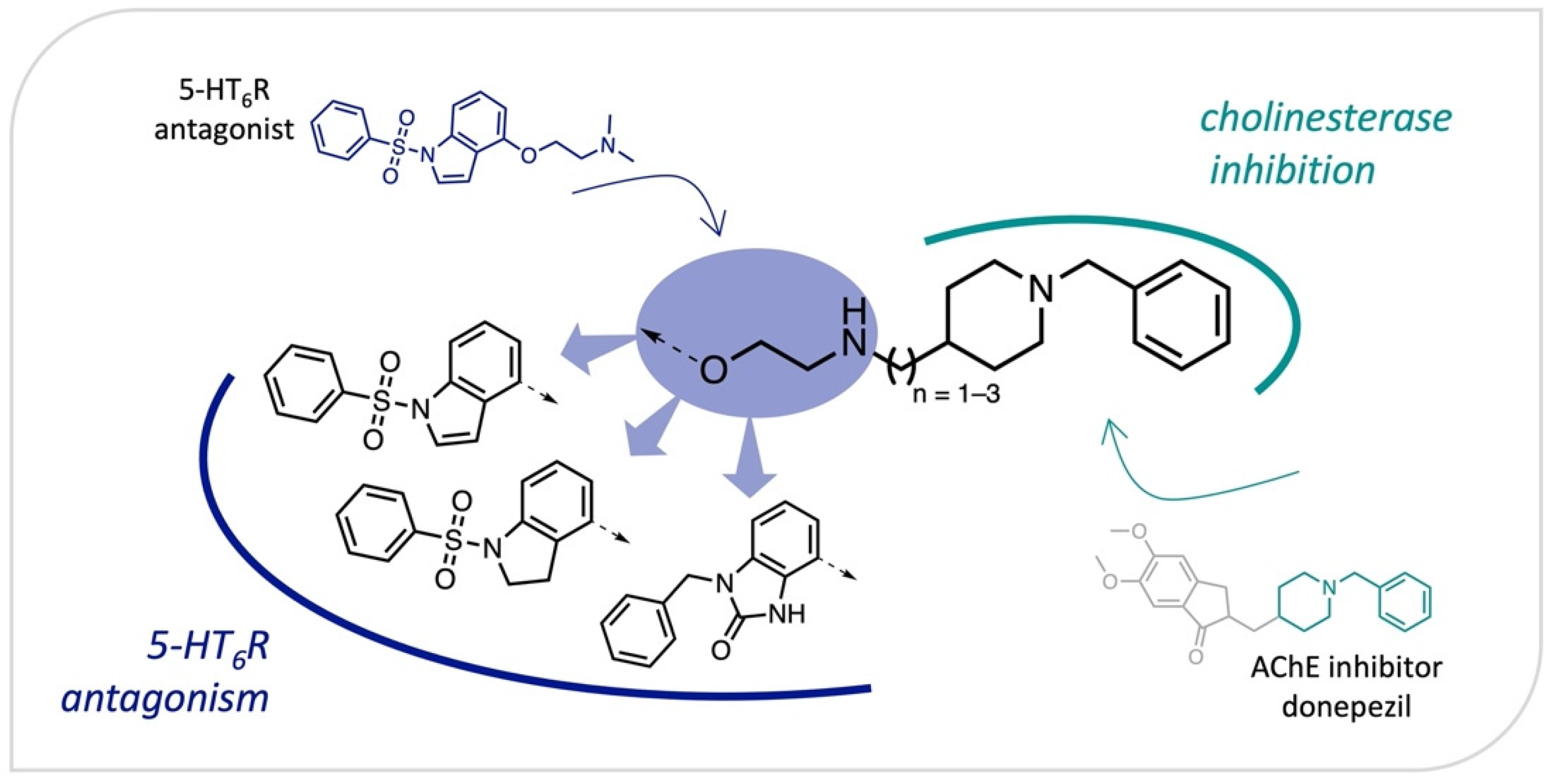
2.1. Design
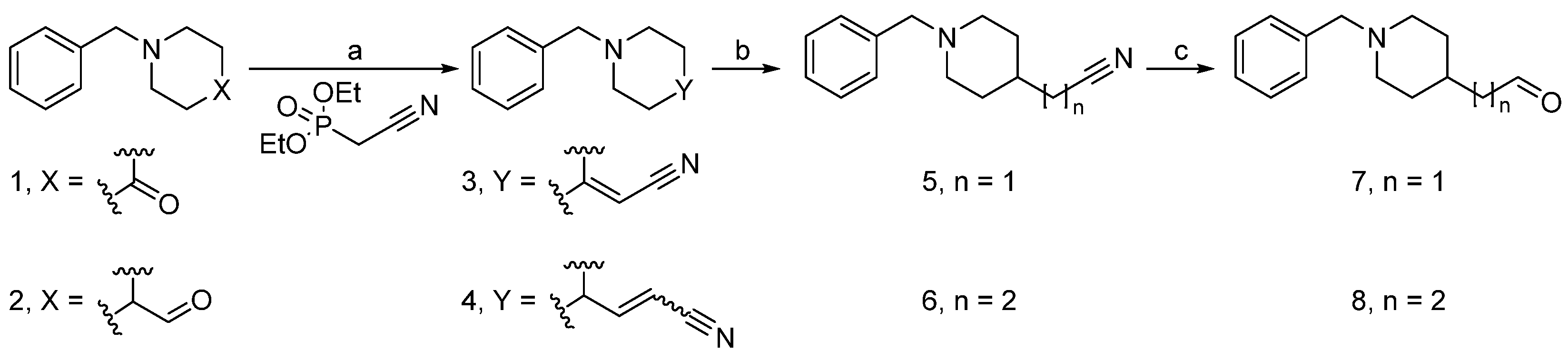
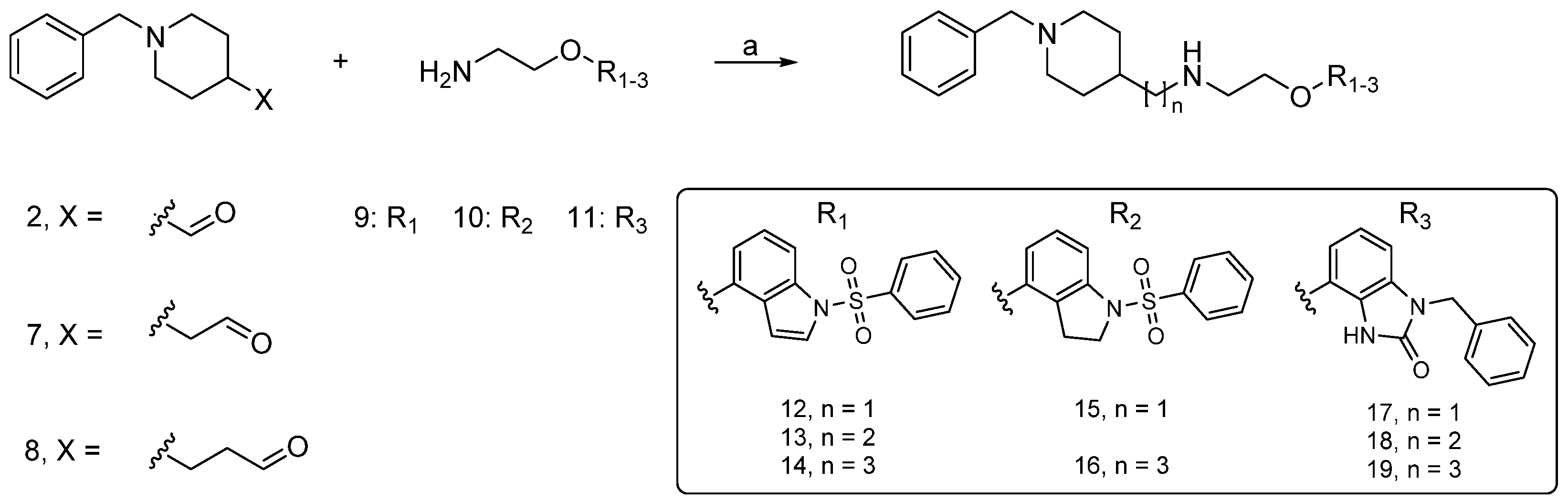
2.2. Synthesis
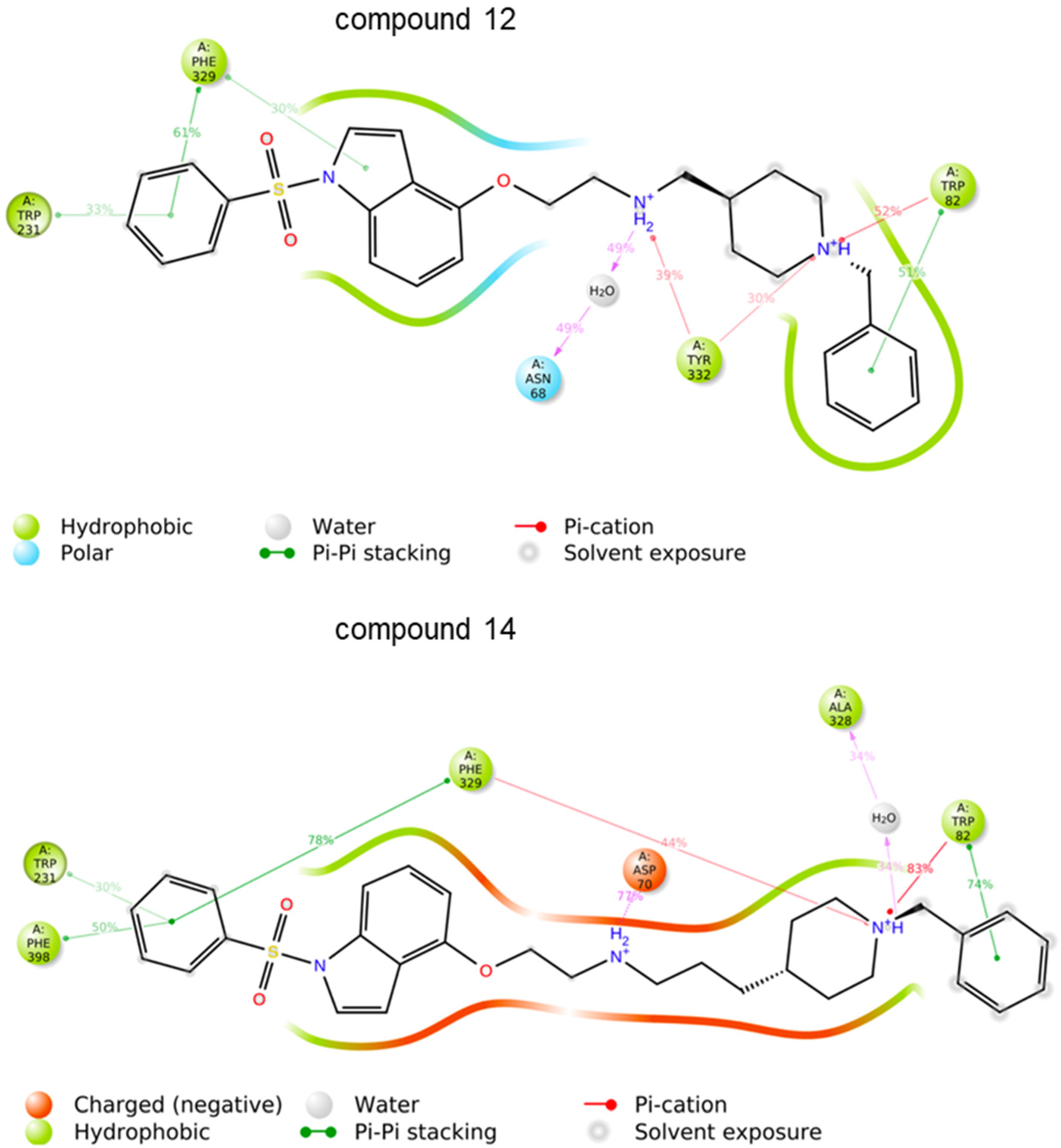
2.3. Biological Evaluation, SAR Analysis
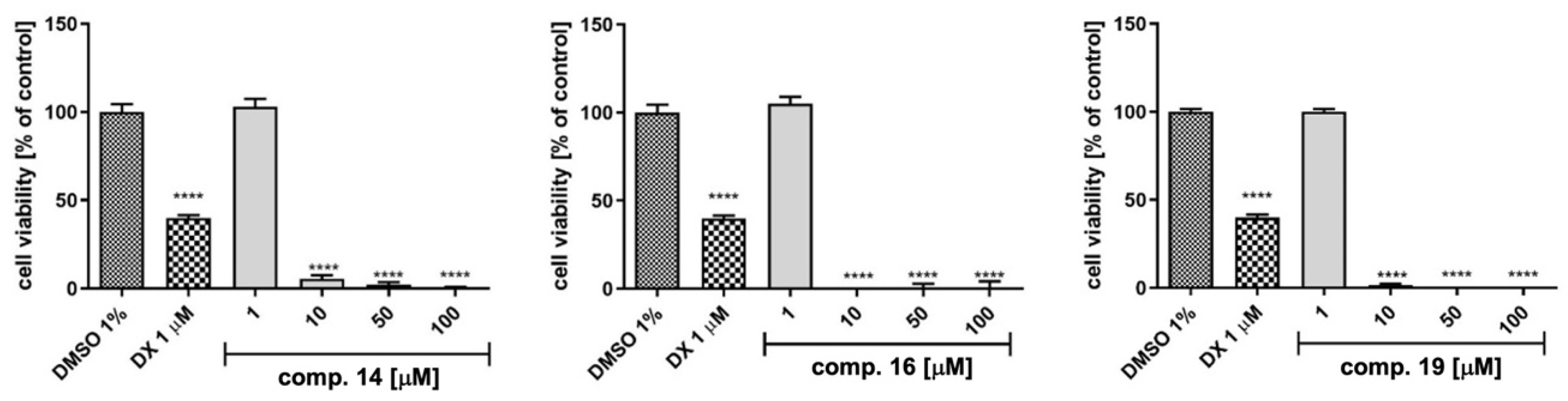
2.4. Preliminary In Vitro ADMET Studies
3. Materials and Methods
3.1. Chemistry
3.1.1. Previously Reported Compounds
3.1.2. General Procedure for the Synthesis of Nitriles 3 and 4 (GP1)
2-(1-Benzylpiperidin-4-ylidene)acetonitrile (3)
3-(1-Benzylpiperidin-4-yl)acrylonitrile (4)
3.1.3. General Procedure for the Synthesis of Nitriles 5 and 6 (GP2)
2-(1-Benzylpiperidin-4-yl)acetonitrile (5)
3-(1-Benzylpiperidin-4-yl)propanenitrile (6)
3.1.4. General Procedure for the Synthesis of Aldehydes 7 and 8 (GP3)
2-(1-Benzylpiperidin-4-yl)acetaldehyde (7)
3-(1-Benzylpiperidin-4-yl)propanal (8)
3.1.5. General Procedure for the Synthesis of Compounds 12–19 (GP4)
N-((1-Benzylpiperidin-4-yl)methyl)-2-((1-(phenylsulfonyl)-1H-indol-4-yl)oxy)ethan-1-amine (12)
2-(1-Benzylpiperidin-4-yl)-N-(2-((1-(phenylsulfonyl)-1H-indol-4-yl) oxy)ethyl)ethan-1-amine (13)
3-(1-Benzylpiperidin-4-yl)-N-(2-((1-(phenylsulfonyl)-1H-indol-4-yl)oxy)ethyl)propan-1-amine (14)
N-((1-Benzylpiperidin-4-yl) methyl)-2-((1-(phenylsulfonyl) indolin-4-yl)oxy)ethan-1-amine (15)
3-(1-Benzylpiperidin-4-yl)-N-(2-((1-(phenylsulfonyl) indolin-4-yl)oxy)ethyl)propan-1-amine (16)
1-Benzyl-4-(2-(((1-benzylpiperidin-4-yl)methyl)amino)ethoxy)-1,3-dihydro-2H-benzo[d]imidazol-2-one (17)
1-Benzyl-4-(2-((2-(1-benzylpiperidin-4-yl)ethyl)amino)ethoxy)-1,3-dihydro-2H-benzo[d]imidazol-2-one (18)
1-Benzyl-4-(2-((3-(1-benzylpiperidin-4-yl)propyl)amino)ethoxy)-1,3-dihydro-2H-benzo[d]imidazol-2-one (19)
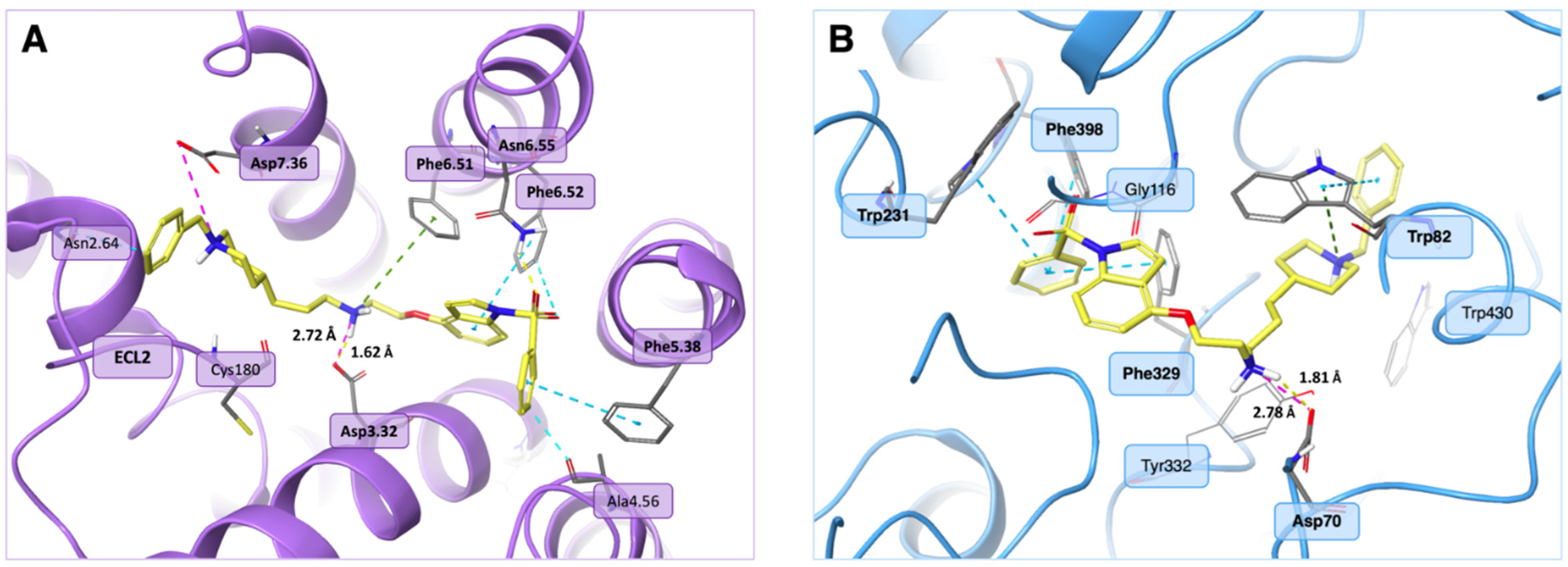
3.2. Molecular Modelling
3.3. Radioligand Binding Assay
3.3.1. Preparation of Solutions of Test and Reference Compounds
3.3.2. 5-HT6 Receptor Binding Assay
3.4. Human Acetylcholinesterase and Butyrylcholinesterase In Vitro Inhibitory Activity
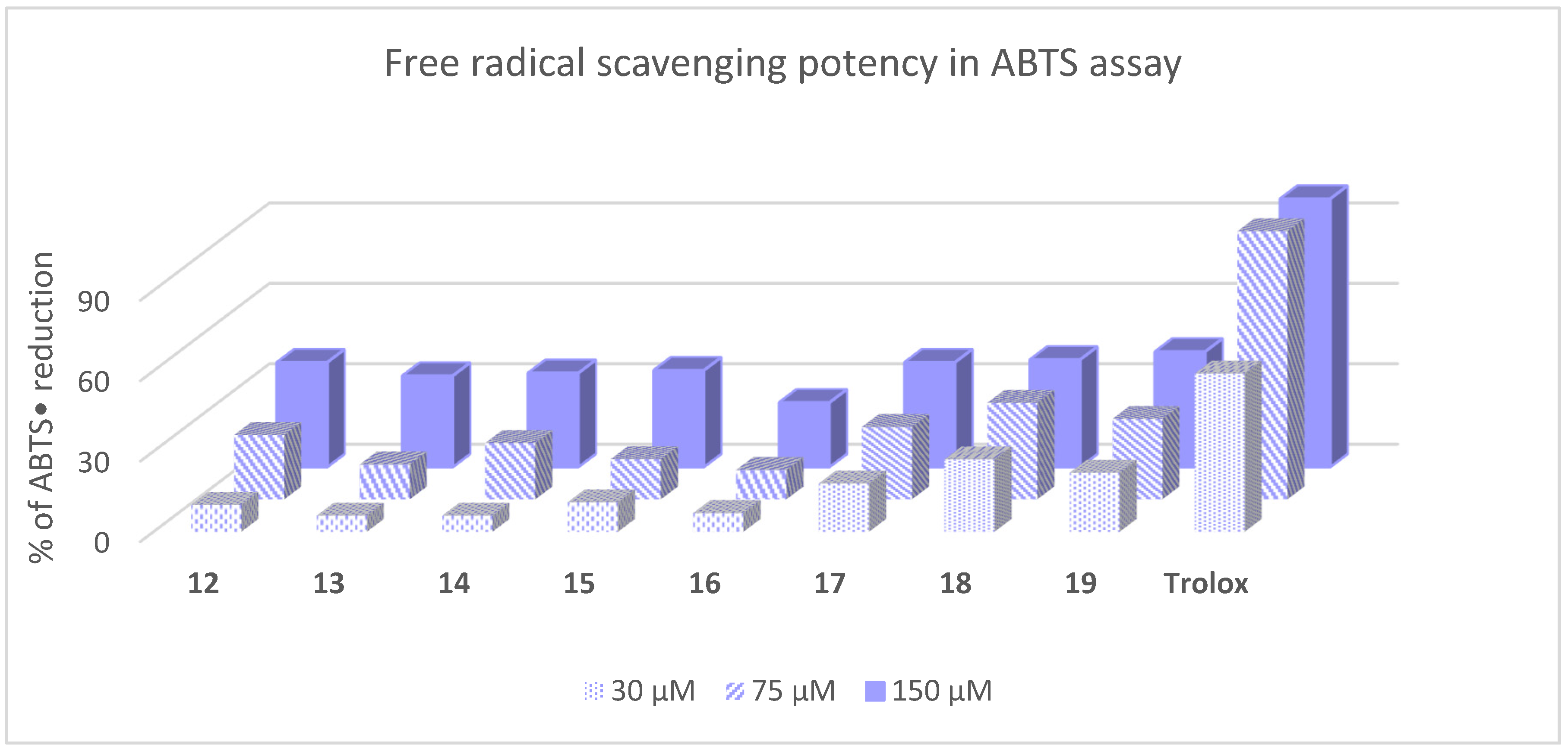
3.5. Free Radical Scavenging In Vitro Activity
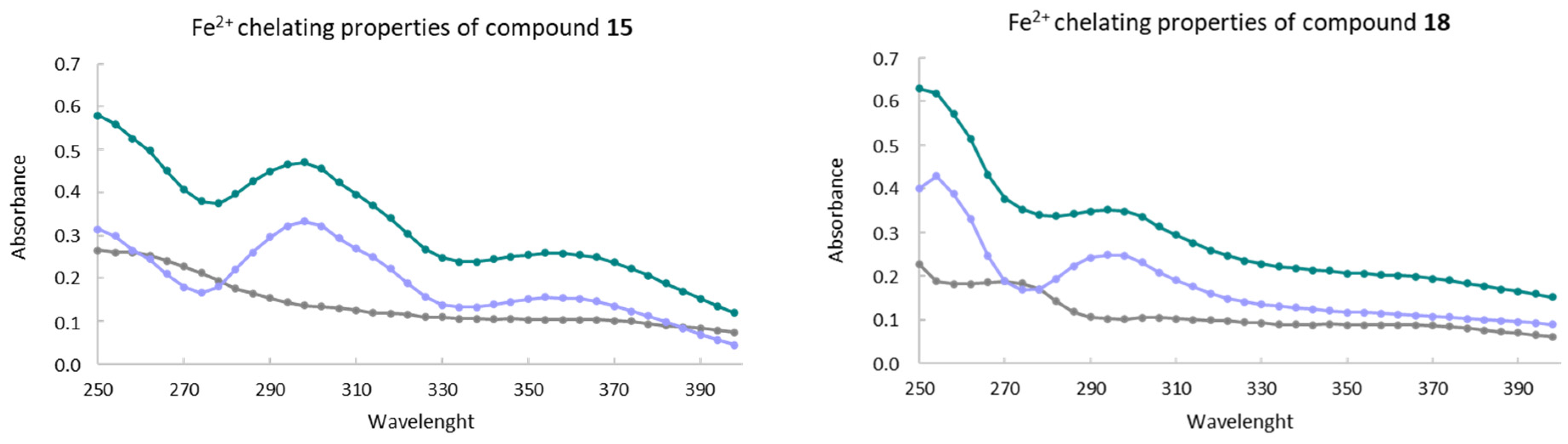
3.6. Metal-Chelating In Vitro Activity
3.7. ADME-Tox Parameters
3.7.1. Metabolic Stability in Human Liver Microsomes
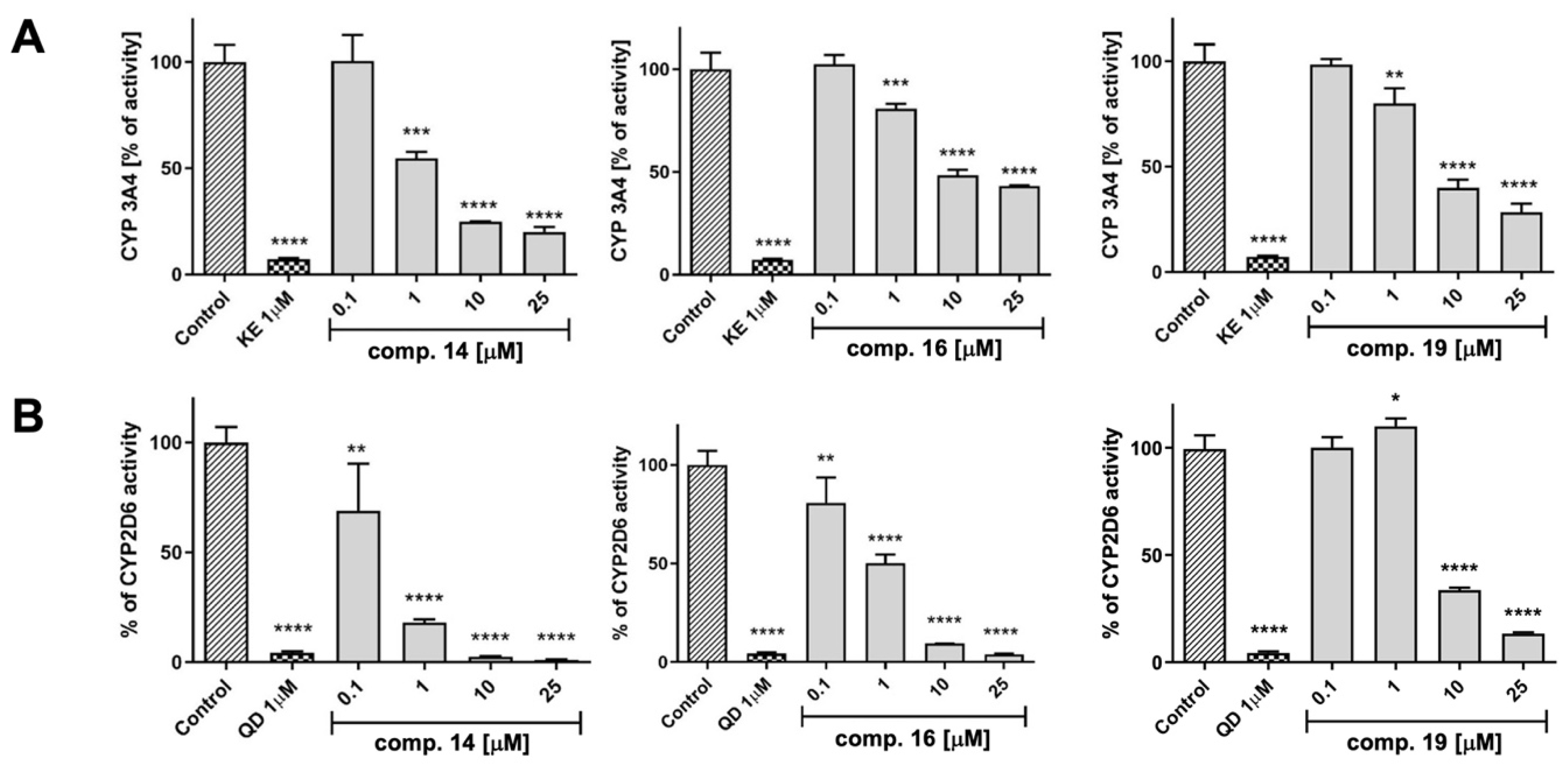
3.7.2. Influence on CYP P450 Activity
3.7.3. Hepatotoxicity in HepG2 Cells
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Alzheimer’s Association. 2021 Alzheimer’s Disease Facts and Figures. Alzheimer′s Dement. 2021, 17, 327–406. [Google Scholar] [CrossRef] [PubMed]
- Serrano-Pozo, A.; Frosch, M.P.; Masliah, E.; Hyman, B.T. Neuropathological Alterations in Alzheimer Disease. Cold Spring Harb. Perspect. Med. 2011, 1, a006189. [Google Scholar] [CrossRef] [PubMed]
- Simunkova, M.; Alwasel, S.H.; Alhazza, I.M.; Jomova, K.; Kollar, V.; Rusko, M.; Valko, M. Management of Oxidative Stress and Other Pathologies in Alzheimer’s Disease. Arch. Toxicol. 2019, 93, 2491–2513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jasiecki, J.; Targońska, M.; Wasąg, B. The Role of Butyrylcholinesterase and Iron in the Regulation of Cholinergic Network and Cognitive Dysfunction in Alzheimer’s Disease Pathogenesis. Int. J. Mol. Sci. 2021, 22, 2033. [Google Scholar] [CrossRef] [PubMed]
- Cui, C.-C.; Sun, Y.; Wang, X.-Y.; Zhang, Y.; Xing, Y. The Effect of Anti-Dementia Drugs on Alzheimer Disease-Induced Cognitive Impairment: A Network Meta-Analysis. Medicine 2019, 98, e16091. [Google Scholar] [CrossRef]
- Haake, A.; Nguyen, K.; Friedman, L.; Chakkamparambil, B.; Grossberg, G.T. An Update on the Utility and Safety of Cholinesterase Inhibitors for the Treatment of Alzheimer’s Disease. Expert Opin. Drug Saf. 2020, 19, 147–157. [Google Scholar] [CrossRef]
- Wang, N.; Qiu, P.; Cui, W.; Yan, X.; Zhang, B.; He, S. Recent Advances in Multi-Target Anti-Alzheimer Disease Compounds (2013 Up to the Present). Curr. Med. Chem. 2019, 26, 5684–5710. [Google Scholar] [CrossRef]
- Wang, H.; Zhang, H. Reconsideration of Anticholinesterase Therapeutic Strategies against Alzheimer’s Disease. ACS Chem. Neurosci. 2019, 10, 852–862. [Google Scholar] [CrossRef]
- Makhaeva, G.F.; Lushchekina, S.V.; Kovaleva, N.V.; Astakhova, T.Y.; Boltneva, N.P.; Rudakova, E.V.; Serebryakova, O.G.; Proshin, A.N.; Serkov, I.V.; Trofimova, T.P.; et al. Amiridine-Piperazine Hybrids as Cholinesterase Inhibitors and Potential Multitarget Agents for Alzheimer’s Disease Treatment. Bioorg. Chem. 2021, 112, 104974. [Google Scholar] [CrossRef]
- Carreiras, M.; Mendes, E.; Perry, M.; Francisco, A.; Marco-Contelles, J. The Multifactorial Nature of Alzheimer’s Disease for Developing Potential Therapeutics. Curr. Top. Med. Chem. 2013, 13, 1745–1770. [Google Scholar] [CrossRef]
- Hartmann, J.; Kiewert, C.; Duysen, E.G.; Lockridge, O.; Greig, N.H.; Klein, J. Excessive Hippocampal Acetylcholine Levels in Acetylcholinesterase-Deficient Mice Are Moderated by Butyrylcholinesterase Activity. J. Neurochem. 2007, 100, 1421–1429. [Google Scholar] [CrossRef] [PubMed]
- Macdonald, I.R.; Maxwell, S.P.; Reid, G.A.; Cash, M.K.; DeBay, D.R.; Darvesh, S. Quantification of Butyrylcholinesterase Activity as a Sensitive and Specific Biomarker of Alzheimer’s Disease. J. Alzheimer’s Dis. 2017, 58, 491–505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeBay, D.R.; Reid, G.A.; Macdonald, I.R.; Mawko, G.; Burrell, S.; Martin, E.; Bowen, C.V.; Darvesh, S. Butyrylcholinesterase-Knockout Reduces Fibrillar β-Amyloid and Conserves (18)FDG Retention in 5XFAD Mouse Model of Alzheimer’s Disease. Brain Res. 2017, 1671, 102–110. [Google Scholar] [CrossRef] [PubMed]
- Atri, A.; Frölich, L.; Ballard, C.; Tariot, P.N.; Molinuevo, J.L.; Boneva, N.; Windfeld, K.; Raket, L.L.; Cummings, J.L. Effect of Idalopirdine as Adjunct to Cholinesterase Inhibitors on Change in Cognition in Patients with Alzheimer Disease: Three Randomized Clinical Trials. JAMA 2018, 319, 130–142. [Google Scholar] [CrossRef] [PubMed]
- Khoury, R.; Grysman, N.; Gold, J.; Patel, K.; Grossberg, G.T. The Role of 5 HT6-Receptor Antagonists in Alzheimer’s Disease: An Update. Expert Opin. Investig. Drugs 2018, 27, 523–533. [Google Scholar] [CrossRef]
- Marazziti, D.; Baroni, S.; Pirone, A.; Giannaccini, G.; Betti, L.; Testa, G.; Schmid, L.; Palego, L.; Borsini, F.; Bordi, F.; et al. Serotonin Receptor of Type 6 (5-HT6) in Human Prefrontal Cortex and Hippocampus Post-Mortem: An Immunohistochemical and Immunofluorescence Study. Neurochem. Int. 2013, 62, 182–188. [Google Scholar] [CrossRef]
- Helboe, L.; Egebjerg, J.; de Jong, I.E.M. Distribution of Serotonin Receptor 5-HT6 MRNA in Rat Neuronal Subpopulations: A Double in Situ Hybridization Study. Neuroscience 2015, 310, 442–454. [Google Scholar] [CrossRef]
- Li, X.; Gao, L.; Liu, J.; Zhang, H.; Chen, H.; Yang, L.; Wu, M.; Li, C.; Zhu, X.; Ding, Y.; et al. Safety, Tolerability and Pharmacokinetics of the Serotonin 5-HT6 Receptor Antagonist, HEC30654, in Healthy Chinese Subjects. Front. Pharmacol. 2021, 12, 726536. [Google Scholar] [CrossRef]
- Wicke, K.; Haupt, A.; Bespalov, A. Investigational Drugs Targeting 5-HT6 Receptors for the Treatment of Alzheimer’s Disease. Expert Opin. Investig. Drugs 2015, 24, 1515–1528. [Google Scholar] [CrossRef]
- Sheline, Y.I.; Snider, B.J.; Beer, J.C.; Seok, D.; Fagan, A.M.; Suckow, R.F.; Lee, J.-M.; Waligorska, T.; Korecka, M.; Aselcioglu, I.; et al. Effect of Escitalopram Dose and Treatment Duration on CSF Aβ Levels in Healthy Older Adults: A Controlled Clinical Trial. Neurology 2020, 95, e2658–e2665. [Google Scholar] [CrossRef]
- West, P.J.; Marcy, V.R.; Marino, M.J.; Schaffhauser, H. Activation of the 5-HT6 Receptor Attenuates Long-Term Potentiation and Facilitates GABAergic Neurotransmission in Rat Hippocampus. Neuroscience 2009, 164, 692–701. [Google Scholar] [CrossRef]
- Sun, Z.; Wang, B.; Chen, C.; Li, C.; Zhang, Y. 5-HT6R Null Mutatrion Induces Synaptic and Cognitive Defects. Aging Cell 2021, 20, e13369. [Google Scholar] [CrossRef] [PubMed]
- Brouard, J.T.; Schweimer, J.V.; Houlton, R.; Burnham, K.E.; Quérée, P.; Sharp, T. Pharmacological Evidence for 5-HT6 Receptor Modulation of 5-HT Neuron Firing In Vivo. ACS Chem. Neurosci. 2015, 6, 1241–1247. [Google Scholar] [CrossRef] [PubMed]
- Dayer, A.G.; Jacobshagen, M.; Chaumont-Dubel, S.; Marin, P. 5-HT6 Receptor: A New Player Controlling the Development of Neural Circuits. ACS Chem. Neurosci. 2015, 6, 951–960. [Google Scholar] [CrossRef] [PubMed]
- Butterfield, D.A.; Halliwell, B. Oxidative Stress, Dysfunctional Glucose Metabolism and Alzheimer Disease. Nat. Rev. Neurosci. 2019, 20, 148–160. [Google Scholar] [CrossRef]
- Jadiya, P.; Kolmetzky, D.W.; Tomar, D.; Di Meco, A.; Lombardi, A.A.; Lambert, J.P.; Luongo, T.S.; Ludtmann, M.H.; Praticò, D.; Elrod, J.W. Impaired Mitochondrial Calcium Efflux Contributes to Disease Progression in Models of Alzheimer′s Disease. Nat. Commun. 2019, 10, 3885. [Google Scholar] [CrossRef] [PubMed]
- Ganguly, U.; Kaur, U.; Chakrabarti, S.S.; Sharma, P.; Agrawal, B.K.; Saso, L.; Chakrabarti, S. Oxidative Stress, Neuroinflammation, and NADPH Oxidase: Implications in the Pathogenesis and Treatment of Alzheimer’s Disease. Oxid. Med. Cell. Longev. 2021, 2021, 7086512. [Google Scholar] [CrossRef]
- Das, N.; Raymick, J.; Sarkar, S. Role of Metals in Alzheimer’s Disease. Metab. Brain Dis. 2021, 36, 1627–1639. [Google Scholar] [CrossRef]
- Ayton, S.; Wang, Y.; Diouf, I.; Schneider, J.A.; Brockman, J.; Morris, M.C.; Bush, A.I. Brain Iron Is Associated with Accelerated Cognitive Decline in People with Alzheimer Pathology. Mol. Psychiatry 2020, 25, 2932–2941. [Google Scholar] [CrossRef]
- Raven, E.P.; Lu, P.H.; Tishler, T.A.; Heydari, P.; Bartzokis, G. Increased Iron Levels and Decreased Tissue Integrity in Hippocampus of Alzheimer’s Disease Detected In Vivo with Magnetic Resonance Imaging. J. Alzheimer’s Dis. 2013, 37, 127–136. [Google Scholar] [CrossRef]
- van Rooden, S.; Doan, N.T.; Versluis, M.J.; Goos, J.D.C.; Webb, A.G.; Oleksik, A.M.; van der Flier, W.M.; Scheltens, P.; Barkhof, F.; Weverling-Rynsburger, A.W.E.; et al. 7T T2*-Weighted Magnetic Resonance Imaging Reveals Cortical Phase Differences between Early- and Late-Onset Alzheimer’s Disease. Neurobiol. Aging 2015, 36, 20–26. [Google Scholar] [CrossRef] [PubMed]
- Derry, P.J.; Hegde, M.L.; Jackson, G.R.; Kayed, R.; Tour, J.M.; Tsai, A.-L.; Kent, T.A. Revisiting the Intersection of Amyloid, Pathologically Modified Tau and Iron in Alzheimer’s Disease from a Ferroptosis Perspective. Prog. Neurobiol. 2020, 184, 101716. [Google Scholar] [CrossRef] [PubMed]
- Ashraf, A.; Jeandriens, J.; Parkes, H.G.; So, P.-W. Iron Dyshomeostasis, Lipid Peroxidation and Perturbed Expression of Cystine/Glutamate Antiporter in Alzheimer’s Disease: Evidence of Ferroptosis. Redox Biol. 2020, 32, 101494. [Google Scholar] [CrossRef] [PubMed]
- Bao, W.-D.; Pang, P.; Zhou, X.-T.; Hu, F.; Xiong, W.; Chen, K.; Wang, J.; Wang, F.; Xie, D.; Hu, Y.-Z.; et al. Loss of Ferroportin Induces Memory Impairment by Promoting Ferroptosis in Alzheimer’s Disease. Cell Death Differ. 2021, 28, 1548–1562. [Google Scholar] [CrossRef] [PubMed]
- Hambright, W.S.; Fonseca, R.S.; Chen, L.; Na, R.; Ran, Q. Ablation of Ferroptosis Regulator Glutathione Peroxidase 4 in Forebrain Neurons Promotes Cognitive Impairment and Neurodegeneration. Redox Biol. 2017, 12, 8–17. [Google Scholar] [CrossRef]
- Huang, L.; McClatchy, D.B.; Maher, P.; Liang, Z.; Diedrich, J.K.; Soriano-Castell, D.; Goldberg, J.; Shokhirev, M.; Yates, J.R.; Schubert, D.; et al. Intracellular Amyloid Toxicity Induces Oxytosis/Ferroptosis Regulated Cell Death. Cell Death Dis. 2020, 11, 828. [Google Scholar] [CrossRef]
- Masaldan, S.; Bush, A.I.; Devos, D.; Rolland, A.S.; Moreau, C. Striking while the Iron is Hot: Iron Metabolism and Ferroptosis in Neurodegeneration. Free Radic. Biol. Med. 2019, 133, 221–233. [Google Scholar] [CrossRef]
- Więckowska, A.; Kołaczkowski, M.; Bucki, A.; Godyń, J.; Marcinkowska, M.; Więckowski, K.; Zaręba, P.; Siwek, A.; Kazek, G.; Głuch-Lutwin, M.; et al. Novel Multi-Target-Directed Ligands for Alzheimer’s Disease: Combining Cholinesterase Inhibitors and 5-HT6 Receptor Antagonists. Design, Synthesis and Biological Evaluation. Eur. J. Med. Chem. 2016, 124, 63–81. [Google Scholar] [CrossRef]
- Więckowska, A.; Wichur, T.; Godyń, J.; Bucki, A.; Marcinkowska, M.; Siwek, A.; Więckowski, K.; Zarȩba, P.; Knez, D.; Głuch-Lutwin, M.; et al. Novel Multitarget-Directed Ligands Aiming at Symptoms and Causes of Alzheimer’s Disease. ACS Chem. Neurosci. 2018, 9, 1195–1214. [Google Scholar] [CrossRef]
- Wichur, T.; Godyń, J.; Góral, I.; Latacz, G.; Bucki, A.; Siwek, A.; Głuch-Lutwin, M.; Mordyl, B.; Śniecikowska, J.; Walczak, M.; et al. Development and Crystallography-Aided SAR Studies of Multifunctional BuChE Inhibitors and 5-HT6R Antagonists with β-Amyloid Anti-Aggregation Properties. Eur. J. Med. Chem. 2021, 225, 113792. [Google Scholar] [CrossRef]
- Wichur, T.; Pasieka, A.; Godyń, J.; Panek, D.; Góral, I.; Latacz, G.; Honkisz-Orzechowska, E.; Bucki, A.; Siwek, A.; Głuch-Lutwin, M.; et al. Discovery of 1-(Phenylsulfonyl)-1H-Indole-Based Multifunctional Ligands Targeting Cholinesterases and 5-HT(6) Receptor with Anti-Aggregation Properties against Amyloid-Beta and Tau. Eur. J. Med. Chem. 2021, 225, 113783. [Google Scholar] [CrossRef] [PubMed]
- Marcinkowska, M.; Bucki, A.; Panek, D.; Siwek, A.; Fajkis, N.; Bednarski, M.; Zygmunt, M.; Godyń, J.; Del Rio Valdivieso, A.; Kotańska, M.; et al. Anti-Alzheimer’s Multitarget-Directed Ligands with Serotonin 5-HT(6) Antagonist, Butyrylcholinesterase Inhibitory, and Antioxidant Activity. Arch. Pharm. 2019, 352, e1900041. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.G.; Robichaud, A.J. 5-HT6 Antagonists as Potential Treatment for Cognitive Dysfunction. Drug Dev. Res. 2009, 70, 145–168. [Google Scholar] [CrossRef]
- Kryger, G.; Silman, I.; Sussman, J.L. Structure of Acetylcholinesterase Complexed with E2020 (Aricept): Implications for the Design of New Anti-Alzheimer Drugs. Structure 1999, 7, 297–307. [Google Scholar] [CrossRef] [Green Version]
- Kryger, G.; Silman, I.; Sussman, J.L. Three-Dimensional Structure of a Complex of E2020 with Acetylcholinesterase from Torpedo Californica. J. Physiol. 1998, 92, 191–194. [Google Scholar] [CrossRef]
- Umukoro, S.; Adewole, F.A.; Eduviere, A.T.; Aderibigbe, A.O.; Onwuchekwa, C. Free Radical Scavenging Effect of Donepezil as the Possible Contribution to Its Memory Enhancing Activity in Mice. Drug Res. 2014, 64, 236–239. [Google Scholar] [CrossRef]
- Contreras, J.M.; Rival, Y.M.; Chayer, S.; Bourguignon, J.J.; Wermuth, C.G. Aminopyridazines as Acetylcholinesterase Inhibitors. J. Med. Chem. 1999, 42, 730–741. [Google Scholar] [CrossRef]
- Zhou, P.; Yan, Y.; Bernotas, R.; Harrison, B.L.; Huryn, D.; Robichaud, A.J.; Zhang, G.M.; Smith, D.L.; Schechter, L.E. 4-(2-Aminoethoxy)-N-(Phenylsulfonyl)Indoles as Novel 5-HT6 Receptor Ligands. Bioorg. Med. Chem. Lett. 2005, 15, 1393–1396. [Google Scholar] [CrossRef]
- Ellman, G.L.; Courtney, K.D.; Andres, V.; Featherstone, R.M. A New and Rapid Colorimetric Determination of Acetylcholinesterase Activity. Biochem. Pharmacol. 1961, 7, 88–95. [Google Scholar] [CrossRef]
- de la Fuente, T.; Martín-Fontecha, M.; Sallander, J.; Benhamú, B.; Campillo, M.; Medina, R.A.; Pellissier, L.P.; Claeysen, S.; Dumuis, A.; Pardo, L.; et al. Benzimidazole Derivatives as New Serotonin 5-HT6 Receptor Antagonists. Molecular Mechanisms of Receptor Inactivation. J. Med. Chem. 2010, 53, 1357–1369. [Google Scholar] [CrossRef]
- Rosenberry, T.L.; Brazzolotto, X.; Macdonald, I.R.; Wandhammer, M.; Trovaslet-Leroy, M.; Darvesh, S.; Nachon, F. Comparison of the Binding of Reversible Inhibitors to Human Butyrylcholinesterase and Acetylcholinesterase: A Crystallographic, Kinetic and Calorimetric Study. Molecules 2017, 22, 2098. [Google Scholar] [CrossRef] [Green Version]
- Dighe, S.N.; Deora, G.S.; De la Mora, E.; Nachon, F.; Chan, S.; Parat, M.-O.; Brazzolotto, X.; Ross, B.P. Discovery and Structure-Activity Relationships of a Highly Selective Butyrylcholinesterase Inhibitor by Structure-Based Virtual Screening. J. Med. Chem. 2016, 59, 7683–7689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sinyor, B.; Mineo, J.; Ochner, C. Alzheimer’s Disease, Inflammation, and the Role of Antioxidants. J. Alzheimer’s Dis. Rep. 2020, 4, 175–183. [Google Scholar] [CrossRef]
- Wichur, T.; Więckowska, A.; Więckowski, K.; Godyń, J.; Jończyk, J.; Valdivieso, Á.R.; Panek, D.; Pasieka, A.; Sabaté, R.; Knez, D.; et al. 1-Benzylpyrrolidine-3-Amine-Based BuChE Inhibitors with Anti-Aggregating, Antioxidant and Metal-Chelating Properties as Multifunctional Agents against Alzheimer’s Disease. Eur. J. Med. Chem. 2020, 187, 111916. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Yin, Y.-L.; Liu, X.-Z.; Shen, P.; Zheng, Y.-G.; Lan, X.-R.; Lu, C.-B.; Wang, J.-Z. Current Understanding of Metal Ions in the Pathogenesis of Alzheimer’s Disease. Transl. Neurodegener. 2020, 9, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angelova, D.M.; Brown, D.R. Microglia and the Aging Brain: Are Senescent Microglia the Key to Neurodegeneration? J. Neurochem. 2019, 151, 676–688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McIntosh, A.; Mela, V.; Harty, C.; Minogue, A.M.; Costello, D.A.; Kerskens, C.; Lynch, M.A. Iron Accumulation in Microglia Triggers a Cascade of Events That Leads to Altered Metabolism and Compromised Function in APP/PS1 Mice. Brain Pathol. 2019, 29, 606–621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, F.; Wang, J.; Shen, Y.; Li, H.; Rausch, W.-D.; Huang, X. Iron Dyshomeostasis and Ferroptosis: A New Alzheimer’s Disease Hypothesis? Front. Aging Neurosci. 2022, 14, 830569. [Google Scholar] [CrossRef]
- Gleason, A.; Bush, A.I. Iron and Ferroptosis as Therapeutic Targets in Alzheimer’s Disease. Neurother. J. Am. Soc. Exp. Neurother. 2021, 18, 252–264. [Google Scholar] [CrossRef]
- Lynch, T.; Price, A. The Effect of Cytochrome P450 Metabolism on Drug Response, Interactions, and Adverse Effects. Am. Fam. Physician 2007, 76, 391–396. [Google Scholar]
- Więckowska, A.; Szałaj, N.; Góral, I.; Bucki, A.; Latacz, G.; Kiec-Kononowicz, K.; Bautista-Aguilera, Ò.M.; Romero, A.; Ramos, E.; Egea, J.; et al. In Vitro and In Silico ADME-Tox Profiling and Safety Significance of Multifunctional Monoamine Oxidase Inhibitors Targeting Neurodegenerative Diseases. ACS Chem. Neurosci. 2020, 11, 3793–3801. [Google Scholar] [CrossRef] [PubMed]
- Haeberlein, S.B.; Aisen, P.S.; Barkhof, F.; Chalkias, S.; Chen, T.; Cohen, S.; Dent, G.; Hansson, O.; Harrison, K.; von Hehn, C.; et al. Two Randomized Phase 3 Studies of Aducanumab in Early Alzheimer’s Disease. J. Prev. Alzheimer′s Dis. 2022, 9, 197–210. [Google Scholar] [CrossRef] [PubMed]
- Beshir, S.A.; Aadithsoorya, A.M.; Parveen, A.; Goh, S.S.L.; Hussain, N.; Menon, V.B. Aducanumab Therapy to Treat Alzheimer’s Disease: A Narrative Review. Int. J. Alzheimer’s Dis. 2022, 2022, 9343514. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
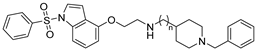
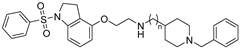
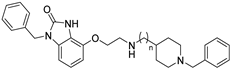
| Cmpd. | n | h5-HT6R Ki (µM) a | hAChE IC50 (µM) b | hBuChE IC50 (µM) b |
|---|---|---|---|---|
 | ||||
| 12 | 1 | 0.021 ± 0.001 | 3.151 ± 0.081 | 2.325 ± 0.059 |
| 13 | 2 | 0.017 ± 0.002 | 2.265 ± 0.091 | 1.115 ± 0.031 |
| 14 | 3 | 0.022 ± 0.002 | 0.930 ± 0.035 | 0.016 ± 0.001 |
 | ||||
| 15 | 1 | 0.267 ± 0.009 | 8.207 ± 0.225 | 5.405 ± 0.100 |
| 16 | 3 | 0.598 ± 0.019 | 0.821 ± 0.014 | 0.487 ± 0.012 |
 | ||||
| 17 | 1 | 0.136 ± 0.022 | 39.5% ± 1.3 c | 44.3% ± 2.0 c |
| 18 | 2 | 0.142 ± 0.020 | 2.856 ± 0.068 | 1.158 ± 0.024 |
| 19 | 3 | 0.480 ± 0.048 | 0.544 ± 0.015 | 0.613 ± 0.011 |
| Donepezil | - | 0.006 ± 0.0001 | - | |
| Tacrine | - | 0.131 ± 0.002 | 0.034 ± 0.0004 | |
| Mianserin | 0.056 ± 0.012 | - | - | |
| HLMs | MLMs | ||||||
|---|---|---|---|---|---|---|---|
| Comp. | (m/z) | % Left | Metabolite (m/z) | Metabolic Pathway * | % Left | Metabolite (m/z) | Metabolic Pathway * |
| 14 | 532.35 | 84.69 | 317.23 (M1) | ox. deamination | 75.54 | 317.23 (M1) | ox. deamination |
| 442.30 (M2) | ox. deamination | 442.37 (M2) | ox. deamination | ||||
| 16 | 534.41 | 84.40 | 444.30 (M1) | ox. deamination | 84.48 | 444.36 (M1) | ox. deamination |
| 550.37 (M2) | hydroxylation | 550.56 (M2) | hydroxylation | ||||
| 19 | 499.39 | 94.00 | 409.41 (M1) | ox. deamination | 79.38 | 409.41 (M1) | ox. deamination |
| 284.27 (M2) | ox. deamination | ||||||
| Verapamil | 455.31 | 30.84 | 441.35 (M1) | demethylation | 23.93 | 441.42 (M1) | demethylation |
| 291.28 (M2) | decomposition | 441.42 (M2) | demethylation | ||||
| 165.09 (M3) | decomposition | 291.35 (M3) | decomposition | ||||
| 441.29 (M4) | demethylation | 293.34 (M4) | decomp./hydrox. | ||||
| 427.33 (M5) | double demethylation | 277.33 (M5) | decomposition | ||||
| 277.26 (M6) | decomposition |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Więckowski, K.; Szałaj, N.; Gryzło, B.; Wichur, T.; Góral, I.; Sługocka, E.; Sniecikowska, J.; Latacz, G.; Siwek, A.; Godyń, J.; et al. Serotonin 5-HT6 Receptor Ligands and Butyrylcholinesterase Inhibitors Displaying Antioxidant Activity—Design, Synthesis and Biological Evaluation of Multifunctional Agents against Alzheimer’s Disease. Int. J. Mol. Sci. 2022, 23, 9443. https://doi.org/10.3390/ijms23169443
Więckowski K, Szałaj N, Gryzło B, Wichur T, Góral I, Sługocka E, Sniecikowska J, Latacz G, Siwek A, Godyń J, et al. Serotonin 5-HT6 Receptor Ligands and Butyrylcholinesterase Inhibitors Displaying Antioxidant Activity—Design, Synthesis and Biological Evaluation of Multifunctional Agents against Alzheimer’s Disease. International Journal of Molecular Sciences. 2022; 23(16):9443. https://doi.org/10.3390/ijms23169443
Chicago/Turabian StyleWięckowski, Krzysztof, Natalia Szałaj, Beata Gryzło, Tomasz Wichur, Izabella Góral, Emilia Sługocka, Joanna Sniecikowska, Gniewomir Latacz, Agata Siwek, Justyna Godyń, and et al. 2022. "Serotonin 5-HT6 Receptor Ligands and Butyrylcholinesterase Inhibitors Displaying Antioxidant Activity—Design, Synthesis and Biological Evaluation of Multifunctional Agents against Alzheimer’s Disease" International Journal of Molecular Sciences 23, no. 16: 9443. https://doi.org/10.3390/ijms23169443