
CDNF Interacts with ER Chaperones and Requires UPR Sensors to Promote Neuronal Survival

 , , , and
, , , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
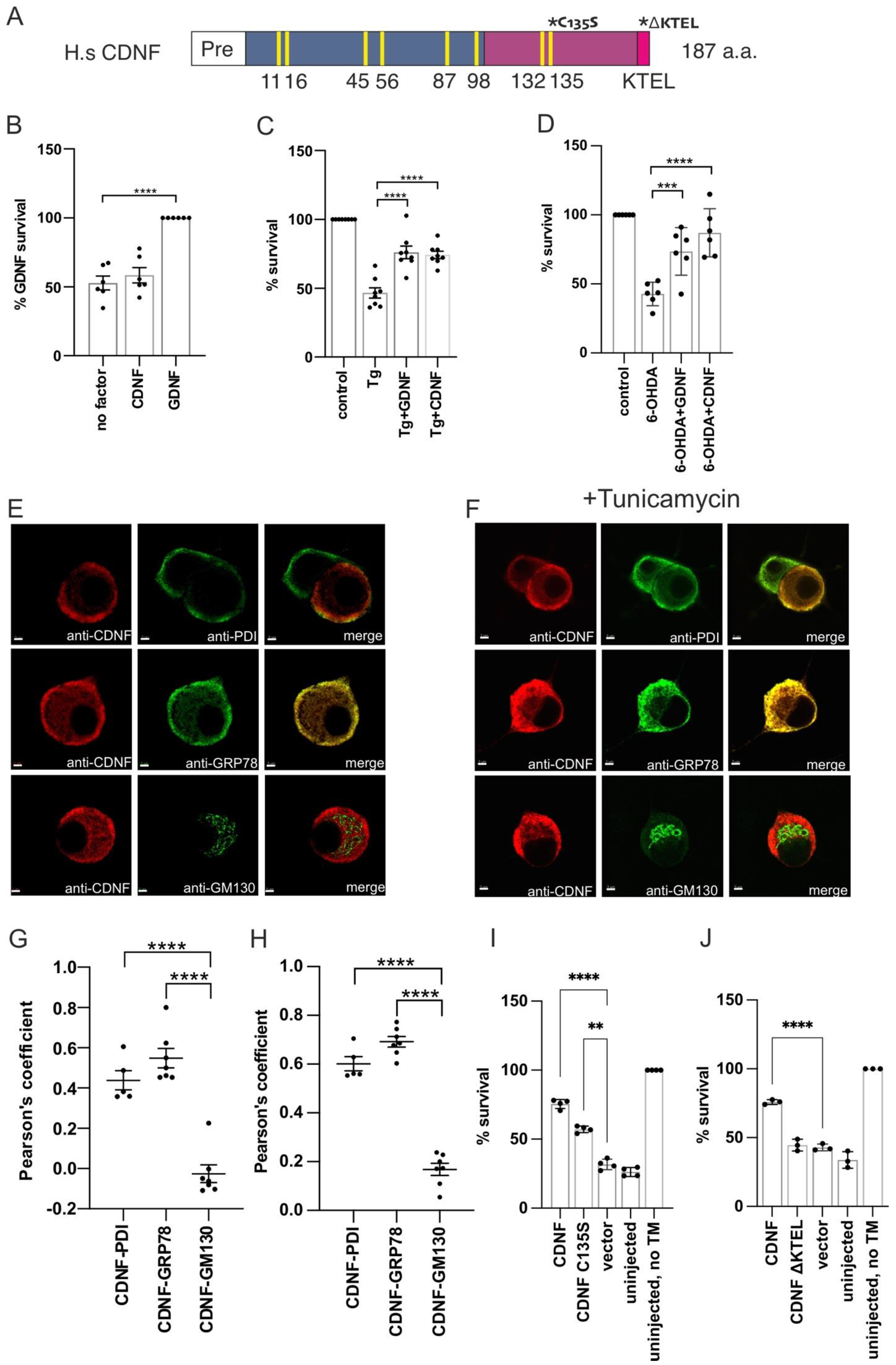
2.1. Extracellularly Added CDNF Does Not Promote the Survival of Naïve Dopamine Neurons, but Rescues Thapsigargin-Treated Neurons from Apoptosis
2.2. Overexpressed CDNF Localizes to the ER and Protects Sympathetic Neurons against ER Stress-Induced Apoptosis
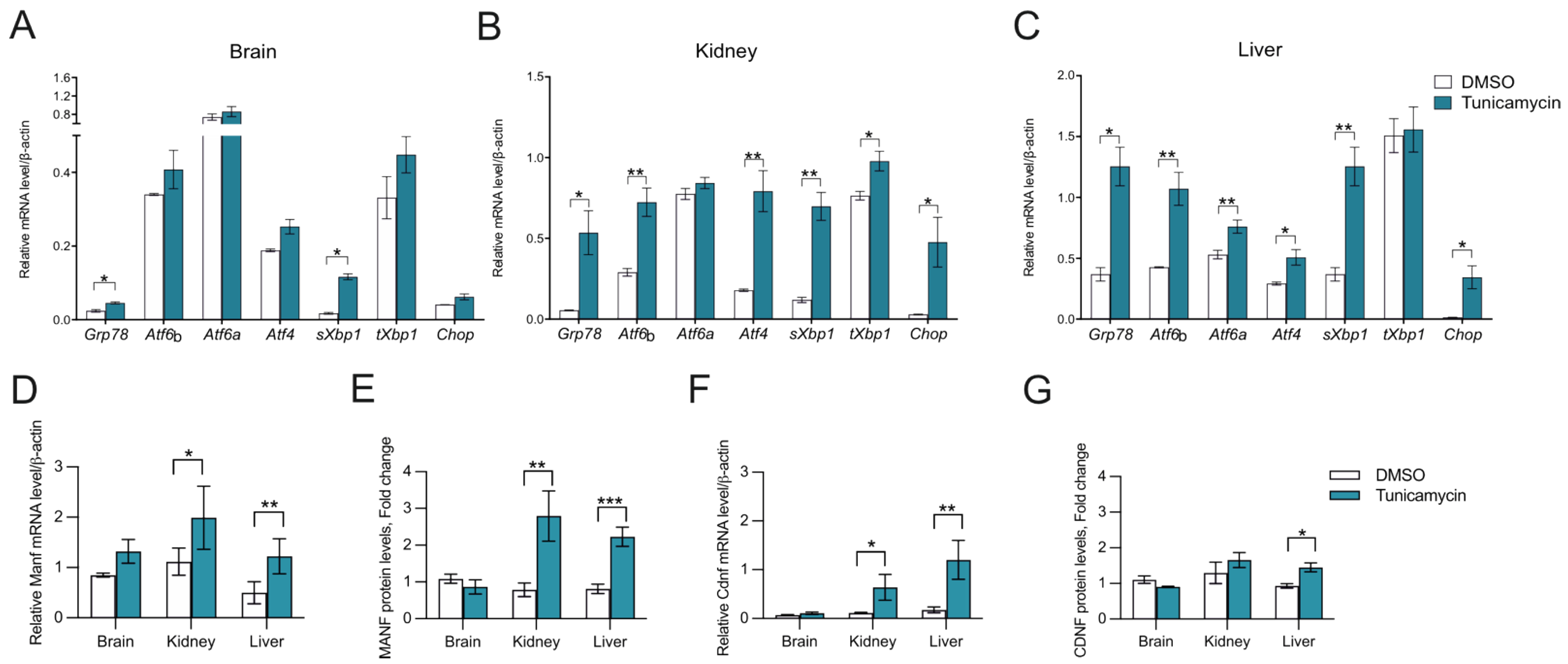
2.3. CDNF Is an ER Stress-Induced Protein In Vivo
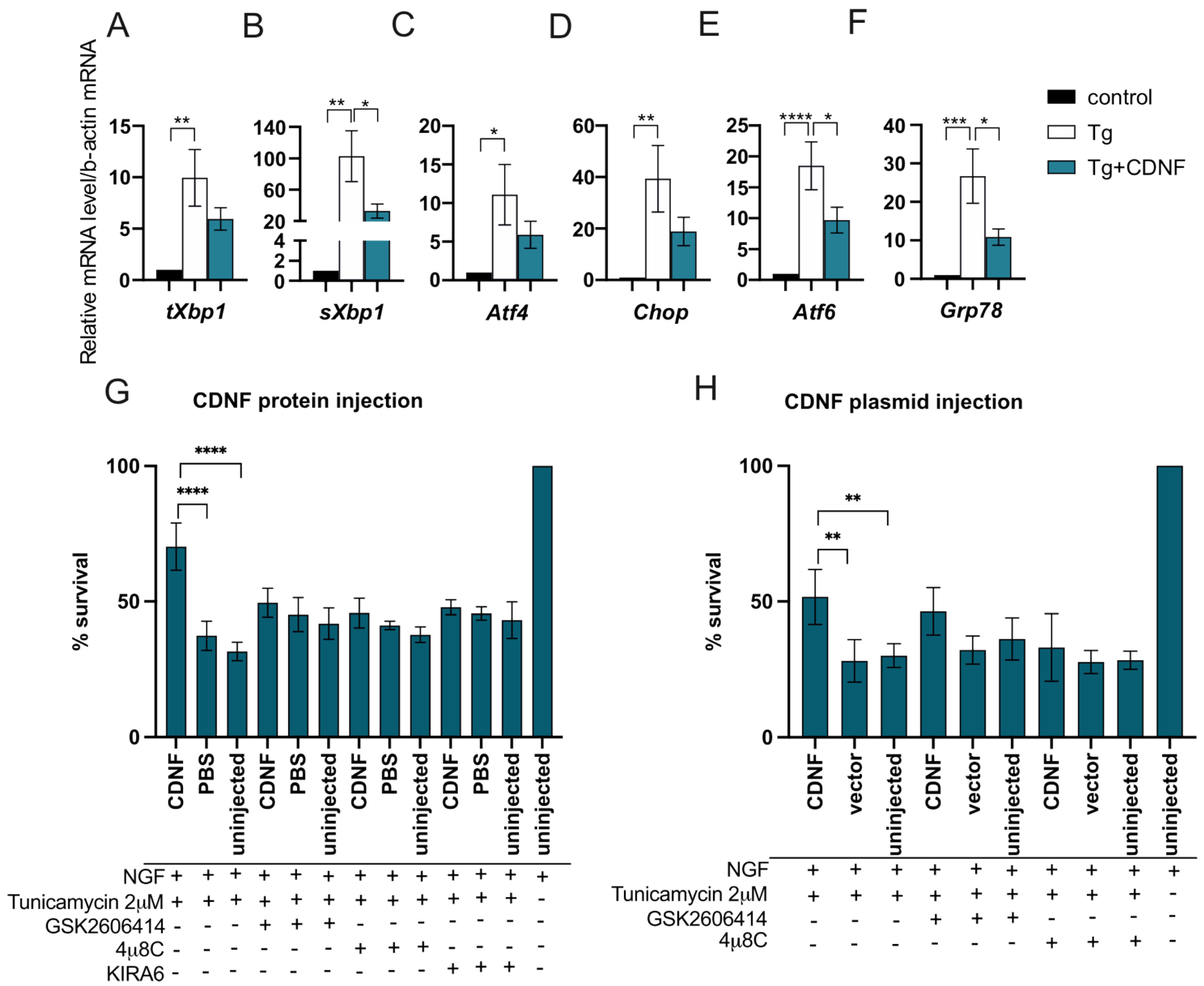
2.4. Extracellularly Applied CDNF Regulates UPR Signaling Pathways in ER-Stressed Dopamine Neuron Cultures
2.5. The Anti-Apoptotic Effect of Intracellularly Applied CDNF Relies on the Activity of IRE1α and PERK Pathways
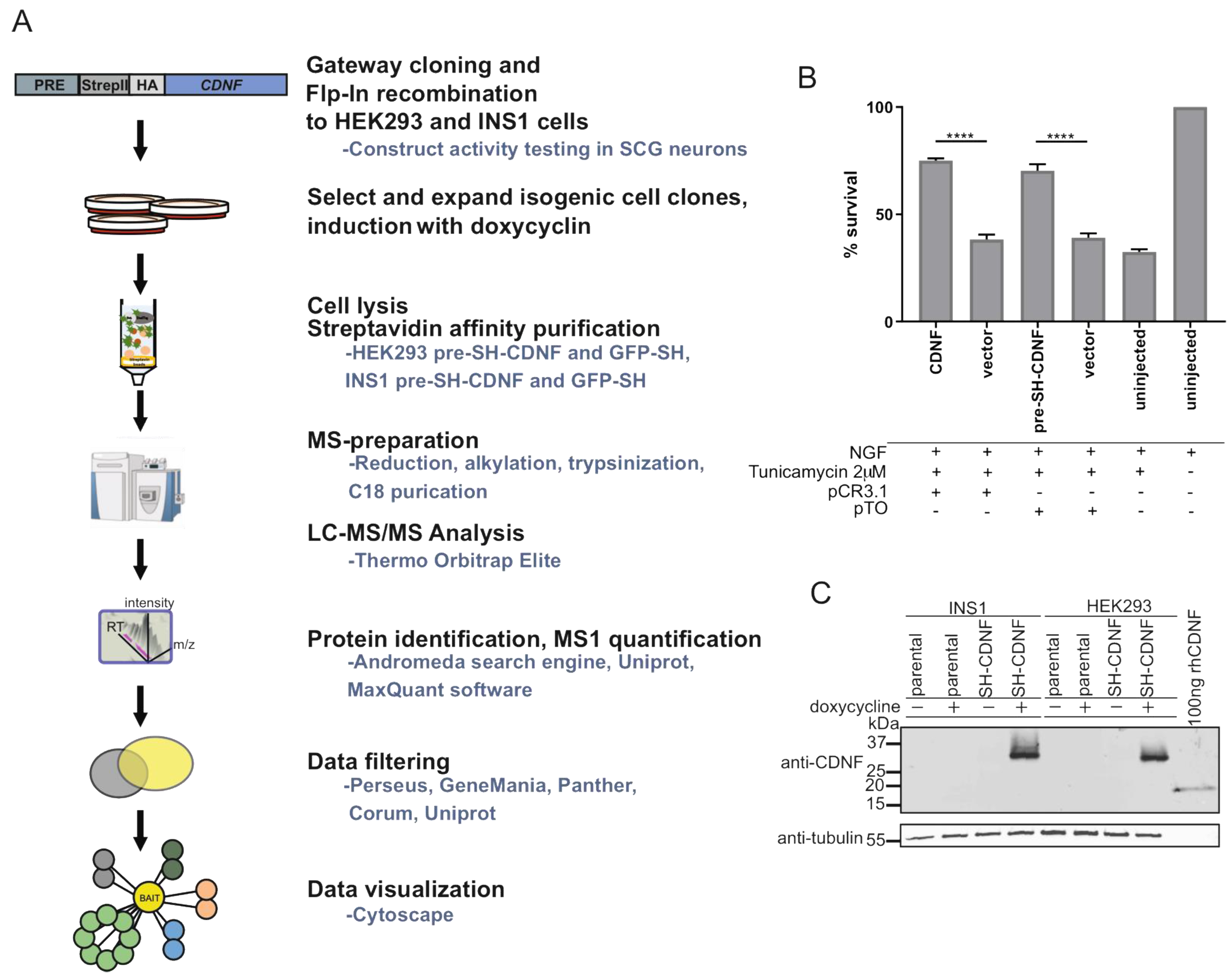
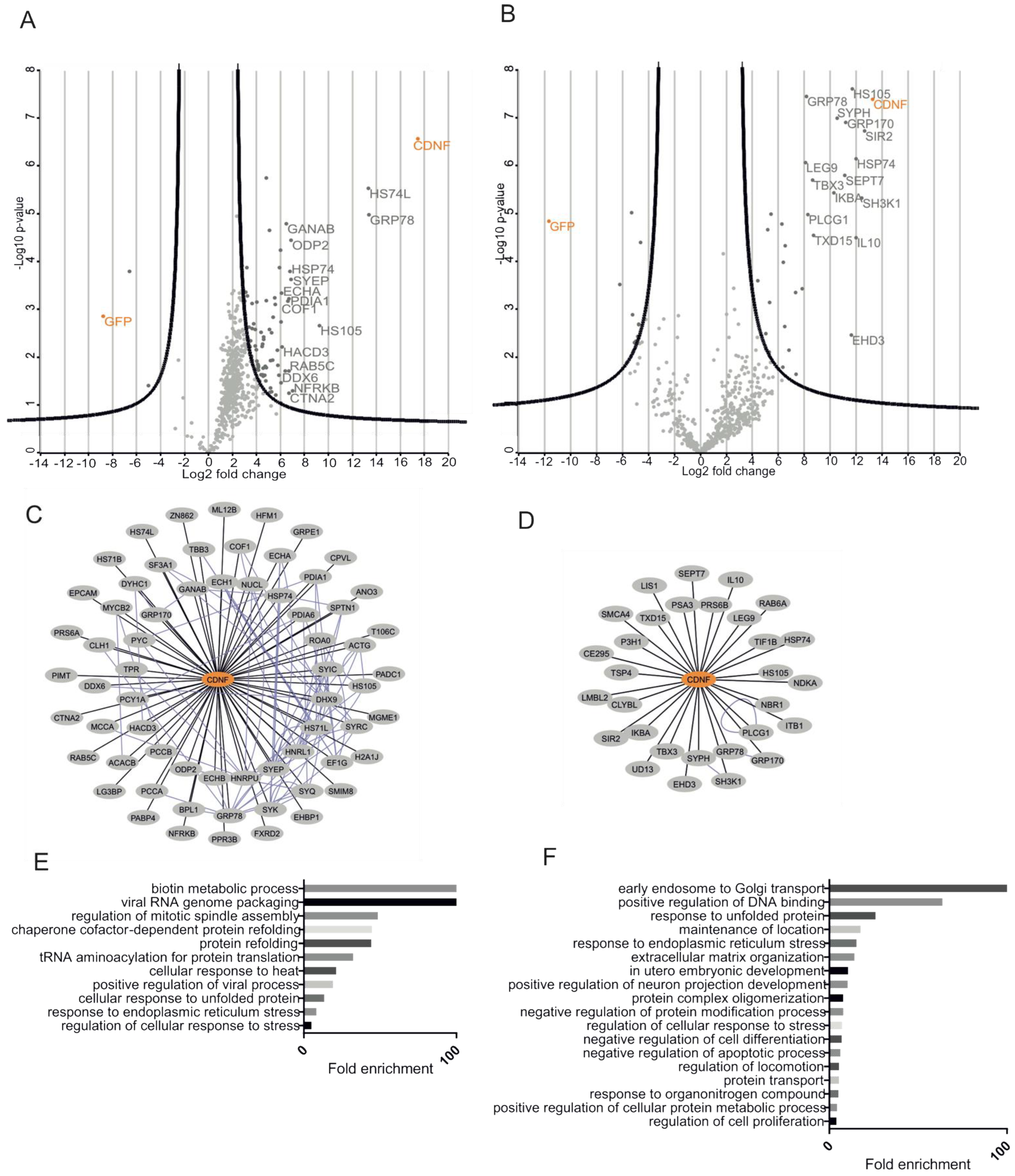
2.6. Characterization of CDNF Expression Constructs for Affinity Purification Coupled to Mass Spectrometry
2.7. CDNF Interactomes in HEK293 and INS1 Cell Lines
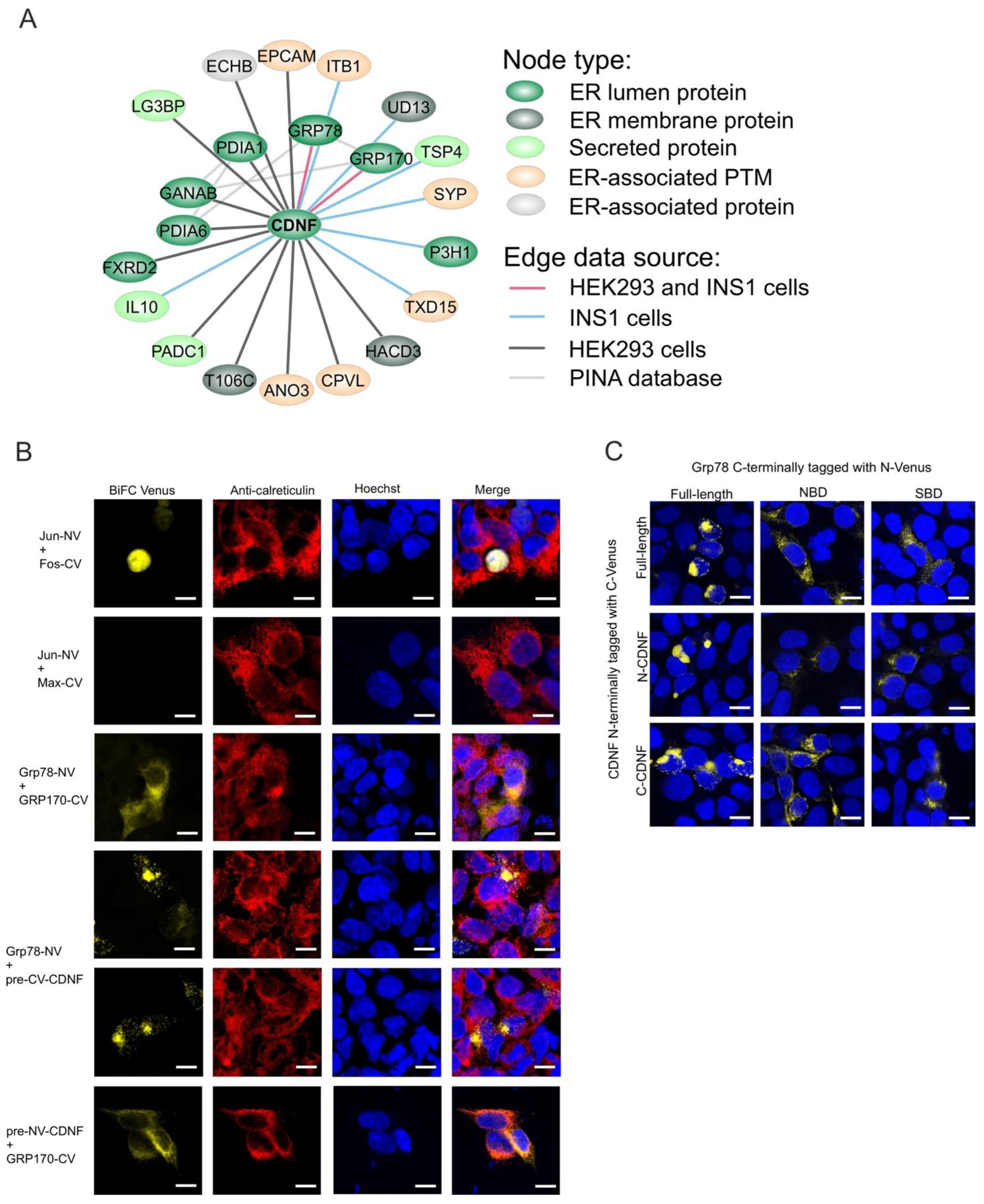
2.8. The ER-Localized Interactome of CDNF
2.9. C-Terminal Domain of CDNF (C-CDNF) Preferentially Interacts with the Nucleotide-Binding Domain of GRP78
3. Discussion
4. Materials and Methods
4.1. Mice and Treatment with Tunicamycin
4.2. RNA Isolation, Reverse Transcription Quantitative PCR (RT-qPCR)
4.3. Protein Extraction and Enzyme Linked Immunosorbent Assay (ELISA)
4.4. CDNF Expression Plasmids
4.5. Generation of Stable Isogenic Doxycycline-Inducible Cell Lines and CDNF Immunoblotting
4.6. Expression Plasmids for Bimolecular Fluorescence Complementation Assay (BiFC)
4.7. Sympathetic Neuronal Cell Culture and Microinjection
4.8. Dopamine Neuron Culture, RNA Isolation, Reverse Transcription, and Quantitative PCR
4.9. Affinity Purification
4.10. Preparation for Mass Spectrometry Analysis
4.11. Mass Spectrometry Analysis
4.12. Protein Identification and Quantification
4.13. MS Data Filtering and Analysis
4.14. Bimolecular Fluorescence Complementation Assay
4.15. Imaging
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lindholm, P.; Voutilainen, M.H.; Laurén, J.; Peränen, J.; Leppänen, V.-M.; Andressoo, J.-O.; Lindahl, M.; Janhunen, S.; Kalkkinen, N.; Timmusk, T.; et al. Novel Neurotrophic Factor CDNF Protects and Rescues Midbrain Dopamine Neurons in Vivo. Nature 2007, 448, 73–77. [Google Scholar] [CrossRef] [PubMed]
- Hellman, M.; Arumäe, U.; Yu, L.Y.; Lindholm, P.; Peränen, J.; Saarma, M.; Permi, P. Mesencephalic Astrocyte-Derived Neurotrophic Factor (MANF) Has a Unique Mechanism to Rescue Apoptotic Neurons. J. Biol. Chem. 2011, 286, 2675–2680. [Google Scholar] [CrossRef] [PubMed]
- Latge, C.; Cabral, K.M.S.; de Oliveira, G.A.P.; Raymundo, D.P.; Freitas, J.A.; Johanson, L.; Romão, L.F.; Palhano, F.L.; Herrmann, T.; Almeida, M.S.; et al. The Solution Structure and Dynamics of Full-Length Human Cerebral Dopamine Neurotrophic Factor and Its Neuroprotective Role against α-Synuclein Oligomers. J. Biol. Chem. 2015, 290, 20527–20540. [Google Scholar] [CrossRef]
- Parkash, V.; Lindholm, P.; Peränen, J.; Kalkkinen, N.; Oksanen, E.; Saarma, M.; Leppänen, V.M.; Goldman, A. The Structure of the Conserved Neurotrophic Factors MANF and CDNF Explains Why They Are Bifunctional. Protein Eng. Des. Sel. 2009, 22, 233–241. [Google Scholar] [CrossRef] [PubMed]
- Hoseki, J.; Sasakawa, H.; Yamaguchi, Y.; Maeda, M.; Kubota, H.; Kato, K.; Nagata, K. Solution Structure and Dynamics of Mouse ARMET. FEBS Lett. 2010, 584, 1536–1542. [Google Scholar] [CrossRef]
- Petrova, P.; Raibekas, A.; Pevsner, J.; Vigo, N.; Anafi, M.; Moore, M.K.; Peaire, A.E.; Shridhar, V.; Smith, D.I.; Kelly, J.; et al. MANF: A New Mesencephalic, Astrocyte-Derived Neurotrophic Factor with Selectivity for Dopaminergic Neurons. J. Mol. Neurosci. 2003, 20, 173–188. [Google Scholar] [CrossRef]
- Lindahl, M.; Saarma, M.; Lindholm, P. Unconventional Neurotrophic Factors CDNF and MANF: Structure, Physiological Functions and Therapeutic Potential. Neurobiol. Dis. 2017, 97, 90–102. [Google Scholar] [CrossRef]
- Huttunen, H.J.; Saarma, M. CDNF Protein Therapy in Parkinson’s Disease. Cell Transplant. 2019, 28, 349–366. [Google Scholar] [CrossRef]
- Lindholm, P.; Saarma, M. Cerebral Dopamine Neurotrophic Factor Protects and Repairs Dopamine Neurons by Novel Mechanism. Mol. Psychiatry 2022, 27, 1310–1321. [Google Scholar] [CrossRef]
- Airaksinen, M.S.; Saarma, M. The GDNF Family: Signalling, Biological Functions and Therapeutic Value. Nat. Rev. Neurosci. 2002, 3, 383–394. [Google Scholar] [CrossRef]
- Sidorova, Y.A.; Saarma, M. Can Growth Factors Cure Parkinson’s Disease? Trends Pharmacol. Sci. 2020, 41, 909–922. [Google Scholar] [CrossRef] [PubMed]
- Kadry, H.; Noorani, B.; Cucullo, L. A Blood-Brain Barrier Overview on Structure, Function, Impairment, and Biomarkers of Integrity. Fluids Barriers CNS 2020, 17, 69. [Google Scholar] [CrossRef] [PubMed]
- Mizobuchi, N.; Hoseki, J.; Kubota, H.; Toyokuni, S.; Nozaki, J.; Naitoh, M.; Koizumi, A.; Nagata, K. ARMET Is a Soluble ER Protein Induced by the Unfolded Protein Response via ERSE-II Element. Cell Struct. Funct. 2007, 32, 41–50. [Google Scholar] [CrossRef]
- Yagi, T.; Asada, R.; Kanekura, K.; Eesmaa, A.; Lindahl, M.; Saarma, M.; Urano, F. Neuroplastin Modulates Anti-Inflammatory Effects of MANF. iScience 2020, 23, 101810. [Google Scholar] [CrossRef]
- Glembotski, C.C.; Thuerauf, D.J.; Huang, C.; Vekich, J.A.; Gottlieb, R.A.; Doroudgar, S. Mesencephalic Astrocyte-Derived Neurotrophic Factor Protects the Heart from Ischemic Damage and Is Selectively Secreted upon Sarco/Endoplasmic Reticulum Calcium Depletion. J. Biol. Chem. 2012, 287, 25893–25904. [Google Scholar] [CrossRef]
- Brozzi, F.; Eizirik, D.L. ER Stress and the Decline and Fall of Pancreatic Beta Cells in Type 1 Diabetes. Upsala J. Med. Sci. 2016, 121, 133. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, R.; Colon-Negron, K.; Papa, F.R. Endoplasmic Reticulum Stress, Degeneration of Pancreatic Islet β-Cells, and Therapeutic Modulation of the Unfolded Protein Response in Diabetes. Mol. Metab. 2019, 27, S60. [Google Scholar] [CrossRef]
- Medinas, D.B.; Hetz, C. Modeling UPR Adaptive Responses. Nat. Chem. Biol. 2014, 10, 879–880. [Google Scholar] [CrossRef]
- Hetz, C. The Unfolded Protein Response: Controlling Cell Fate Decisions under ER Stress and Beyond. Nat. Rev. Mol. Cell Biol. 2012, 13, 89–102. [Google Scholar] [CrossRef]
- Walter, P.; Ron, D. The Unfolded Protein Response: From Stress Pathway to Homeostatic Regulation. Science 2011, 334, 1081–1086. [Google Scholar] [CrossRef]
- Hetz, C.; Papa, F.R. The Unfolded Protein Response and Cell Fate Control. Mol. Cell 2018, 69, 169–181. [Google Scholar] [CrossRef] [PubMed]
- Urra, H.; Dufey, E.; Lisbona, F.; Rojas-Rivera, D.; Hetz, C. When ER Stress Reaches a Dead End. Biochim. Et Biophys. Acta (BBA)-Mol. Cell Res. 2013, 1833, 3507–3517. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C.; Saxena, S. ER Stress and the Unfolded Protein Response in Neurodegeneration. Nat. Rev. Neurol. 2017, 13, 477–491. [Google Scholar] [CrossRef] [PubMed]
- Lindahl, M.; Danilova, T.; Palm, E.; Lindholm, P.; Võikar, V.; Hakonen, E.; Ustinov, J.; Andressoo, J.O.; Harvey, B.; Otonkoski, T.; et al. MANF Is Indispensable for the Proliferation and Survival of Pancreatic β Cells. Cell Rep. 2014, 7, 366–375. [Google Scholar] [CrossRef]
- Eesmaa, A.; Yu, L.-Y.; Göös, H.; Nõges, K.; Kovaleva, V.; Hellman, M.; Zimmermann, R.; Jung, M.; Permi, P.; Varjosalo, M.; et al. The Cytoprotective Protein MANF Promotes Neuronal Survival Independently from Its Role as a GRP78 Cofactor. J. Biol. Chem. 2021, 296, 100295. [Google Scholar] [CrossRef]
- Apostolou, A.; Shen, Y.; Liang, Y.; Luo, J.; Fang, S. Armet, a UPR-Upregulated Protein, Inhibits Cell Proliferation and ER Stress-Induced Cell Death. Exp. Cell Res. 2008, 314, 2454–2467. [Google Scholar] [CrossRef]
- Voutilainen, M.H.; de Lorenzo, F.; Stepanova, P.; Bäck, S.; Yu, L.-Y.; Lindholm, P.; Pörsti, E.; Saarma, M.; Männistö, P.T.; Tuominen, R.K. Evidence for an Additive Neurorestorative Effect of Simultaneously Administered CDNF and GDNF in Hemiparkinsonian Rats: Implications for Different Mechanism of Action. eNeuro 2017, 4, ENEURO.0117-16.2017. [Google Scholar] [CrossRef]
- Pakarinen, E.; Lindholm, P.; Saarma, M.; Lindahl, M. CDNF and MANF Regulate ER Stress in a Tissue-Specific Manner. Cell. Mol. Life Sci. 2022, 79, 124. [Google Scholar] [CrossRef]
- Arancibia, D.; Zamorano, P.; Andrés, M.E. CDNF Induces the Adaptive Unfolded Protein Response and Attenuates Endoplasmic Reticulum Stress-Induced Cell Death. Biochim. Et Biophys. Acta (BBA)-Mol. Cell Res. 2018, 1865, 1579–1589. [Google Scholar] [CrossRef]
- Zhang, G.L.; Wang, L.H.; Liu, X.Y.; Zhang, Y.X.; Hu, M.Y.; Liu, L.; Fang, Y.Y.; Mu, Y.; Zhao, Y.; Huang, S.H.; et al. Cerebral Dopamine Neurotrophic Factor (CDNF) Has Neuroprotective Effects against Cerebral Ischemia That May Occur through the Endoplasmic Reticulum Stress Pathway. Int. J. Mol. Sci. 2018, 19, 1905. [Google Scholar] [CrossRef]
- Zhou, W.; Chang, L.; Fang, Y.; Du, Z.; Li, Y.; Song, Y.; Hao, F.; Lv, L.; Wu, Y. Cerebral Dopamine Neurotrophic Factor Alleviates Aβ25-35-Induced Endoplasmic Reticulum Stress and Early Synaptotoxicity in Rat Hippocampal Cells. Neurosci. Lett. 2016, 633, 40–46. [Google Scholar] [CrossRef] [PubMed]
- Latgé, C.; Cabral, K.M.S.; Almeida, M.S.; Foguel, D. 1H-, 13C- and 15N-NMR Assignment of the N-Terminal Domain of Human Cerebral Dopamine Neurotrophic Factor (CDNF). Biomol. NMR Assign. 2013, 7, 101–103. [Google Scholar] [CrossRef] [PubMed]
- Mätlik, K.; Yu, L.-Y.; Eesmaa, A.; Hellman, M.; Lindholm, P.; Peränen, J.; Galli, E.; Anttila, J.; Saarma, M.; Permi, P.; et al. Role of Two Sequence Motifs of Mesencephalic Astrocyte-Derived Neurotrophic Factor in Its Survival-Promoting Activity. Cell Death Dis. 2015, 6, e2032. [Google Scholar] [CrossRef] [PubMed]
- Henderson, M.J.; Richie, C.T.; Airavaara, M.; Wang, Y.; Harvey, B.K. Mesencephalic Astrocyte-Derived Neurotrophic Factor (MANF) Secretion and Cell Surface Binding Are Modulated by KDEL Receptors. J. Biol. Chem. 2013, 288, 4209–4225. [Google Scholar] [CrossRef]
- Galli, E.; Lindholm, P.; Kontturi, L.-S.; Saarma, M.; Urtti, A.; Yliperttula, M. Characterization of CDNF-Secreting ARPE-19 Cell Clones for Encapsulated Cell Therapy. Cell Transplant. 2019, 28, 413–424. [Google Scholar] [CrossRef]
- Norisada, J.; Hirata, Y.; Amaya, F.; Kiuchi, K.; Oh-hashi, K. A Comparative Analysis of the Molecular Features of MANF and CDNF. PLoS ONE 2016, 11, e0146923. [Google Scholar] [CrossRef]
- Albert, K.; Raymundo, D.P.; Panhelainen, A.; Eesmaa, A.; Shvachiy, L.; Araújo, G.R.; Chmielarz, P.; Yan, X.; Singh, A.; Cordeiro, Y.; et al. Cerebral Dopamine Neurotrophic Factor Reduces α-Synuclein Aggregation and Propagation and Alleviates Behavioral Alterations in Vivo. Mol. Ther. 2021, 29, 2821–2840. [Google Scholar] [CrossRef]
- Lindahl, M.; Chalazonitis, A.; Palm, E.; Pakarinen, E.; Danilova, T.; Pham, T.D.; Setlik, W.; Rao, M.; Võikar, V.; Huotari, J.; et al. Cerebral Dopamine Neurotrophic Factor–Deficiency Leads to Degeneration of Enteric Neurons and Altered Brain Dopamine Neuronal Function in Mice. Neurobiol. Dis. 2020, 134, 104696. [Google Scholar] [CrossRef]
- Chalazonitis, A.; Li, Z.S.; Pham, T.D.; Chen, J.; Rao, M.; Lindholm, P.; Saarma, M.; Lindahl, M.; Gershon, M.D. Cerebral Dopamine Neurotrophic Factor Is Essential for Enteric Neuronal Development, Maintenance, and Regulation of Gastrointestinal Transit. J. Comp. Neurol. 2020, 528, 2420–2444. [Google Scholar] [CrossRef]
- Lin, L.F.; Doherty, D.H.; Lile, J.D.; Bektesh, S.; Collins, F. GDNF: A Glial Cell Line-Derived Neurotrophic Factor for Midbrain Dopaminergic Neurons. Science 1993, 260, 1130–1132. [Google Scholar] [CrossRef]
- Treiman, M.; Caspersen, C.; Christensen, S.B. A Tool Coming of Age: Thapsigargin as an Inhibitor of Sarco-Endoplasmic Reticulum Ca2+-ATPases. Trends Pharmacol. Sci. 1998, 19, 131–135. [Google Scholar] [CrossRef]
- Ryu, E.J.; Harding, H.P.; Angelastro, J.M.; Vitolo, O.V.; Ron, D.; Greene, L.A. Endoplasmic Reticulum Stress and the Unfolded Protein Response in Cellular Models of Parkinson’s Disease. J. Neurosci. 2002, 22, 10690–10698. [Google Scholar] [CrossRef] [PubMed]
- Airavaara, M.; Harvey, B.K.; Voutilainen, M.H.; Shen, H.; Chou, J.; Lindholm, P.; Lindahl, M.; Tuominen, R.K.; Saarma, M.; Hoffer, B.; et al. CDNF Protects the Nigrostriatal Dopamine System and Promotes Recovery After MPTP Treatment in Mice. Cell Transplant. 2012, 21, 1213–1223. [Google Scholar] [CrossRef] [PubMed]
- Voutilainen, M.H.; Bäck, S.; Peränen, J.; Lindholm, P.; Raasmaja, A.; Männistö, P.T.; Saarma, M.; Tuominen, R.K. Chronic Infusion of CDNF Prevents 6-OHDA-Induced Deficits in a Rat Model of Parkinson’s Disease. Exp. Neurol. 2011, 228, 99–108. [Google Scholar] [CrossRef] [PubMed]
- Oslowski, C.M.; Urano, F. Measuring ER Stress and the Unfolded Protein Response Using Mammalian Tissue Culture System. Methods Enzym. 2011, 490, 71–92. [Google Scholar] [CrossRef]
- Ettlinger, C.; Schindler, J.; Lehle, L. Cell-Cycle Arrest of Plant Suspension Cultures by Tunicamycin. Planta 1986, 168, 101–105. [Google Scholar] [CrossRef]
- Parodi, A.J. Role of N-Oligosaccharide Endoplasmic Reticulum Processing Reactions in Glycoprotein Folding and Degradation. Biochem. J. 2000, 348 Pt 1, 1–13. [Google Scholar] [CrossRef]
- Wang, H.; Wang, X.; Ke, Z.-J.; Comer, A.L.; Xu, M.; Frank, J.A.; Zhang, Z.; Shi, X.; Luo, J. Tunicamycin-Induced Unfolded Protein Response in the Developing Mouse Brain. Toxicol. Appl. Pharmacol. 2015, 283, 157–167. [Google Scholar] [CrossRef]
- Kim, Y.; Lee, H.; Manson, S.R.; Lindahl, M.; Evans, B.; Miner, J.H.; Urano, F.; Chen, Y.M. Mesencephalic Astrocyte-Derived Neurotrophic Factor as a Urine Biomarker for Endoplasmic Reticulum Stress-Related Kidney Diseases. J. Am. Soc. Nephrol. 2016, 27, 2974–2982. [Google Scholar] [CrossRef]
- Axten, J.M.; Medina, J.R.; Feng, Y.; Shu, A.; Romeril, S.P.; Grant, S.W.; Li, W.H.H.; Heerding, D.A.; Minthorn, E.; Mencken, T.; et al. Discovery of 7-Methyl-5-(1-{[3-(Trifluoromethyl)Phenyl]Acetyl}-2,3-Dihydro-1H-Indol-5-Yl)-7H-Pyrrolo[2,3-d ]Pyrimidin-4-Amine (GSK2606414), a Potent and Selective First-in-Class Inhibitor of Protein Kinase R (PKR)-like Endoplasmi. J. Med. Chem. 2012, 55, 7193–7207. [Google Scholar] [CrossRef]
- Cross, B.C.S.; Bond, P.J.; Sadowski, P.G.; Jha, B.K.; Zak, J.; Goodman, J.M.; Silverman, R.H.; Neubert, T.A.; Baxendale, I.R.; Ron, D.; et al. The Molecular Basis for Selective Inhibition of Unconventional MRNA Splicing by an IRE1-Binding Small Molecule. Proc. Natl. Acad. Sci. USA 2012, 109, E869–E878. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, R.; Wang, L.; Wang, E.S.; Perera, B.G.K.; Igbaria, A.; Morita, S.; Prado, K.; Thamsen, M.; Caswell, D.; Macias, H.; et al. Allosteric Inhibition of the IRE1α RNase Preserves Cell Viability and Function during Endoplasmic Reticulum Stress. Cell 2014, 158, 534–548. [Google Scholar] [CrossRef] [PubMed]
- Thomas, H.; Senkel, S.; Erdmann, S.; Arndt, T.; Turan, G.; Klein-Hitpass, L.; Ryffel, G.U. Pattern of Genes Influenced by Conditional Expression of the Transcription Factors HNF6, HNF4alpha and HNF1beta in a Pancreatic Beta-Cell Line. Nucleic Acids Res. 2004, 32, e150. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Yan, Y.; Rato, C.; Rohland, L.; Preissler, S.; Ron, D. MANF Antagonizes Nucleotide Exchange by the Endoplasmic Reticulum Chaperone BiP. Nat. Commun. 2019, 10, 541. [Google Scholar] [CrossRef]
- Hu, C.-D.; Chinenov, Y.; Kerppola, T.K. Visualization of Interactions among BZIP and Rel Family Proteins in Living Cells Using Bimolecular Fluorescence Complementation. Mol. Cell 2002, 9, 789–798. [Google Scholar] [CrossRef]
- Melnyk, A.; Rieger, H.; Zimmermann, R. Co-Chaperones of the Mammalian Endoplasmic Reticulum; Springer: Cham, Switzerland, 2015; pp. 179–200. [Google Scholar]
- Lindholm, P.; Saarma, M. Novel CDNF/MANF Family of Neurotrophic Factors. Dev. Neurobiol. 2010, 70, 360–371. [Google Scholar] [CrossRef]
- Yu, L.Y.; Saarma, M.; Arumäe, U. Death Receptors and Caspases but Not Mitochondria Are Activated in the GDNF- or BDNF-Deprived Dopaminergic Neurons. J. Neurosci. 2008, 28, 7467–7475. [Google Scholar] [CrossRef]
- Hellman, M.; Peränen, J.; Saarma, M.; Permi, P. 1H, 13C and 15N Resonance Assignments of the Human Mesencephalic Astrocyte-Derived Neurotrophic Factor. Biomol. NMR Assign. 2010, 4, 215–217. [Google Scholar] [CrossRef]
- Bai, M.; Vozdek, R.; Hnízda, A.; Jiang, C.; Wang, B.; Kuchar, L.; Li, T.; Zhang, Y.; Wood, C.; Feng, L.; et al. Conserved Roles of C. Elegans and Human MANFs in Sulfatide Binding and Cytoprotection. Nat. Commun. 2018, 9, 897. [Google Scholar] [CrossRef]
- Lindström, R.; Lindholm, P.; Kallijärvi, J.; Yu, L.-y.; Piepponen, T.P.; Arumäe, U.; Saarma, M.; Heino, T.I. Characterization of the Structural and Functional Determinants of MANF/CDNF in Drosophila In Vivo Model. PLoS ONE 2013, 8, e73928. [Google Scholar] [CrossRef]
- Maciel, L.; de Oliveira, D.F.; Mesquita, F.; da Silva Souza, H.A.; Oliveira, L.; Christie, M.L.A.; Palhano, F.L.; de Carvalho, A.C.C.; Nascimento, J.H.M.; Foguel, D. New Cardiomyokine Reduces Myocardial Ischemia/Reperfusion Injury by PI3K-AKT Pathway Via a Putative KDEL-Receptor Binding. J. Am. Heart Assoc. 2021, 10, e019685. [Google Scholar] [CrossRef]
- Berndt, C.; Lillig, C.H.; Holmgren, A. Thioredoxins and Glutaredoxins as Facilitators of Protein Folding. Biochim. Et Biophys. Acta (BBA)-Mol. Cell Res. 2008, 1783, 641–650. [Google Scholar] [CrossRef] [PubMed]
- Hartley, C.L.; Edwards, S.; Mullan, L.; Bell, P.A.; Fresquet, M.; Boot-Handford, R.P.; Briggs, M.D. Armet/Manf and Creld2 Are Components of a Specialized ER Stress Response Provoked by Inappropriate Formation of Disulphide Bonds: Implications for Genetic Skeletal Diseases. Hum. Mol. Genet. 2013, 22, 5262–5275. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Yu, C.; Yu, H.; Zhong, L.; Wang, Y.; Liu, J.; Zhang, S.; Sun, J.; Duan, L.; Gong, L.; et al. Cerebral Dopamine Neurotrophic Factor Protects H9c2 Cardiomyocytes from Apoptosis. Herz 2018, 43, 346–351. [Google Scholar] [CrossRef]
- DuRose, J.B.; Tam, A.B.; Niwa, M. Intrinsic Capacities of Molecular Sensors of the Unfolded Protein Response to Sense Alternate Forms of Endoplasmic Reticulum Stress. Mol. Biol. Cell 2006, 17, 3095–3107. [Google Scholar] [CrossRef]
- Raina, K.; Noblin, D.J.; Serebrenik, Y.V.; Adams, A.; Zhao, C.; Crews, C.M. Targeted Protein Destabilization Reveals an Estrogen-Mediated ER Stress Response. Nat. Chem. Biol. 2014, 10, 957–962. [Google Scholar] [CrossRef]
- Bergmann, T.J.; Fregno, I.; Fumagalli, F.; Rinaldi, A.; Bertoni, F.; Boersema, P.J.; Picotti, P.; Molinari, M. Chemical Stresses Fail to Mimic the Unfolded Protein Response Resulting from Luminal Load with Unfolded Polypeptides. J. Biol. Chem. 2018, 293, 5600–5612. [Google Scholar] [CrossRef]
- Blaszczak, E.; Lazarewicz, N.; Sudevan, A.; Wysocki, R.; Rabut, G. Protein-Fragment Complementation Assays for Large-Scale Analysis of Protein-Protein Interactions. Biochem. Soc. Trans. 2021, 49, 1337–1348. [Google Scholar] [CrossRef]
- Liao, J.; Madahar, V.; Dang, R.; Jiang, L. Quantitative FRET (QFRET) Technology for the Determination of Protein-Protein Interaction Affinity in Solution. Molecules 2021, 26, 6339. [Google Scholar] [CrossRef]
- Verweij, E.W.E.; Bosma, R.; Gao, M.; van den Bor, J.; al Araaj, B.; de Munnik, S.M.; Ma, X.; Leurs, R.; Vischer, H.F. BRET-Based Biosensors to Measure Agonist Efficacies in Histamine H 1 Receptor-Mediated G Protein Activation, Signaling and Interactions with GRKs and β-Arrestins. Int. J. Mol. Sci. 2022, 23, 3184. [Google Scholar] [CrossRef]
- Zhang, L.; Zhou, J.; Xu, M.; Yuan, G. Exploration of the Hsa-MiR-1587-Protein Interaction and the Inhibition to CASK. Int. J. Mol. Sci. 2021, 22, 10716. [Google Scholar] [CrossRef] [PubMed]
- Varjosalo, M.; Sacco, R.; Stukalov, A.; van Drogen, A.; Planyavsky, M.; Hauri, S.; Aebersold, R.; Bennett, K.L.; Colinge, J.; Gstaiger, M.; et al. Interlaboratory Reproducibility of Large-Scale Human Protein-Complex Analysis by Standardized AP-MS. Nat. Methods 2013, 10, 307–314. [Google Scholar] [CrossRef] [PubMed]
- Varjosalo, M.; Keskitalo, S.; Van Drogen, A.; Nurkkala, H.; Vichalkovski, A.; Aebersold, R.; Gstaiger, M. The Protein Interaction Landscape of the Human CMGC Kinase Group. Cell Rep. 2013, 3, 1306–1320. [Google Scholar] [CrossRef] [PubMed]
- Danilova, T.; Galli, E.; Pakarinen, E.; Palm, E.; Lindholm, P.; Saarma, M.; Lindahl, M. Mesencephalic Astrocyte-Derived Neurotrophic Factor (MANF) Is Highly Expressed in Mouse Tissues With Metabolic Function. Front. Endocrinol. 2019, 10, 765. [Google Scholar] [CrossRef] [PubMed]
- Kovaleva, V.; Yu, L.-Y.; Ivanova, L.; Nam, J.; Eesmaa, A.; Kumpula, E.-P.; Huiskonen, J.; Lindholm, P.; Voutilainen, M.; Karelson, M.; et al. MANF Regulates Unfolded Protein Response and Neuronal Survival through Its ER-Located Receptor IRE1α. bioRxiv 2020. [Google Scholar] [CrossRef]
- Donaldson, A.E.; Marshall, C.E.; Yang, M.; Suon, S.; Iacovitti, L. Purified Mouse Dopamine Neurons Thrive and Function after Transplantation into Brain but Require Novel Glial Factors for Survival in Culture. Mol. Cell. Neurosci. 2005, 30, 108–117. [Google Scholar] [CrossRef]
- Kessler, M.A.; Yang, M.; Gollomp, K.L.; Jin, H.; Iacovitti, L. The Human Tyrosine Hydroxylase Gene Promoter. Mol. Brain Res. 2003, 112, 8–23. [Google Scholar] [CrossRef]
- Cao, D.; Ma, X.; Cai, J.; Luan, J.; Liu, A.-J.; Yang, R.; Cao, Y.; Zhu, X.; Zhang, H.; Chen, Y.-X.; et al. ZBTB20 Is Required for Anterior Pituitary Development and Lactotrope Specification. Nat. Commun. 2016, 7, 11121. [Google Scholar] [CrossRef]
- Galli, E.; Rossi, J.; Neumann, T.; Andressoo, J.O.; Drinda, S.; Lindholm, P. Mesencephalic Astrocyte-Derived Neurotrophic Factor Is Upregulated with Therapeutic Fasting in Humans and Diet Fat Withdrawal in Obese Mice. Sci. Rep. 2019, 9, 14318. [Google Scholar] [CrossRef]
- Petersen, T.N.; Brunak, S.; von Heijne, G.; Nielsen, H. SignalP 4.0: Discriminating Signal Peptides from Transmembrane Regions. Nat. Methods 2011, 8, 785–786. [Google Scholar] [CrossRef]
- Yu, L.Y.; Jokitalo, E.; Sun, Y.F.; Mehlen, P.; Lindholm, D.; Saarma, M.; Arumäe, U. GDNF-Deprived Sympathetic Neurons Die via a Novel Nonmitochondrial Pathway. J. Cell Biol. 2003, 163, 987. [Google Scholar] [CrossRef] [PubMed]
- Danilova, T.; Belevich, I.; Li, H.; Palm, E.; Jokitalo, E.; Otonkoski, T.; Lindahl, M. MANF Is Required for the Postnatal Expansion and Maintenance of Pancreatic β-Cell Mass in Mice. Diabetes 2019, 68, 66–80. [Google Scholar] [CrossRef] [PubMed]
- Cox, J.; Neuhauser, N.; Michalski, A.; Scheltema, R.A.; Olsen, J.V.; Mann, M. Andromeda: A Peptide Search Engine Integrated into the MaxQuant Environment. J. Proteome Res. 2011, 10, 1794–1805. [Google Scholar] [CrossRef] [PubMed]
- Tyanova, S.; Temu, T.; Sinitcyn, P.; Carlson, A.; Hein, M.Y.; Geiger, T.; Mann, M.; Cox, J. The Perseus Computational Platform for Comprehensive Analysis of (Prote)Omics Data. Nat. Methods 2016, 13, 731–740. [Google Scholar] [CrossRef]
- Cowley, M.J.; Pinese, M.; Kassahn, K.S.; Waddell, N.; Pearson, J.V.; Grimmond, S.M.; Biankin, A.V.; Hautaniemi, S.; Wu, J. PINA v2.0: Mining Interactome Modules. Nucleic Acids Res. 2012, 40, D862–D865. [Google Scholar] [CrossRef]
- Wu, J.; Vallenius, T.; Ovaska, K.; Westermarck, J.; Mäkelä, T.P.; Hautaniemi, S. Integrated Network Analysis Platform for Protein-Protein Interactions. Nat. Methods 2009, 6, 75–77. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Mi, H.; Huang, X.; Muruganujan, A.; Tang, H.; Mills, C.; Kang, D.; Thomas, P.D. PANTHER Version 11: Expanded Annotation Data from Gene Ontology and Reactome Pathways, and Data Analysis Tool Enhancements. Nucleic Acids Res. 2017, 45, D183–D189. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Eesmaa, A.; Yu, L.-Y.; Göös, H.; Danilova, T.; Nõges, K.; Pakarinen, E.; Varjosalo, M.; Lindahl, M.; Lindholm, P.; Saarma, M. CDNF Interacts with ER Chaperones and Requires UPR Sensors to Promote Neuronal Survival. Int. J. Mol. Sci. 2022, 23, 9489. https://doi.org/10.3390/ijms23169489
Eesmaa A, Yu L-Y, Göös H, Danilova T, Nõges K, Pakarinen E, Varjosalo M, Lindahl M, Lindholm P, Saarma M. CDNF Interacts with ER Chaperones and Requires UPR Sensors to Promote Neuronal Survival. International Journal of Molecular Sciences. 2022; 23(16):9489. https://doi.org/10.3390/ijms23169489
Chicago/Turabian StyleEesmaa, Ave, Li-Ying Yu, Helka Göös, Tatiana Danilova, Kristofer Nõges, Emmi Pakarinen, Markku Varjosalo, Maria Lindahl, Päivi Lindholm, and Mart Saarma. 2022. "CDNF Interacts with ER Chaperones and Requires UPR Sensors to Promote Neuronal Survival" International Journal of Molecular Sciences 23, no. 16: 9489. https://doi.org/10.3390/ijms23169489
APA StyleEesmaa, A., Yu, L.-Y., Göös, H., Danilova, T., Nõges, K., Pakarinen, E., Varjosalo, M., Lindahl, M., Lindholm, P., & Saarma, M. (2022). CDNF Interacts with ER Chaperones and Requires UPR Sensors to Promote Neuronal Survival. International Journal of Molecular Sciences, 23(16), 9489. https://doi.org/10.3390/ijms23169489