Abstract
Graves’ disease, characterized by hyperthyroidism resulting from loss of immune tolerance to thyroid autoantigens, may be attributable to both genetic and environmental factors. Allogeneic hematopoietic stem cell transplantation (HSCT) represents a means to induce immunotolerance via an artificial immune environment. We present a male patient with severe aplastic anemia arising from a germline SAMD9L missense mutation who successfully underwent HSCT from his HLA-haploidentical SAMD9L non-mutated father together with nonmyeloablative conditioning and post-transplant cyclophosphamide at 8 years of age. He did not suffer graft-versus-host disease, but Graves’ disease evolved 10 months post-transplant when cyclosporine was discontinued for one month. Reconstitution of peripheral lymphocyte subsets was found to be transiently downregulated shortly after Graves’ disease onset but recovered upon antithyroid treatment. Our investigation revealed the presence of genetic factors associated with Graves’ disease, including HLA-B*46:01 and HLA-DRB1*09:01 haplotypes carried by the asymptomatic donor and germline FLT3 c.2500C>T mutation carried by both the patient and the donor. Given his current euthyroid state with normal hematopoiesis, the patient has returned to normal school life. This rare event of Graves’ disease in a young boy arising from special HSCT circumstances indicates that both the genetic background and the HSCT environment can prompt the evolution of Graves’ disease.
1. Introduction
Autoimmune Graves’ disease, featuring a failure to tolerate thyroid-derived antigens, is characterized by hyperthyroidism and biochemically elevated thyroxine or suppressed thyrotropin together with the presence of thyroid-stimulating thyrotropin receptor antibodies [1,2]. Genetic predisposition and nongenetic factors have been linked to Graves’ disease, resulting in overproduction of thyroid hormones by thyroid epithelial cells due to overactive thyrotropin receptor. Together with drastically amplified intrathyroidal cytokine production by infiltrating immune cells, thyrotropin receptor overactivity further activates and sustains inflammation to alter the behavior of thyroid epithelial cells [2,3,4]. Allogeneic hematopoietic stem cell transplantation (HSCT) procedures, including cytoreductive therapies to condition and modulate graft–host interactions, represent artificial platforms for immune reconstitution and tolerance induction. However, significantly, chronic graft-versus-host disease (GVHD) may develop if immune tolerance induction is aberrant [5]. Several reports have described Graves’ disease as having evolved following allogeneic HSCT for aplastic anemia, and they proposed associated factors for the rare coincidence including GVHD, use of rabbit antithymocyte globulins (rATG), and adoptive transfer of donor pathogenic lymphocytes [6,7,8,9,10,11]. Here, we report a case of coincident Graves’ disease during post-transplant immune reconstitution in a boy displaying bone marrow failure and carrying germline SAMD9L and FLT3 variants. This patient had undergone father-to-son haploidentical peripheral blood stem cell (PBSC) transplantation with nonmyeloablative conditioning and high-dose post-transplant cyclophosphamide (PTCy) treatment.
2. Case Report
The patient, an eight-year-old boy and the sole child of nonconsanguineous parents with Chinese Han ethnicity, was diagnosed with severe aplastic anemia in October 2019. Treatments were initiated soon after diagnosis, including intravenous immunoglobulin (1 g per kg; 2 doses in October 2019), eltrombopag (up to 75 mg/day administered from October 2019 to April 2020), rATG (thymoglobulin 2.5 mg/kg/day for 5 days in October 2019), cyclosporine (administered by titrating the serum level between 200 and 300 ng/mL from October 2019), and intermittent granulocyte-colony-stimulating factors. The hematologic response was deemed inadequate, with a transfusion dependency requiring red blood cells to be provided monthly and platelets biweekly. Persistently low neutrophil counts (ranging between 0.35 and 0.75 × 109/L) were also observed despite bone marrow cellularity increasing from <1% at diagnosis to 5–10% (July 2020). No physical anomalies or laboratory evidence of cytogenetic changes or Fanconi anemia were detected in July 2020 upon transfer to Koo Foundation Sun Yat-Sen Cancer Center, Taipei, although morphological myelodysplasia in the bone marrow was a concern (data not shown). Whole-exome sequencing revealed a germline SAMD9L c.3800G>T; p.(Cys1267Phe) mutation with a very low population allele frequency, which was validated by Sanger sequencing (Figure 1a–c). The patient’s bone marrow failure syndrome was assumed to be linked to this mutation. Both parents displayed normal hematopoiesis despite the patient’s mother also carrying the same SAMD9L mutation. Telomere length was also found to be short in the patient relative to an age-matched control (Figure 1d).
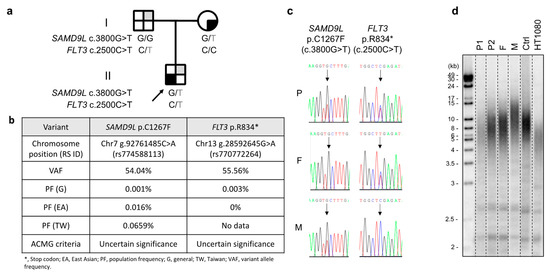
Figure 1.
(a) Pedigree of the affected family. Gray-shaded quarters represent carriers of the FLT3 c.2500C>T mutation, and black-shaded quarters represent carriers of the SAMD9L c.3800G>T mutation. Squares and circle represent male and female subjects, respectively. (b) Characteristics of the variants. American College of Medical Genetics and Genomics (ACMG) criteria for variant interpretation encompass a five-tier classification: Pathogenic, Likely pathogenic, Uncertain significance, Likely benign, and Benign. (c) Sanger DNA sequencing of SAMD9L exon 5 on chromosome 7 and FLT3 exon 20 on chromosome 13 from peripheral blood cells taken from subjects of the studied pedigree. Patient (P), patient’s father (F), patient’s mother (M). (d) Telomere length analysis (using terminal restriction fragment assay) of DNA from leukocytes of the patient pre-transplant (P1) and 1.5 years post-transplant (P2) as well as from the patient’s father (F), patient’s mother (M), a healthy control (Ctrl), and from HT1080 fibrosarcoma cancer cells. The patient pre-transplant (P1) possessed shorter and heterogeneous telomeres compared to the control (Ctrl). However, the telomeres of the patient 1.5 years post-transplant (P2) became more normal and similar to those of his father (F). For illustrative purposes, the blot has been cropped (original provided in the Supplementary Materials; Figure S1).
As HLA-matched donors were unavailable, a decision was made to pursue haploidentical PBSC transplantation into the patient from his father, who lacked the SAMD9L mutation, as salvage therapy. The conditioning treatments consisted of intravenous (IV) fludarabine (30 mg/m2 daily from day −6 to −2) and cyclophosphamide (14.5 mg/kg daily IV from day −6 to −5), as well as 4 Gy total body irradiation on day −1. In August 2020, the boy received PBSCs comprising 14.5 × 108 total nucleated cells/kg, 14.0 × 106 CD34+ cells/kg and 1.4 × 108 CD3+ cells/kg. The donor and patient were HLA-haploidentical (A 11/-, B 46/51, C 01/15, DQ 03/-, DR 09/11 to A 0201/1101, B 3901/5102, Cw 0702/1502, DQB1 0301/-, DRB1 1101/-) and ABO-nonidentical (O to B). For GVHD prophylaxis, the patient received PTCy (cyclophosphamide 50 mg/kg daily IV on days +3 and +4), cyclosporine (for 9 months by adjusting the serum level at 200–300 ng/mL during first month post-transplant and at 150–250 ng/mL thereafter), and mycophenolate mofetil (500 mg orally twice daily until day +30) from day +5. Ursodiol (until day +100) was given to prevent sinusoidal obstruction syndrome, and baktar (for 9 months), valacyclovir (until day +30), entecavir (for 1 year), and letermovir (until day +100) were provided to prevent post-transplant pneumocystis infection or reactivations of herpes simplex, hepatitis B, and cytomegalovirus, respectively. Neutrophils engrafted on day +14, with 100% donor chimerism thereafter. No transfusions were required after day +11. CD4+ T cell reconstitution reached 164 cells/μL on day +63.
The patient’s treatment course was complicated by stomatitis (alleviated by valacyclovir treatment, without recurrence after drug withdrawal on day +68), an episode of uncharacterized low-grade febrile upper airway illness with self-limited azotemia (creatine levels increased to 1.52 mg/dL on day +55, but returned to a baseline level of 0.48 mg/dL 3 days later), and right-side epididymo-orchitis 6 months post-transplant that was resolved via a short course of orally administered cephalosporin. No other significant infections, GVHD, sinusoidal obstruction syndrome, respiratory distress, or other organ dysfunctions were encountered.
However, Graves’ disease arose dramatically 10 months post-transplant, corresponding to 1 month after tapering and discontinuation of cyclosporine treatment (Figure 2). Graves’ disease was diagnosed when the boy presented with restlessness, rapid speech, poor sleep, orbital swelling, and persistent tachycardia (resting pulse of up to 130 beats/min compared to a baseline of 90–100). An investigation of thyroid function revealed markedly elevated free thyroxin (fT4; >7.77 ng/dL, normal range 0.8–2.3) and total triiodothyronine (T3; 488.90 ng/dL, normal range 90–240), but undetectable thyroid-stimulating hormone (TSH; <0.005 μIU/L, normal range 0.7–6.4). Ultrasonography of the thyroid showed heterogeneous parenchymal echotexture with hyperemia throughout the gland (Figure 3), compatible with Graves’ disease. The patient also displayed markedly elevated levels of antibodies to thyroglobulin (479.0 IU/mL, normal < 115), thyroid peroxidase (293.3 IU/mL, normal < 5.61), and thyrotropin receptors (64.0%, normal 0.0–14.0%), all compatible with the diagnosis of Graves’ disease. The patient’s post-transplant course of immune reconstitution was also affected due to downregulation of lymphocyte subsets soon after Graves’ disease diagnosis, which was more prominent for non-B cell lineages (Figure 2). Immunoglobulin levels remained within normal ranges throughout the post-transplant period (data not shown).
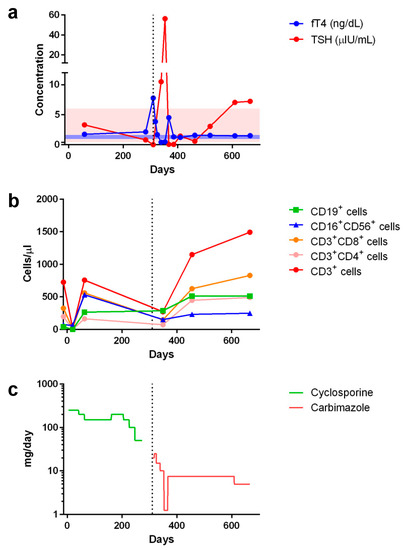
Figure 2.
Peri-transplant changes in serum concentrations of (a) free thyroxin (fT4) and thyroid stimulating hormone (TSH) as well as (b) lymphocyte subset counts in response to (c) cyclosporine and carbimazole treatments (showing the comparative dosages). Shaded areas in (a) indicate normal ranges of fT4 (blue, 0.925–1.615 ng/dL) and TSH (pink, 0.394–6.00 μIU/mL). Day 0 indicates the day of peripheral blood stem cell infusion. Vertical dotted line indicates when Graves’ disease was diagnosed and respective treatment started.
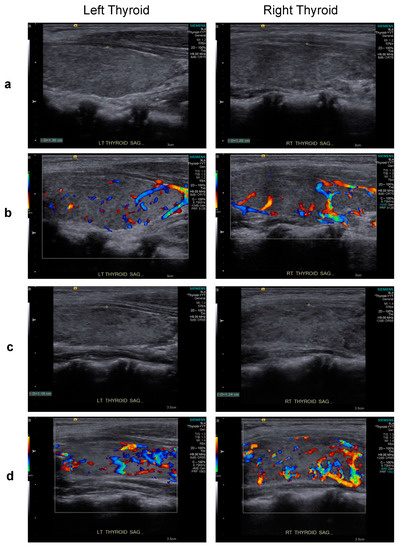
Figure 3.
Upon presentation for hyperthyroidism, thyroid sonography of the patient revealed thyroid dimensions at the upper limit of the normal range and mildly heterogeneous parenchymal echotexture (grayscale sagittal images) (a) and hyperemia throughout the gland (color Doppler images) (b), features characteristic of Graves’ disease. A similar pattern of heterogeneous parenchymal echotexture (c) and hyperemia (d) were noted after 5 months of carbimazole treatment.
In investigating the patient’s genetic predisposition to Graves’ disease, we identified a FLT3 c.2500C>T; p.(Arg834Ter) mutation in both the patient and donor (Figure 1a–c). This mutation occurs at a rare population frequency (Figure 1b) and is predicted to elicit premature termination in tyrosine domain 2, indicating that it may result in a truncated protein. Since the FLT3 stop mutation rs76428106-C, which also results in a truncated protein, has been linked to autoimmune thyroid disease [12], in April 2022, we conducted relevant tests for the donor, which revealed normal telomere length (Figure 1d), fT4 of 1.68 ng/dL, T3 of 121.20 ng/dL, TSH of 1.740 μIU/L, nondetectable antinuclear antibody, normal levels of antibodies to thyroglobulin (19.7 IU/mL), and thyrotropin receptors (6.0%), but borderline elevated antithyroid peroxidase antibodies (7.8 IU/mL). Susceptibility to Graves’ disease among different ethnic groups has been associated with increased frequencies of specific HLA haplotypes [13,14,15,16,17,18,19,20,21], and we identified the donor as carrying HLA-B*46:01 and HLA-DRB1*09:01 and the patient as carrying HLA-C*07 (Table 1). Neither the patient nor the donor hosted HLA haplotypes reported to be protective against Graves’ disease, such as HLA-DRB1*07:01 and HLA-DQA1*02:01 (DR7) (data not shown) [18,19].

Table 1.
HLA haplotypes of the patient and donor (his father) compared to those reportedly susceptible to Graves’ disease.
Initiating and titrating carbimazole (antithyroid) therapy was begun immediately upon detecting hyperthyroidism, but the trialed rapid tapering failed, as shown in Figure 2, and the boy now requires an oral dosage of 2.5 mg carbimazole twice daily to maintain his euthyroid status. Moreover, follow-up thyroid ultrasonography at 5 months (Figure 3) and 1 year (data not shown) of carbimazole treatment revealed characteristics similar to those at the time of Graves’ disease diagnosis. Follow-up at 6 and 12 months of carbimazole treatment, respectively, indicated that levels of antithyroglobulin antibodies (1062.0 and >4000 IU/mL), thyroid peroxidase (214.6 and 632.1 IU/mL), and thyrotropin receptors (54.0% and 24.0%) remained high compared to those measured when Graves’ disease was diagnosed. No proptosis, periorbital edema, scleral injection, or lid retraction was observed, evidencing inactive or mild orbitopathy. Furthermore, there were no indications of dermatopathy or acropathy. Absolute numbers of lymphocyte subsets recovered during carbimazole treatment (Figure 2), indicating a revival of immune reconstitution. The patient’s telomere lengths improved 1.5 years post-transplant compared to those of a pre-transplant sample (Figure 1d). The boy has returned to school and has remained transfusion-independent since August 2020, with hematological assessment in 2022 revealing a neutrophil count of 2.07–2.95 × 109/L, hemoglobin of 13.0–13.8 mg/dL, and a platelet count of 204–209 × 109/L.
3. Discussion
Here, we report the case of a young boy diagnosed with aplastic anemia who developed Graves’ disease after rATG-containing immunosuppressive treatments and haploidentical HSCT from his father. Co-occurrence of aplastic anemia and Graves’ disease, both rare disorders that display similar autoimmune pathogeneses, has been reported previously on several occasions, albeit under different circumstances. For instance, aplastic anemia can arise after Graves’ disease and with or without administration of antithyroid medication [22,23]. Alternatively, Graves’ disease may develop after aplastic anemia, with different mechanisms having been proposed [24,25,26,27]. For example, rATG, which has proven effective in treating steroid-resistant Graves’ orbitopathy [28], has been implicated in promoting the development of Graves’ disease following treatment for aplastic anemia with and without HSCT [24,26]. Adoptive transfer of a pathogenic clone from a donor suffering Graves’ disease [6,7,8], as well as the process associated with GVHD [9], have been postulated as plausible mechanisms by which Graves’ disease evolves upon allogeneic HSCT for aplastic anemia. Nevertheless, neither of those proposed mechanisms seem applicable in the case reported herein; although the donor did display a low titer of antithyroid peroxidase, he exhibited a euthyroid state and an absence of other tested autoantibodies. Indeed, Graves’ disease may evolve 10 months to 8 years following HSCT through variable conditioning regimens, stem cell sources, treatment types, and with or without GVHD (Table 2). We believe that HSCT platforms must lower the threshold level of stimulating anti-thyroid autoimmunity, meaning that, in association with other predisposing factors, Graves’ disease evolves more frequently during immune reconstitution. Interestingly, in our patient’s case, reconstitution of lymphocyte subsets was transiently downregulated as Graves’ disease evolved, as shown in Figure 2.
Table 2.
Characteristics of patients who have developed Graves’ disease following allogeneic HSCT for severe aplastic anemia.
The pathogenesis of post-HSCT autoimmunity is believed to be multifactorial, involving genetic, infectious, hormonal, and other environmental factors [29]. Many variants of genes involved in immune responses (e.g., HLA, PTPN22, CTLA4, and IL2RA), thyroid function (e.g., TSHR, FOXE1), and other processes (e.g., LPP, TRIB2) have been linked to autoimmune thyroid dysfunctions [3]. A large single-center study focused on post-HSCT thyroid autoimmunity, including Graves’ disease and Hashimoto thyroiditis, reported a 2.9% 5-year actuarial rate for autoimmune thyroid dysfunction post-allogeneic HSCT [20]. Thus, investigation of the genetic factors potentially contributing to the development of Graves’ disease in our patient was warranted. We identified germline variants of FLT3 (carried by both the patient and his HSCT donor) and SAMD9L (carried solely by the patient), as well as HLA-B*46:01 and HLA-DRB1*09:01 (carried by the donor) and HLA-C*07 (carried by the patient) haplotypes, together potentially representing genetic predispositions for the evolution of Graves’ disease post-HSCT treatment for aplastic anemia.
FLT3 encodes fms-related tyrosine kinase 3, a receptor that regulates differentiation and proliferation of hematopoietic progenitor and dendritic cells [30,31]. Notably, mutation at FLT3 residue D835 has been linked to an increased risk of developing acute myeloid leukemia [32,33]. Both our patient and his father carried a germline FLT3 p.R834* variant, which introduces a premature stop mutation at tyrosine kinase domain 2 and thus represents a loss-of-function germline mutation. Interestingly, the germline intronic rs76428106-C variant of FLT3, also representing a loss-of-function mutation, engenders the greatest risk of autoimmune thyroid disease [12]. It has been proposed that this latter intronic variant introduces a novel splice site to curtail FLT3 protein length, resulting in a compensatory increase in levels of its ligand so that it also behaves like a gain-of-function mutation [12]. It would be illuminating to establish if the germline FLT3 p.R834* variant identified in our patient induces similar effects.
Specific HLA alleles have been associated with incidences of Graves’ disease in different ethnic populations [13,14,15,16,17,18,19,20,21]. In Table 1, we summarize HLA haplotypes reportedly susceptible to Graves’ disease in various ethnic groups. Our HSCT donor possesses HLA-B*46:01 and HLA-DRB1*09:01 haplotypes, both of which have been linked to de novo Graves’ disease and/or to post-HSCT autoimmune thyroid disease in the ethnic Chinese Han population [13,14,20]. Our patient also carries the HLA-C*07 haplotype, although Graves’ disease susceptibility associated with this haplotype has only been documented previously for Caucasians [18,19].
Consistent with reported pediatric myelodysplastic and bone marrow failure syndromes [34,35], our patient also carried a germline SAMD9L p.C1267F mutation, which is ultra-rare in terms of population allele frequency (Figure 1b). Gain-of-function SAMD9L mutations have been shown to impair multiple pathways and result in enhanced proliferative inhibition and repression of protein translation elongation in primary hematopoietic cells [35,36]. HSCT treatment, either from unrelated cord blood or matched sibling/unrelated bone marrow, has been reported as successful for four of six cases of pediatric myelodysplastic syndromes with germline SAMD9L mutations, including one with reduced intensity conditioning [37]. In our patient, bone marrow failure was successfully corrected by nonmyeloablative haploidentical PBSC transplantation from his father, who does not have the SAMD9L p.C1267F variant. It remains to be determined how interactions between the host cells carrying SAMD9L p.C1267F/FLT3 p.R834* mutations and the donor cells carrying the FLT3 p.R834* mutation resulted in Graves’s disease evolving post-HSCT. The donor, who carries both FLT3 p.R834* mutation and HLA-B*46:01/HLA-DRB1*09:01 haplotypes, has not shown any manifestation of Graves’s disease to date.
In conclusion, the interplay between genetic (the FLT3 variant carried by the patient and the FLT3/HLA-B*46:01/HLA-DRB1*09:01 variants carried by his PBSC donor) and environmental factors arising from the process of immune reconstitution post-haploidentical PBSC transplantation may prompt loss of immune tolerance to thyroid autoantigens, which may have resulted in the development of Graves’ disease in our patient.
4. Materials and Methods
4.1. Identification of Variants by Whole-Exome Sequencing (WES)
Informed consent was explained and obtained from both the patient and his parents prior to this study. Peripheral blood from the patient and his parents was collected before PBSC transplantation of the patient. One additional peripheral blood sample was obtained from the patient 1.5 years after PBSC transplantation. Genomic DNA from whole blood was isolated using a Wizard genomic DNA purification kit (Promega, Madison, WI, USA) for WES. A DNA library was prepared using a Roche KAPA HyperExome kit (Roche, Basel, Switzerland), and sequencing was performed using a NovaSeq 6000 system (Illumina, San Diego, CA, USA). The sequencing data were aligned to the GRCh37 (hg19) reference genome to identify genetic variants. Mutations were verified by Sanger sequencing. A fragment of the SAMD9L gene containing the SAMD9L c.3800G>T mutation was amplified by PCR using 5′-TCTCCTAGAAGCTGCGGAAA-3′ and 5′-TGCTGCAGTAGGAAGGCATA-3′ primers, with the former also used for Sanger sequencing. A fragment of the FLT3 gene containing the FLT3 c.2500C>T mutation was amplified by PCR using 5′-CACAAAGAACTGCAGCCACC-3′ and 5′-GCCCAAGGACAGATGTGATG-3′ primers. The DNA fragment of the correct size (559 bp) was isolated from the agarose gel for Sanger sequencing using the 5′-CACAAAGAACTGCAGCCACC-3′ primer.
4.2. Determination of HLA Haplotypes
Clinical HLA types including low-resolution 2-digit (the donor) and high-resolution 4-digit (the patient) were obtained by Micro SSP™ HLA DNA Typing Trays from LinkSeqTM HLA-ABCDRDQB1 kit with Applied BiosystemsTM Standard Block (One Lambda, Inc. West Hills, CA, USA). Next-generation sequencing-based HLA genotypes were extracted from donor and patient WES data using the HLAscan application [38]. In brief, WES sequence reads were aligned with human genome reference hg19 (UCSC browser). Each read in aligned results was classified into a different HLA gene classes via HLAscan application using exon sequence from ImMunoGeneTics project (IMGT)/HLA database (https://www.ebi.ac.uk/ipd/imgt/hla/ (accessed on 4 December 2019)). Reads were aligned with exons 2, 3, 4, and 5 of HLA class I genes, and exons 2, 3, and 4 of HLA class II genes. Allele types were then determined based on the numbers and distribution patterns of the reads for each reference target.
4.3. Terminal Restriction Fragment Assay
Leukocyte telomere length was analyzed using terminal restriction fragment assay, as described previously [39]. In brief, leukocyte genomic DNA was digested by Rsa I and Hinf I restriction enzymes and then resolved by pulsed-field gel electrophoresis. Telomeric DNA was detected by in-gel hybridization using a [32P]-labeled telomeric probe.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/ijms23169494/s1.
Author Contributions
P.P.I., R.-L.C. and L.-Y.C. designed the study. P.P.I., L.-H.F., Y.-L.S., K.-C.T., M.-T.L., L.-Y.J., L.-Y.C. and R.-L.C. carried out the experiments and performed data analysis. P.P.I., R.-L.C. and L.-Y.C. wrote the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Family Association for Children with Serious Illness, Taiwan (Grant no.: FACSI 211001).
Institutional Review Board Statement
The study was conducted in accordance with the Declaration of Helsinki, and approved by the Institutional Review Board (or Ethics Committee) of Koo Foundation Sun Yat-Sen Cancer Center (protocol code 20210625A/20324 and date of approval 26 January 2022).
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Acknowledgments
The authors thank Hsin-Nan Lin from the bioinformatics core at the Institute of Molecular Biology, Academia Sinica, Taipei, Taiwan, for data analysis.
Conflicts of Interest
The authors declare no conflict of interest.
References
- De Leo, S.; Lee, S.Y.; Braverman, L.E. Hyperthyroidism. Lancet 2016, 388, 906–918. [Google Scholar] [CrossRef]
- Smith, T.J.; Hegedus, L. Graves’ Disease. N. Engl. J. Med. 2016, 375, 1552–1565. [Google Scholar] [CrossRef]
- Boguslawska, J.; Godlewska, M.; Gajda, E.; Piekielko-Witkowska, A. Cellular and molecular basis of thyroid autoimmunity. Eur. Thyroid J. 2022, 11, e210024. [Google Scholar] [CrossRef]
- Tomer, Y. Mechanisms of autoimmune thyroid diseases: From genetics to epigenetics. Annu. Rev. Pathol. 2014, 9, 147–156. [Google Scholar] [CrossRef]
- Sarantopoulos, S.; Ritz, J. Aberrant B-cell homeostasis in chronic GVHD. Blood 2015, 125, 1703–1707. [Google Scholar] [CrossRef]
- Drabko, K.; Winnicka, D.; Gaworczyk, A.; Ben-Skowronek, I.; Skomra, D.; Kowalczyk, J.R. Donor origin of Graves disease in a BMT recipient: Evidence from FISH studies of thyroid tissue. Bone Marrow Transpl. 2006, 37, 789–791. [Google Scholar] [CrossRef][Green Version]
- Holland, F.J.; McConnon, J.K.; Volpe, R.; Saunders, E.F. Concordant Graves’ disease after bone marrow transplantation: Implications for pathogenesis. J. Clin. Endocrinol. Metab. 1991, 72, 837–840. [Google Scholar] [CrossRef]
- Ichihashi, T.; Yoshida, H.; Kiyoi, H.; Fukutani, H.; Kubo, K.; Yamauchi, T.; Naoe, T.; Ohno, R. Development of hyperthyroidism in donor and recipient after allogeneic bone marrow transplantation. Bone Marrow Transpl. 1992, 10, 397–398. [Google Scholar]
- Mulligan, S.P.; Joshua, D.E.; Joasoo, A.; Kronenberg, H. Autoimmune hyperthyroidism associated with chronic graft-versus-host disease. Transplantation 1987, 44, 463–464. [Google Scholar]
- Paketci, A.; Demir, K.; Tufekci, O.; Acar, S.; Abaci, A.; Yilmaz, S.; Bober, E. Graves’ disease following allogenic hematopoietic stem cell transplantation for severe aplastic anemia: Case report and literature review. J. Pediatr. Endocrinol. Metab. 2018, 31, 589–593. [Google Scholar] [CrossRef]
- Shimazaki, S.; Kazukawa, I.; Minagawa, M. Autoimmune thyroid disease following hematopoietic stem cell transplantation in childhood cancer survivors. Clin. Pediatr. Endocrinol. 2022, 31, 54–58. [Google Scholar] [CrossRef]
- Saevarsdottir, S.; Olafsdottir, T.A.; Ivarsdottir, E.V.; Halldorsson, G.H.; Gunnarsdottir, K.; Sigurdsson, A.; Johannesson, A.; Sigurdsson, J.K.; Juliusdottir, T.; Lund, S.H.; et al. FLT3 stop mutation increases FLT3 ligand level and risk of autoimmune thyroid disease. Nature 2020, 584, 619–623. [Google Scholar] [CrossRef]
- Chen, P.L.; Fann, C.S.; Chu, C.C.; Chang, C.C.; Chang, S.W.; Hsieh, H.Y.; Lin, M.; Yang, W.S.; Chang, T.C. Comprehensive genotyping in two homogeneous Graves’ disease samples reveals major and novel HLA association alleles. PLoS ONE 2011, 6, e16635. [Google Scholar] [CrossRef]
- Hawkins, B.R.; Ma, J.T.; Lam, K.S.; Wang, C.C.; Yeung, R.T. Association of HLA antigens with thyrotoxic Graves’ disease and periodic paralysis in Hong Kong Chinese. Clin. Endocrinol. 1985, 23, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Vita, R.; Lapa, D.; Trimarchi, F.; Vita, G.; Fallahi, P.; Antonelli, A.; Benvenga, S. Certain HLA alleles are associated with stress-triggered Graves’ disease and influence its course. Endocrine 2017, 55, 93–100. [Google Scholar] [CrossRef]
- Badenhoop, K.; Walfish, P.G.; Rau, H.; Fischer, S.; Nicolay, A.; Bogner, U.; Schleusener, H.; Usadel, K.H. Susceptibility and resistance alleles of human leukocyte antigen (HLA) DQA1 and HLA DQB1 are shared in endocrine autoimmune disease. J. Clin. Endocrinol. Metab. 1995, 80, 2112–2117. [Google Scholar] [CrossRef]
- Ban, Y.; Davies, T.F.; Greenberg, D.A.; Concepcion, E.S.; Osman, R.; Oashi, T.; Tomer, Y. Arginine at position 74 of the HLA-DR beta1 chain is associated with Graves’ disease. Genes Immun. 2004, 5, 203–208. [Google Scholar] [CrossRef]
- Chen, Q.Y.; Huang, W.; She, J.X.; Baxter, F.; Volpe, R.; Maclaren, N.K. HLA-DRB1*08, DRB1*03/DRB3*0101, and DRB3*0202 are susceptibility genes for Graves’ disease in North American Caucasians, whereas DRB1*07 is protective. J. Clin. Endocrinol. Metab. 1999, 84, 3182–3186. [Google Scholar] [CrossRef][Green Version]
- Simmonds, M.J.; Howson, J.M.; Heward, J.M.; Cordell, H.J.; Foxall, H.; Carr-Smith, J.; Gibson, S.M.; Walker, N.; Tomer, Y.; Franklyn, J.A.; et al. Regression mapping of association between the human leukocyte antigen region and Graves disease. Am. J. Hum. Genet. 2005, 76, 157–163. [Google Scholar] [CrossRef]
- Au, W.Y.; Lie, A.K.; Kung, A.W.; Liang, R.; Hawkins, B.R.; Kwong, Y.L. Autoimmune thyroid dysfunction after hematopoietic stem cell transplantation. Bone Marrow Transpl. 2005, 35, 383–388. [Google Scholar] [CrossRef]
- Mangklabruks, A.; Cox, N.; DeGroot, L.J. Genetic factors in autoimmune thyroid disease analyzed by restriction fragment length polymorphisms of candidate genes. J. Clin. Endocrinol. Metab. 1991, 73, 236–244. [Google Scholar] [CrossRef]
- Das, P.K.; Wherrett, D.; Dror, Y. Remission of aplastic anemia induced by treatment for Graves disease in a pediatric patient. Pediatr. Blood Cancer 2007, 49, 210–212. [Google Scholar] [CrossRef] [PubMed]
- Thomas, D.; Moisidis, A.; Tsiakalos, A.; Alexandraki, K.; Syriou, V.; Kaltsas, G. Antithyroid drug-induced aplastic anemia. Thyroid 2008, 18, 1043–1048. [Google Scholar] [CrossRef] [PubMed]
- Choi, I.S.; Kim, H.K.; Han, D.K.; Baek, H.J.; Jang, H.I.; Kim, C.J.; Kook, H. Graves disease following rabbit antithymocyte globulin treatment of severe aplastic anemia in a Korean child. Korean J. Pediatr. 2015, 58, 267–269. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.; Goldman, J. Severe aplastic anaemia and Grave’s disease in a paediatric patient. Br. J. Haematol. 2002, 118, 327–329. [Google Scholar] [CrossRef]
- Todd, A.; Todd, J. Graves’ disease following successful treatment of severe aplastic anaemia with antilymphocyte globulin. Clin. Lab. Haematol. 1999, 21, 69–70. [Google Scholar] [CrossRef]
- Zhang, W.; Shao, Z. Grave’s disease following aplastic anemia: Predisposition or coincidence? Indian Pediatr. 2015, 52, 347–348. [Google Scholar]
- Sarnat-Kucharczyk, M.; Swierkot, M.; Handzlik, G.; Kulawik, G.; Jagoda, K.; Grochola-Malecka, I.; Fryzewska, J.; Mrukwa-Kominek, E.; Chudek, J. Antithymocyte globulin as second-line therapy in Graves orbitopathy-preliminary results from a prospective single-center study. Front. Endocrinol. 2022, 13, 871009. [Google Scholar] [CrossRef]
- Sherer, Y.; Shoenfeld, Y. Autoimmune diseases and autoimmunity post-bone marrow transplantation. Bone Marrow Transpl. 1998, 22, 873–881. [Google Scholar] [CrossRef]
- Brasel, K.; Escobar, S.; Anderberg, R.; de Vries, P.; Gruss, H.J.; Lyman, S.D. Expression of the flt3 receptor and its ligand on hematopoietic cells. Leukemia 1995, 9, 1212–1218. [Google Scholar]
- Turner, A.M.; Lin, N.L.; Issarachai, S.; Lyman, S.D.; Broudy, V.C. FLT3 receptor expression on the surface of normal and malignant human hematopoietic cells. Blood 1996, 88, 3383–3390. [Google Scholar] [CrossRef] [PubMed]
- Abu-Duhier, F.M.; Goodeve, A.C.; Wilson, G.A.; Care, R.S.; Peake, I.R.; Reilly, J.T. Identification of novel FLT-3 Asp835 mutations in adult acute myeloid leukaemia. Br. J. Haematol. 2001, 113, 983–988. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, Y.; Kiyoi, H.; Nakano, Y.; Suzuki, R.; Kodera, Y.; Miyawaki, S.; Asou, N.; Kuriyama, K.; Yagasaki, F.; Shimazaki, C.; et al. Activating mutation of D835 within the activation loop of FLT3 in human hematologic malignancies. Blood 2001, 97, 2434–2439. [Google Scholar] [CrossRef]
- Sahoo, S.S.; Pastor, V.B.; Goodings, C.; Voss, R.K.; Kozyra, E.J.; Szvetnik, A.; Noellke, P.; Dworzak, M.; Stary, J.; Locatelli, F.; et al. Clinical evolution, genetic landscape and trajectories of clonal hematopoiesis in SAMD9/SAMD9L syndromes. Nat. Med. 2021, 27, 1806–1817. [Google Scholar] [CrossRef]
- Thomas, M.E., 3rd; Abdelhamed, S.; Hiltenbrand, R.; Schwartz, J.R.; Sakurada, S.M.; Walsh, M.; Song, G.; Ma, J.; Pruett-Miller, S.M.; Klco, J.M. Pediatric MDS and bone marrow failure-associated germline mutations in SAMD9 and SAMD9L impair multiple pathways in primary hematopoietic cells. Leukemia 2021, 35, 3232–3244. [Google Scholar] [CrossRef] [PubMed]
- Tesi, B.; Davidsson, J.; Voss, M.; Rahikkala, E.; Holmes, T.D.; Chiang, S.C.C.; Komulainen-Ebrahim, J.; Gorcenco, S.; Nilsson, A.R.; Ripperger, T.; et al. Gain-of-function SAMD9L mutations cause a syndrome of cytopenia, immunodeficiency, MDS, and neurological symptoms. Blood 2017, 129, 2266–2279. [Google Scholar] [CrossRef]
- Ahmed, I.A.; Farooqi, M.S.; Vander Lugt, M.T.; Boklan, J.; Rose, M.; Friehling, E.D.; Triplett, B.; Lieuw, K.; Saldana, B.D.; Smith, C.M.; et al. Outcomes of hematopoietic cell transplantation in patients with germline SAMD9/SAMD9L mutations. Biol Blood Marrow Transpl. 2019, 25, 2186–2196. [Google Scholar] [CrossRef]
- Ka, S.; Lee, S.; Hong, J.; Cho, Y.; Sung, J.; Kim, H.N.; Kim, H.L.; Jung, J. HLAscan: Genotyping of the HLA region using next-generation sequencing data. BMC Bioinform. 2017, 18, 258. [Google Scholar] [CrossRef]
- Chen, L.Y.; Majerska, J.; Lingner, J. Molecular basis of telomere syndrome caused by CTC1 mutations. Genes Dev. 2013, 27, 2099–2108. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).